

Abstract
There are three classes of promoters for flagellar operons in Salmonella. Class 2 promoters are transcribed by σ70 RNA polymerase in the presence of an essential activator, FlhD4C2, and activated by an auxiliary regulator, FliZ. Class 3 promoters are transcribed by σ28 RNA polymerase and repressed by an anti-σ28 factor, FlgM. σ28 (FliA) and FliZ are encoded by the fliA and fliZ genes, respectively, which together constitute an operon transcribed in this order. This operon is transcribed from both class 2 and class 3 promoters, suggesting that it should be activated by its own product, σ28, even in the absence of FlhD4C2. However, σ28-dependent transcription occurs in vivo only in the presence of FlhD4C2, indicating that transcription from the class 2 promoter is a prerequisite to that from the class 3 promoter. In this study, we examined the effects of variously modified versions of the fliA regulatory region on transcription and translation of the fliA gene. We showed that FliA is not significantly translated from the class 3 transcript. In contrast, the 5′-terminal AU-rich sequence found in the class 2 transcript confers efficient fliA translation. Replacement of the Shine-Dalgarno sequence of the fliA gene with a better one improved fliA translation from the class 3 transcript. These results suggest that the 5′-terminal AU-rich sequence of the class 2 transcript may assist ribosome binding. FliZ was shown to be expressed from both the class 2 and class 3 transcripts.
INTRODUCTION
Bacterial flagellum consists of three structural parts, a basal body, a hook, and a filament. More than 50 genes are specifically required for flagellar formation and function in Salmonella enterica serovar Typhimurium and Escherichia coli (1, 30). These flagellar genes are organized into at least 15 operons (21, 33), and their expression forms a highly organized cascade called a flagellar regulon (22, 32). There are three classes of promoters responsible for transcription of the flagellar operons (28, 32). In the flagellar regulon, flhDC is the sole operon under the control of the class 1 promoter, which is transcribed by σ70 RNA polymerase and responds to various global regulators such as cyclic AMP-Crp, H-NS, and RcsB (25, 50, 52). The class 1 promoter is also under an autogenous control from FlhD and FlhC (25). The class 2 promoter is transcribed by σ70 RNA polymerase in the presence of the class 1 gene products FlhD and FlhC (15, 36). These two proteins assemble into an FlhD4C2 heterohexamer, which binds to the FlhD4C2 binding site located upstream of the class 2 promoter (15, 36, 47). The FlhD4C2 binding site shows an imperfect symmetry comprising two 17- or 18-bp inverted repeats, called the FlhD4C2 box, separated by a 10- or 11-bp spacer (7, 35, 49). The class 2 promoter is also under positive and negative control from FliZ and FliT, respectively, both of which are encoded by the genes within the flagellar regulon (28). FliT was shown to act as an anti-FlhD4C2 factor which inhibits FlhD4C2 from binding to the class 2 promoter (51). Recently, FliZ was shown to be a transcriptional repressor of the nonflagellar gene ydiV (46), which encodes another anti-FlhD4C2 factor responsible for repression of flagellar expression in poor medium (45). Most of the genes transcribed from the class 2 promoter are responsible for construction of the hook-basal body structure. The class 3 promoter is transcribed by σ28 RNA polymerase and under negative control of an anti-σ28 factor, FlgM (39, 40). The σ28 and anti-σ28 factors are encoded by the fliA and flgM genes, respectively, both of which are within the flagellar regulon (10, 14, 17, 24, 31, 39, 40). Class 3 promoters transcribe the genes for filament assembly, flagellar rotation, and chemotaxis.
The fliA and fliZ genes together constitute an operon and are transcribed in this order (Fig. 1A) (14, 38). The upstream region of this operon contains both class 2 and class 3 promoters, called P1 and P2, respectively. Downstream of these two genes exists another gene, STM1954, which can be transcribed together with these two genes. However, STM1954 is also expressed from its own nonflagellar promoter and encodes a protein unrelated to flagellar formation and function (14, 28). Therefore, though this gene has been called fliY (38), it is unlikely to be a member of the flagellar genes.
Fig. 1.

Expression control of the fliAZ operon and nucleotide sequence of its regulatory region. (A) The structure of the fliAZ operon on the Salmonella chromosome is drawn with the model of its expression control according to previously published reports (1, 14, 27, 28, 31, 45, 46). STM1954 has also been called fliY (38). (B) The nucleotide sequence of the regulatory region of the fliAZ operon is shown with the translation initiation codon of the fliA gene being numbered 1. Various control signals of the fliA gene are underlined. (C) The structures of the regulatory region of the fliAZ operon in the various mutants constructed in this study are shown with the nucleotide sequences of the 5′ ends of their transcripts. Closed boxes, sequences responsible for the P1 promoter; open boxes, sequences responsible for the P2 promoter. Altered or inserted nucleotides in the mutant constructs are underlined. ×××, 3-nucleotide substitution mutation (GCC to CGT) which inactivates the P2 promoter. SD sequences are written in boldface.
In the fliAZ operon, the P1 and P2 promoters are overlapping with each other, as shown in Fig. 1B. Primer extension analysis of mRNAs extracted from the wild-type cells revealed that this operon is transcribed from both of these promoters in vivo (14). The transcript from the P1 promoter (P1 mRNA) was present in the fliA mutant and absent in the flhD mutant, whereas the transcript from the P2 promoter (P2 mRNA) was absent from both the fliA and flhD mutants. In vitro transcription analysis with purified proteins showed that the P1 promoter was transcribed by σ70 RNA polymerase in the presence of the FlhD4C2 complex, whereas the P2 promoter was transcribed in the presence of σ28 RNA polymerase (14). The P2 transcript was produced in vitro even in the absence of FlhD4C2 (14), indicating that transcription from the P2 promoter by σ28 RNA polymerase does not require FlhD4C2. This raised the possibility that, in the absence of the anti-σ28 factor FlgM, the fliAZ operon may be autogenously activated by FliA in vivo even in the flhDC-negative background. However, the expression of this operon was never observed in vivo in the flhDC mutant, irrespective of the flgM genotype (27). Therefore, we hypothesize that Salmonella cells may have some mechanism suppressing FliA-mediated autogenous activation of the fliAZ operon. The work described here was carried out to test this hypothesis.
Here we report evidence that P2 mRNA is translationally inert in the expression of the fliA gene. We show that FliA is expressed efficiently only from P1 mRNA, whereas FliZ is expressed from both P1 and P2 mRNAs. This indicates that the FlhD4C2-mediated transcription is a prerequisite to FliA-mediated autogenous activation in the expression of the fliAZ operon. In the course of this study, Wozniak et al. reported the observation that the mutant defective in the P1 promoter did not express the fliAZ operon and could not support swimming and swarming motility in Salmonella (48). Our findings reported here can also account for their observation.
MATERIALS AND METHODS
Bacterial strains, plasmids, chemicals, and culture media.
The Salmonella strains and plasmids used in this study are listed in Tables 1 and 2, respectively. All the Salmonella strains used were derivatives of standard laboratory strain LT2. Their construction procedures are described below. P22-mediated transduction was performed as described previously (33). Unless otherwise specified, all the chemicals and DNA enzymes used were purchased from Nacalai Tesque (Kyoto, Japan) and Toyobo (Osaka, Japan), respectively. Oligonucleotide primers used were purchased from Life Technologies (Tokyo, Japan) and are summarized in Table 3. The rich and poor media used were Luria broth (LB) and M9 minimal medium (37) supplemented with 0.2% glycerol and 0.2% Casamino Acids (MGC), respectively (45). Hard and motility agar plates were prepared by adding 1.2% and 0.25% agar (Shoei, Tokyo, Japan), respectively, to LB or MGC. Ampicillin, kanamycin, and tetracycline were used at final concentrations of 50, 50, and 20 μg/ml, respectively. Where required, arabinose or isopropyl-β-d-thiogalactopyranoside (IPTG) was added to the culture medium at final concentrations of 0.2% and 0.1 mM, respectively.
Table 1.
Strains used in this study
| Strain | Relevant characteristics | Source or reference |
|---|---|---|
| KK1004 | LT2 Δ(hin-fljBA) ΔFels-2 | 29, 33 |
| KK1110 | KK1004 fliC-lacamp | 27 |
| KK1004hDC | KK1004 flhD2140::Tn10 | 33, 46 |
| KK1004gM | KK1004 ΔflgM::FRT (in frame) | This study |
| KK1004iA | KK1004 ΔfliA::FRT (in frame) | 46 |
| KK1004iZ | KK1004 ΔfliZ::FRT (in frame) | 46 |
| KK1004iAZ | KK1004 ΔfliAZ::FRT (in frame) | 46 |
| KK1004Δlon | KK1004 Δlon::FRT | This study |
| KK1004AF | KK1004 fliA-3×FLAG FRT | This study |
| KK1004AFZF | KK1004 fliA-3×FLAG FRT fliZ-3×FLAG FRT::kan::FRT | This study |
Table 2.
Plasmids used in this study
| Plasmid | Descriptiona | Source or reference |
|---|---|---|
| pKD4 | FRT-kan-FRT template, Ampr | 9 |
| pKD13 | FRT-kan-FRT template, Ampr | 9 |
| pKD46 | Phage Red expression plasmid, Ampr | 9 |
| pCP20 | Flp expression plasmid, Ampr Catr | 9 |
| pSUB11 | 3×FLAG tag FRT-kan-FRT template, Ampr | 44 |
| pKK1064 | pBR322 fliB-IS200-fliA-fliZ′ | 39 |
| pBAD18kan | ParaBAD expression vector, Kanr | 11 |
| pBAD-P1-fliA | pBAD18kan fliA (−155 to +720), P2 negative (−33CGT−31) | This study |
| pQE80L | His6-tagged protein expression vector, Ampr | Qiagen |
| pQE-fliA | pQE80L fliA (+1 to +720), His6-FliA | This study |
| pTrc99A | Ptrc expression vector, Ampr | 2 |
| pTrc99K | pTrc99A amp::kan, Amps Kanr | Laboratory stock |
| pTrc-fliA | pTrc99K fliA (−19 to +720) | This study |
| pTrc-P1/P2-fliA | pTrc99K fliA (−115 to +720) | This study |
| pTrc-P2-fliA | pTrc99K fliA (−57 to +720) | This study |
| pTrc-P2c-fliA | pTrc99K fliA (−57 to +720), P2 consensus (−52TAAAGTTT−45) | This study |
| pFZY1 | Transcriptional lacZ fusion vector, Ampr | 23 |
| pF-P1/P2 | pFZY1 fliA (−115 to +399) | pFZY1-PfliA-lacZ in reference 46 |
| pF-P1/P2-fliA | pFZY1 fliA (−115 to +720) | This study |
| pF-P1-fliA | pFZY1 fliA (−115 to +720), P2 negative (−33CGT−31) | This study |
| pF-P1(P2)-fliA | pFZY1 fliA (−115 to +720), −29 to −19 deleted | This study |
| pF-P2-fliA | pFZY1 fliA (−57 to +720) | This study |
| pF-P2c-fliA | pFZY1 fliA (−57 to +720), P2 consensus (−52TAAAGTTT−45) | This study |
| pF-P2(P1)-fliA | pFZY1 fliA (−57 to +720), −29 to −19 duplicated | This study |
| pF-P1/P2-fliAZ | pFZY1 fliA (−155)-fliZ (+552) | This study |
| pF-P1-fliAZ | pFZY1 fliA (−155)-fliZ (+552), P2 negative (−33CGT−31) | This study |
| pF-P1(P2)-fliAZ | pFZY1 fliA (−155)-fliZ (+552), −29 to −19 deleted | This study |
| pF-P2-fliAZ | pFZY1 fliA (−57)-fliZ (+552) | This study |
| pF-P2(P1)-fliAZ | pFZY1 fliA (−57)-fliZ (+552), −29 to −19 duplicated | This study |
| pMC1403 | Translational lacZ fusion vector, Ampr | 5 |
| pMC-P1 | pMC1403 fliA (−115 to +398), P2 negative (−33CGT−31) | This study |
| pMC-P1(P2) | pMC1403 fliA (−115 to +398), −29 to −19 deleted | This study |
| pMC-P1(GC*) | pMC1403 fliA (−115 to +398), P2 negative (−33CGT−31), GC* (−28CGG−26) | This study |
| pMC-P2 | pMC1403 fliA (−57 to +398) | This study |
| pMC-P2(P1) | pMC1403 fliA (−57 to +398), −29 to −19 duplicated | This study |
| pMC-P2(SD*) | pMC1403 fliA (−57 to +398), 5′ UTR from pQE80L | This study |
The DNA regions inserted in the plasmids are shown with the translation initiation codon of the fliA or fliZ gene being numbered 1.
Table 3.
Oligonucleotide primers used in this study
| Use and primer name | Nucleotide sequence (5′ → 3′) |
|---|---|
| Construction of chromosomal deletion mutants | |
| lon-H1P1 | CAGCTATACTATCTGATTACCTGGCGGACACTAAACTAAGAGAGAGCTCTGTGTAGGCTGGAGCTGCTTC |
| lon-H2P2 | CGAAATAGCCTGCCAGCCCTGTTTTTATTAGCGCTATTTGCGCGAGGTCACATATGAATATCCTCCTTAG |
| flgMH1P1 | TGATGGTAGCTGGCCGCTACAACGTAACCCTCGATGAGGATAAATAAATGGTGTAGGCTGGAGCTGCTTC |
| flgMH2P4 | AGGACGGTGGTCATCTGGTCAAGTATTTCTGACAAACGAGTCATACGCTTATTCCGGGGATCCGTCGACC |
| Construction of chromosomal genes encoding 3×FLAG fusion proteins | |
| fliA-FLAG-Fw | AGTTGCATAGTCAGGCCATCAAACGATTACGCACCAAACTGGGTAAGTTA GACTACAAAGACCATGACGG |
| fliA-FLAG-Rv | CTGGTAGTCTATACGTTGTGCGGCACTTTTCGGGTGCGATCATGCGCGAC CATATGAATATCCTCCTTAG |
| fliZ-FLAG-Fw | AGATTGCGCCCAGACAGAAATCCCCCTTTACCGCCAGTTCTGATATATAT GACTACAAAGACCATGACGG |
| fliZ-FLAG-Rv | GAAGGTTTGCCACGTTTCACCAACACGACTCTGCTACATCTTATGCTTTT CATATGAATATCCTCCTTAG |
| Construction of plasmids | |
| BamHIfliAR | GGGGGATCCCTATAACTTACCCAGTTTGGT |
| BamHIfliAR2 | GGGGGATCCTGACGATACTCCGCAACAGGG |
| fliApF | ATCGTATGCGCCTGTTAGGG |
| fliAP1-P2mRNAF | CGGATAATCATGCCGAACGCAGGGCTGTTT |
| fliAP1-P2mRNAR | AAACAGCCCTGCGTTCGGCATGATTATCCG |
| fliAp2sF | ACGGATAATCATCGTGATAACTCATTTAAC |
| fliAp2sR | GTTAAATGAGTTATCACGATGATTATCCGT |
| fliASDqF | GATAATCATGCCGACGGCTCATTTAACGCA |
| fliASDqR | TCACGATAAATTTCTCCTCTTTAAATGAGT |
| IAE1X | GGGTCTAGACTATAACTTACCCAGTTTGGT |
| IAf1B | GGGGGGATCCGTGAATTCACTGTATACCGC |
| IAr1H | GGGGAACCTTCTATAACTTACCCAGTTTGG |
| ITf1 | CATACTCCCGCCATTCAGAGAAGAAACC |
| IZP4 | GGGGATCCTTAATATATATCAGAACTGG |
| KpnIfliA0 | GGGGGTACCGCTACAGGTTACATAA |
| KpnIfliA1 | GGGGGTACCCTCTGTAGAAACGGAT |
| KpnIfliA1c | GGGGGTACCCTCTAAAGTTTCGGATAATCATGCCG |
| KpnIfliA3 | GGGGGATCCTAACGCAGGGCTGTTT |
| KpnIfliAP2-P1mRNAF | GGGGGTACCCTCTGTAGAAACGGATAATCATGCCGATAACTCATTTATAACTCATTTAACGCAGGGC |
| P1m(taa-cgg)fliAF | GATAATCATGCCGACGGCTCATTTAACGCA |
| P1m(taa-cgg)fliAR | TGCGTTAAATGAGCCGTCGGCATGATTATC |
| SmaIfliA0 | GGGCCCGGGCGCTACAGGTTACATAA |
Construction of chromosomal deletion mutants.
The bacteriophage λ Red recombinase system described by Datsenko and Wanner (9) was used for construction of gene disruption mutants of Salmonella.
A lon disruption mutant, KK1004Δlon, was constructed as follows. The kanamycin resistance (kan) FLP recombination target (FRT) cassette was PCR amplified from pKD4 with primers lon-H1P1 and lon-H2P2, homologous to the 5′ and 3′ flanking regions of the lon gene, respectively. The amplified product was introduced into KK1004 harboring pKD46 by electroporation, and kanamycin-resistant colonies were selected on LB agar plates containing kanamycin and arabinose. After correct replacement of the lon gene by the kan gene cassette was confirmed, this mutation was moved to fresh KK1004 via P22-mediated transduction. The kan gene cassette was removed through site-specific recombination between the flanking FRT sequences by transient exposure of the cells to pCP20.
A mutant with an in-frame deletion of the flgM gene, KK1004gM, was constructed as follows. The kan FRT cassette was PCR amplified from pKD13 with primers flgMH1P1 and flgMH2P4, which possess sequences homologous to the 5′ and 3′ flanking regions of the flgM gene, respectively. The amplified product was introduced into KK1004 harboring pKD46 by electroporation, and kanamycin-resistant colonies were selected as described above. After correct replacement of the flgM gene by the kan gene cassette was confirmed, this mutation was moved to fresh KK1004 and the kan gene cassette was removed as described above.
Construction of strains carrying fusion genes on the chromosome.
Chromosomal genes expressing the C-terminally 3×FLAG-tagged proteins were created according to the method described by Uzzau et al. (44). A DNA fragment encoding a 3×FLAG tag-encoding sequence and the kan gene was PCR amplified from pSUB11 using primers fliA-FLAG-Fw and fliA-FLAG-Rv. The amplified product was introduced into KK1004 harboring pKD46 by electroporation, and kanamycin-resistant colonies were selected. After correct construction of the fliA-3×FLAG gene on the chromosome was confirmed, the kan gene cassette was excised using pCP20 to obtain strain KK1004AF. Next, a DNA fragment encoding the 3×FLAG tag-encoding sequence and the kan gene was PCR amplified from pSUB11 with primers fliZ-FLAG-Fw and fliZ-FLAG-Rv. The amplified product was introduced into KK1004AF harboring pKD46 by electroporation, and kanamycin-resistant colonies were selected. After correct construction of the fliA-3×FLAG and fliZ-3×FLAG genes was confirmed, the DNA region containing these genes was moved to fresh KK1004 to obtain strain KK1004AFZF. This strain expresses both the FliA-3×FLAG and FliZ-3×FLAG proteins.
Construction of FliA expression plasmids.
The fliA coding sequence was PCR amplified from pKK1064 with primers IAf1B and IAr1H. The amplified product was digested with BamHI and HindIII and inserted into the corresponding site of pQE80L to obtain pQE-fliA. On the other hand, a DNA fragment containing the fliA gene was PCR amplified from the genomic DNA of KK1004 with primers KpnIfliA3 and IAE1X. The amplified product was digested with KpnI and XbaI and inserted into the corresponding site of pTrc99K to obtain pTrc-fliA.
Construction of plasmids carrying fliA-lacZ transcriptional fusion genes.
The fliA-lacZ transcriptional fusion genes were constructed on a single-copy plasmid, pFZY1. The structures of the regulatory region of the fliA gene on the recombinant plasmids are summarized in Fig. 1C. Unless otherwise specified, the template used for PCR amplification was the genomic DNA from KK1004.
A DNA fragment containing the fliAZ operon with the P1 and P2 promoters was PCR amplified with primers KpnIfliA0 and IZP4. The amplified product was digested with KpnI and BamHI and inserted into the corresponding site of pFZY1 to obtain pF-P1/P2-fliAZ. A DNA fragment containing the fliA gene with the P1 and P2 promoters was PCR amplified with primers KpnIfliA0 and IAE1X. The amplified product was digested with KpnI and XbaI and inserted into the corresponding site of pTrc99K to obtain pTrc-P1/P2-fliA. From this plasmid, a DNA fragment containing the fliA gene and the P1 and P2 promoters was excised with KpnI and SphI and inserted into the corresponding site of pFZY1 to obtain pF-P1/P2-fliA.
A DNA fragment containing the P1 and P2 promoters was PCR amplified with primers fliApF and fliAp2sR. Similarly, a DNA fragment containing the P2 promoter and the fliAZ genes was PCR amplified with primers fliAp2sF and IZP4. The primers fliAp2sR and fliAp2sF are complementary to each other and contain base substitution mutations GCC to CGT in the −10 sequence of the P2 promoter. These two amplified products were mixed and used for further PCR amplification with primers KpnIfliA0 and IZP4. The final amplified product was digested with KpnI and BamHI and inserted into the corresponding site of pFZY1 to obtain pF-P1-fliAZ. A DNA fragment containing the P1 and P2 promoters was PCR amplified as described above, while a DNA fragment containing the P2 promoter and the fliA gene was PCR amplified with primers fliAp2sF and IAE1X. These two amplified products were mixed and used for further PCR amplification with primers KpnIfliA0 and IAE1X. The final amplified product was digested with KpnI and XbaI and inserted into the corresponding site of pBAD18kan to obtain pBAD-P1-fliA. From this plasmid, a DNA fragment containing the fliA gene and the P1 promoter was excised with KpnI and SphI and inserted into the corresponding site of pFZY1 to obtain pF-P1-fliA.
A DNA fragment containing the P2 promoter and the fliAZ genes was PCR amplified with primers KpnIfliA1 and IZP4. The amplified product was digested with KpnI and BamHI and inserted into the corresponding site of pFZY1 to obtain pF-P2-fliAZ. A DNA fragment containing the P2 promoter and the fliA gene was PCR amplified with primers KpnIfliA1 and IAE1X. The amplified product was digested with KpnI and XbaI and inserted into the corresponding site of pTrc99K to obtain pTrc-P2-fliA. From this plasmid, a DNA fragment containing the P2 promoter and the fliA gene was excised with KpnI and SphI and inserted into the corresponding site of pFZY1 to obtain pF-P2-fliA. Plasmid pF-P2c-fliA, which carries the consensus sequence of the σ28-dependent promoter in place of the native P2 promoter, was constructed via pTrc-P2c-fliA through the same procedure described above using primers KpnIfliA1c and IAE1X.
A DNA fragment containing the P2 promoter was PCR amplified with primers fliApF and fliAP1-P2mRNAR. Similarly, a DNA fragment containing the P2 promoter and the fliA gene was PCR amplified with primers fliAP1-P2mRNAF and BamHIfliAR. The primers fliAP1-P2mRNAR and fliAP1-P2mRNAF are complementary to each other and have deletions of 11 nucleotides from the transcriptional start site of the P1 promoter to that of the P2 promoter. These two amplified products were mixed and used for further PCR amplification with primers KpnIfliA0 and BamHIfliAR. The final amplified product was digested with KpnI and BamHI and inserted into the corresponding site of pFZY1 to obtain pF-P1(P2)-fliA. A DNA fragment containing the P2 promoter was PCR amplified as described above, while a DNA fragment containing the P2 promoter and the fliAZ genes was PCR amplified with primers fliAP1-P2mRNAF and IZP4. These two amplified products were mixed and used for further PCR amplification with primers KpnIfliA0 and IZP4. The final amplified product was digested with KpnI and BamHI and inserted into the corresponding site of pFZY1 to obtain pF-P1(P2)-fliAZ.
A DNA fragment containing the P2 promoter and the fliA gene was PCR amplified with primers KpnIfliAP2-P1mRNAF and BamHIfliAR. Primer KpnIfliAP2-P1mRNAF contains a duplication of 11 nucleotides from the transcriptional start site of the P1 promoter to that of the P2 promoter. The amplified product was digested with KpnI and BamHI and inserted into the corresponding site of pFZY1 to obtain pF-P2(P1)-fliA. Similarly, a DNA fragment containing the P2 promoter and the fliAZ genes was PCR amplified with primers KpnIfliAP2-P1mRNAF and IZP4. The amplified product was digested with KpnI and BamHI and inserted into the corresponding site of pFZY1 to obtain pF-P2(P1)-fliAZ.
Construction of plasmids carrying fliA-lacZ translational fusion genes.
The fliA-lacZ translational fusion genes were constructed on pMC1403. Sequences of the 5′ untranslated regions (UTRs) of the fliA transcripts from the recombinant plasmids are summarized in Fig. 1C. Unless otherwise specified, the template used for PCR amplification was the genomic DNA from KK1004.
A DNA fragment containing the intact P1 and inactivated P2 promoters together with the first 398 nucleotides of the fliA coding region was PCR amplified from pBAD-P1-fliA with primers SmaIfliA0 and BamHIfliAR2. The amplified product was digested with SmaI and BamHI and inserted into the corresponding site of pMC1403 to obtain pMC-P1. A DNA fragment containing the intact P2 promoter and the first 398 nucleotides of the fliA coding region was PCR amplified with primers KpnIfliA1 and BamHIfliAR2. The amplified product was digested with BamHI and inserted into the SmaI-BamHI site of pMC1403 to obtain pMC-P2.
A DNA fragment containing the P2 promoter was PCR amplified with primers fliApF and fliAP1-P2mRNAR. Similarly, a DNA fragment containing the P2 promoter and the fliA gene was PCR amplified with primers fliAP1-P2mRNAF and IAE1X. These two amplified products were mixed and used for further PCR amplification with primers SmaIfliA0 and BamHIfliAR2. The final amplified product was digested with SmaI and BamHI and inserted into the corresponding site of pMC1403 to obtain pMC-P1(P2). A DNA fragment containing the P2 promoter and the fliA gene was PCR amplified with primers KpnIfliAP2-P1mRNAF and BamHIfliAR2. The amplified product was digested with BamHI and inserted into the SmaI-BamHI site of pMC1403 to obtain pMC-P2(P1).
A DNA fragment containing the intact P1 and inactivated P2 promoters and the 5′ UTR of the fliA gene was PCR amplified from pBAD-P1-fliA with primers ITf1 and P1m(taa-cgg)fliAR. Similarly, a DNA fragment containing the fliA gene and its 5′ UTR was PCR amplified from pBAD-P1-fliA with primers P1m(taa-cgg)fliAF and IAE1X. The primers P1m(taa-cgg)fliAR and P1m(taa-cgg)fliAF are complementary to each other and possess a 3-nucleotide substitution in the 5′ UTR of the fliA gene. These two amplified products were mixed and used for further PCR amplification with primers SmaIfliA0 and BamHIfliAR2. The final amplified product was digested with SmaI and BamHI and inserted into the corresponding site of pMC1403 to obtain pMC-P1(GC*).
A DNA fragment containing the P2 promoter and the 5′ UTR of the fliA gene was PCR amplified with primers fliApF and fliASDqR. Similarly, a DNA fragment containing the fliA gene and its 5′ UTR was PCR amplified with primers fliASDqF and IAE1X. The primers fliASDqR and fliASDqF are complementary to each other and possess a sequence identical to the 5′ UTR sequence of the His6-tagged fusion protein gene from pQE80L in place of that of the fliA gene. These two amplified products were mixed and used for further PCR amplification with primers KpnIfliA1 and BamHIfliAR2. The final amplified product was digested with BamHI and inserted into the SmaI-BamHI site of pMC1403 to obtain pMC-P2(SD*).
Motility and β-galactosidase enzyme assays.
Motility of cells was assayed as follows. A single colony formed on an MGC agar plate was stabbed onto an MGC motility agar plate and incubated at 37°C for 8 h.
β-Galactosidase activity was assayed as described previously (31) using cells grown aerobically to exponential phase at 37°C in MGC containing appropriate antibiotics (45, 46). Each sample was assayed in triplicate.
Protein analysis.
Cells were grown to exponential phase at 37°C in MGC, and the cultures were directly subjected to sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis (PAGE). SDS-PAGE and Western blotting of proteins were performed according to the method described previously (31). FLAG-tagged proteins were detected with a horseradish peroxidase-conjugated anti-FLAG M2 monoclonal antibody (Sigma, MO) using an ECL Plus Western blotting detection system (GE Healthcare, NJ).
RESULTS
Transcription of class 3 operon in flhD mutant background.
In order to assess the FliA activity in various genetic backgrounds, we examined class 3 expression using the chromosomal fliC-lac fusion gene as a reporter (Table 4). As reported previously (27), the flhD mutant showed reduced β-galactosidase activity in both the flgM-positive and flgM-negative backgrounds. Interestingly, even in the flhD mutant, β-galactosidase activity was 4.1-fold higher in the flgM-negative background than in the flgM-positive background. This difference was not observed in the fliA-negative background, suggesting that the FliA protein should be produced, though in a small amount, even in the absence of FlhD4C2. Nevertheless, even in the flgM mutant, β-galactosidase activity was much lower in the flhD-negative background than in the flhD-positive background, suggesting that the FliA protein is not produced in large enough amounts to activate class 3 transcription in the absence of FlhD4C2. These results suggest the absence of the autogenous activation of the fliA gene. However, as described previously (27), when FliA was overproduced from the expression plasmid pTrc-fliA, the fliC-lacZ gene was expressed at a high level in the flhD mutant (Table 4), indicating that FlhD4C2 per se is not required for FliA-mediated class 3 transcription. This is consistent with our previous result showing that the fliA gene was transcribed in vitro by σ28 RNA polymerase in the absence of FlhD4C2 (14).
Table 4.
Class 3 transcription in various genetic backgroundsa
| Genotypeb |
Expression plasmid | LacZ expression (Miller units)c | ||
|---|---|---|---|---|
| flhD | flgM | fliA | ||
| + | + | + | − | 230 ± 16 |
| + | − | + | − | 1,100 ± 28 |
| + | + | − | − | 9.4 ± 2.0 |
| + | − | − | − | 6.9 ± 0.78 |
| − | + | + | − | 8.3 ± 1.5 |
| − | − | + | − | 34 ± 2.7 |
| − | + | − | − | 8.1 ± 1.9 |
| − | − | − | − | 9.2 ± 2.2 |
| − | − | − | pTrc99K | 6.4 ± 1.9 |
| − | − | − | pTrc-fliA | 2,500 ± 140 |
Class 3 transcription was assayed using the fliC-lacZ transcriptional fusion gene originated from KK1110.
The flhD, flgM, and fliA mutations used originated from KK1004hDC, KK1004gM, and KK1004iA, respectively.
Cells grown in MGC were used.
Intracellular FliA level in the absence of FlhD4C2.
In order to assess the intracellular level of the FliA protein, total cell proteins from various genetic backgrounds were analyzed by Western blotting. For this purpose, we used strain KK1004AFZF, which encodes the 3×FLAG epitope-tagged FliA (FliA-3×FLAG) and FliZ (FliZ-3×FLAG) proteins. This strain showed a swimming phenotype indistinguishable from that of the wild-type strain (data not shown), indicating that fusion of the 3×FLAG epitope to FliA and FliZ has no significant effect on flagellar expression. Using this strain, we examined the effects of flhD and flgM mutations on the intracellular levels of FliA and FliZ by Western blotting with anti-FLAG antibody (Fig. 2A). As expected, neither FliA-3×FLAG nor FliZ-3×FLAG was detected in the flhD mutant, irrespective of the flgM genotype. Interestingly, in the flhDC-positive background, the flgM mutation increased the FliZ-3×FLAG level, while it had no significant effect on the FliA-3×FLAG level.
Fig. 2.
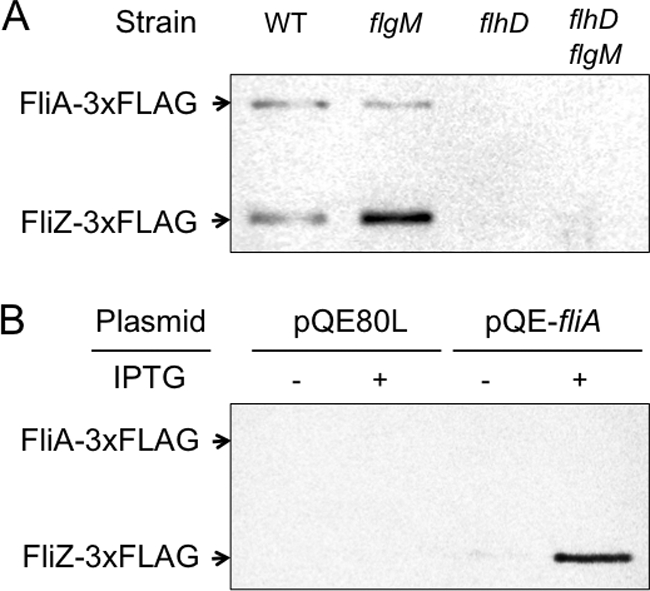
Expression of FliA and FliZ proteins. (A) Cells of strain KK1004AFZF carrying the mutation(s) indicated above the lane were grown to exponential phase at 37°C in MGC. The culture containing 106 cells was directly subjected to SDS-PAGE. FLAG-tagged proteins in the gel were detected by Western blotting using an anti-FLAG M2 monoclonal antibody. WT, wild type. (B) Cells of the flhD::Tn10 mutant strain of KK1004AFZF harboring the plasmid indicated above the lane were cultured as described for panel A in MGC containing ampicillin with or without IPTG. The FLAG-tagged proteins were analyzed as described for panel A.
Barembruch and Hengge reported that, in E. coli, FliA is subjected to proteolysis by Lon protease and FlgM protects FliA against Lon-mediated degradation (3). This raised a possibility that, in the absence of FlgM, FliA may also be rapidly degraded by Lon in Salmonella, which can account for suppression of the FliA-mediated autogenous activation of the fliA gene in the flgM mutant. In order to test this possibility, we introduced a lon mutation into the flgM flhD double mutant carrying the fliC-lac fusion gene, and the resulting lon mutant was examined for class 3 expression. However, the lon mutation could not relieve the inhibition of class 3 expression in the flgM flhD mutant (data not shown). This result indicates that Lon is not responsible for suppression of the autogenous activation of the fliA gene in the absence of FlgM.
Transcriptional activity of the fliA gene.
In order to assess the transcriptional activity of the fliA gene, we used plasmid pF-P1/P2, in which the lacZ gene is transcriptionally fused to the fliA promoters on a single-copy plasmid, pFYZ1. In this plasmid, the lacZ gene with its own Shine-Dalgarno (SD) sequence follows the 115 bp DNA upstream of the fliA gene and the first 399 bp DNA of the fliA coding region and thus is transcribed from both the P1 and P2 promoters (Fig. 1C). This plasmid was introduced by transformation into strains with various genetic backgrounds, and β-galactosidase activity was measured with the resulting transformants grown in MGC (Table 5). As expected, the flgM mutation enhanced enzyme activity, whereas the flhD mutation completely abolished enzyme activity. In the flhD mutant, the flgM mutation had only a marginal effect on enzyme activity, which also suggests the absence of the autogenous activation of the fliA gene. However, in the flhDC-positive background, the fliA mutation drastically reduced enzyme activity, indicating that the FliA protein has a large contribution to fliA transcription. Similarly, the fliZ mutation also drastically reduced enzyme activity, indicating a large contribution of the P1 promoter to fliA transcription and the presence of a synergistic effect of the P1 and P2 promoters on fliA transcription. However, the flgM mutation also enhanced enzyme activity in the fliZ mutant, but the enzyme level was much lower than that in the fliZ-positive background. This indicates that FliZ-enhanced transcription from the P1 promoter is a prerequisite to high-level expression from the P2 promoter. This is consistent with our previous observation that FliZ is required for enhanced expression of the class 2 and class 3 operons in the flgM mutant (28). Expression of β-galactosidase from pF-P1/P2 was also examined in the flhD mutant harboring pTrc-fliA (Table 5). Enzyme activity was high in the absence of FlgM, indicating that FlhD4C2 per se is not required for FliA-mediated transcription of the fliA gene.
Table 5.
Transcriptional activity from the fliA promoter on a single-copy plasmid
| Host genotypea |
Expression plasmid | LacZ expression (Miller units)b |
||||
|---|---|---|---|---|---|---|
| flhD | flgM | fliA | fliZ | pF-P1/P2 | pF-P1/P2-fliA | |
| + | + | + | + | − | 330 ± 35 | 290 ± 37 |
| + | − | + | + | − | 860 ± 85 | 1,300 ± 120 |
| + | + | − | + | − | 17 ± 6.4 | 120 ± 64 |
| + | − | − | + | − | 9.8 ± 1.5 | 770 ± 45 |
| − | + | + | + | − | 0.0 ± 1.4 | 1.2 ± 1.3 |
| − | − | + | + | − | 0.35 ± 1.9 | 6.6 ± 0.84 |
| + | + | + | − | − | 6.9 ± 0.61 | 8.0 ± 1.6 |
| + | − | + | − | − | 27 ± 7.4 | 77 ± 23 |
| − | + | + | + | pTrc99K | 0.0 ± 0.11 | ND |
| − | + | + | + | pTrc-fliA | 6.0 ± 1.2 | ND |
| − | − | + | + | pTrc99K | 0.24 ± 0.18 | ND |
| − | − | + | + | pTrc-fliA | 640 ± 23 | ND |
The flhD, flgM, fliA, and fliZ mutations used originated from KK1004hDC, KK1004gM, KK1004iA, and KK1004iZ, respectively.
In pF-P1/P2, the lacZ gene with its own SD sequence follows the P1 and P2 promoters of the fliA gene. pF-P1/P2-fliA has the same structure as pF-P1/P2, except that it contains an intact fliA gene. Cells grown in MGC were used. ND, not done.
Promoter activity was also analyzed with plasmid pF-P1/P2-fliA, which carries the P1 and P2 promoters together with the intact fliA gene transcriptionally fused to the lacZ gene on pFZY1 (Table 5). As expected, enzyme activity was high in the fliA mutant host strains, and the other expression profiles were similar to those in strains with pF-P1/P2.
Transcriptional activity of P2 promoter of the fliAZ operon.
Next, we constructed plasmid pF-P2-fliA, in which the lacZ gene is transcriptionally fused to the fliA gene with the P2 promoter on pFYZ1. Because this plasmid lacks the FlhD4C2 box of the P1 promoter, the lacZ gene can be transcribed only from the P2 promoter (Fig. 1C). As shown in Table 6, β-galactosidase activity was 15-fold lower than that from pF-P1/P2-fliA (Table 5) in the wild-type (flhD-positive flgM-positive) strain. This suggests that transcription from the P1 promoter makes a much greater contribution to the expression of the fliAZ operon than that from the P2 promoter. This is consistent with the results recently reported by Wozniak et al. (48). As expected, the flhD and flgM mutations reduced and enhanced enzyme activity, respectively. Even in the flgM mutant, enzyme activity was much lower in the absence of FlhD4C2 than in the presence of FlhD4C2. This also suggests the absence of autogenous activation of the fliA gene. Expression of β-galactosidase from pF-P2-fliA was also examined in the flhD mutant harboring pTrc-fliA. As expected, enzyme activity was much higher in the flgM mutant than in the flgM-positive strain.
Table 6.
Transcriptional activity from the P2 promoter of the fliA gene
| Host genotypea |
Expression plasmid | LacZ expression (Miller units)b |
||
|---|---|---|---|---|
| flhD | flgM | pF-P2-fliA | pF-P2c-fliA | |
| + | + | − | 20 ± 1.8 | 270 ± 20 |
| + | − | − | 390 ± 11 | 1,100 ± 35 |
| − | + | − | 3.0 ± 2.1 | 3.7 ± 0.86 |
| − | − | − | 3.5 ± 1.7 | 8.5 ± 0.97 |
| − | + | pTrc99K | 4.1 ± 1.3 | 13 ± 3.9 |
| − | + | pTrc-fliA | 18 ± 2.4 | 300 ± 18 |
| − | − | pTrc99K | 12 ± 2.5 | 34 ± 2.9 |
| − | − | pTrc-fliA | 74 ± 11 | 970 ± 6.5 |
The flhD and flgM mutations used originated from KK1004hDC and KK1004gM, respectively.
In pF-P2-fliA, the lacZ gene is transcribed from the P2 promoter of the fliA gene. In pF-P2c-fliA, the P2 promoter sequence in pF-P2 was replaced with the consensus sequence for the σ28-dependent promoter. Both plasmids carry an intact coding sequence of the fliA gene. Cells grown in MGC were used.
Close examination of the nucleotide sequence of the P2 promoter revealed that its −35 sequence (TGTAGAAA) is divergent from the consensus sequence (TAAAGTTT) of the σ28-dependent promoter (Fig. 1B) (13). This suggested the possibility that transcription from the P2 promoter requires a higher level of FliA than that from the other class 3 promoters, which might render the P2 promoter less active under a condition, such as in the flhD mutant, where only a limited amount of FliA is available. In order to test this possibility, we constructed plasmid pF-P2c-fliA, in which the −35 sequence of the native P2 promoter was replaced with the consensus sequence (Fig. 1C). Compared with pF-P2-fliA, this plasmid produced higher β-galactosidase activity in all genetic backgrounds examined (Table 6). However, enzyme activity was still low in the flhD mutant even in the absence of FlgM. This indicates that suppression of the autogenous activation of the fliA gene is not due to its nonconsensus P2 promoter.
Translation of the fliA gene from P1 and P2 mRNAs.
The above-mentioned results suggested the possibility that the FliA protein, if expressed, can transcribe the fliAZ operon from the P2 promoter but mRNA transcribed from the P2 promoter (P2 mRNA) should be inactive for translation of the fliA gene. To test this possibility, we examined the translation of the fliA gene with plasmids carrying a fliA-lacZ fusion gene with the P1 or P2 promoter and variously engineered 5′ UTR sequences on a transcription-translation probe vector, pMC1403. In these plasmids, the lacZ gene is fused in frame to the 398th nucleotide of the fliA gene.
In plasmid pMC-P1, the P1 promoter is intact, but the P2 promoter is inactive due to the base substitutions in its −10 sequence (Fig. 1C). In this plasmid, transcription from the P1 promoter yields mRNA having a 5′ UTR sequence identical to that of native P1 mRNA. In order to avoid any residual P2 promoter activity, the translational activity of P1 mRNA from pMC-P1 was examined in the ΔfliAZ mutant. On the other hand, pMC-P2 lacks the FlhD4C2 box of the P1 promoter but possesses the intact P2 promoter with its downstream sequence (Fig. 1C). Therefore, in this plasmid, the fliA-lacZ fusion gene can be transcribed only from the P2 promoter by σ28 RNA polymerase, and mRNA thus transcribed has a 5′ UTR sequence identical to that of native P2 mRNA. The translational activity of P2 mRNA from pMC-P2 was examined in the ΔflgM mutant. As expected, pMC-P1 showed a high level of β-galactosidase activity, whereas pMC-P2 showed only a low level of enzyme activity even in the presence of FliA and absence of FlgM (Table 7). This result suggests that the fliA gene is not efficiently translated from P2 mRNA.
Table 7.
Translational activity of the altered versions of fliA mRNA
| Translational fusion plasmid |
Host genotypea | LacZ expression (Miller units)b | ||
|---|---|---|---|---|
| Name | Promoter | 5′ UTRc | ||
| pMC-P1 | P1 | P1 | ΔfliAZ | 1,300 ± 44 |
| pMC-P1(P2) | P1 | P2 | ΔfliAZ | 3.1 ± 0.57 |
| pMC-P1(GC*) | P1 | P1(GC*) | ΔfliAZ | 23 ± 1.5 |
| pMC-P2 | P2 | P2 | ΔflgM | 33 ± 1.5 |
| pMC-P2(P1) | P2 | P1 | ΔflgM | 1,000 ± 160 |
| pMC-P2(SD*) | P2 | P2(SD*) | ΔflgM | 4,700 ± 1,600 |
Host strains used were KK1004iAZ for ΔfliAZ and KK1004gM for ΔflgM.
Cells grown in MGC were used.
GC*, the AT-rich sequence of the 5′ end of P1 mRNA was altered to a GC-rich sequence; SD*, the 5′ UTR sequence of the fliA gene was replaced with a sequence identical to the 5′ UTR sequence of the His6-tagged fusion protein gene on pQE80L, resulting in substitution of the SD sequence of the fliA gene (AGG) with a stronger one (AGGAG).
The structures of P1 and P2 mRNAs are identical, except that P2 mRNA lacks 11 nucleotides present at the 5′ end of P1 mRNA (Fig. 1B and C). We hypothesized that this 11-bp sequence has an important effect on the translational activity of the fliA gene. In order to test this hypothesis, we constructed two plasmids, pMC-P1(P2) and pMC-P2(P1). In plasmid pMC-P1(P2), this 11-bp sequence is deleted, and thus, transcription from the P1 promoter produces mRNA whose 5′ UTR sequence is identical to that of P2 mRNA (Fig. 1C). On the other hand, pMC-P2(P1) has a duplication of this 11-bp sequence, and thus, transcription from the P2 promoter produces mRNA whose 5′ end sequence is identical to that of P1 mRNA (Fig. 1C). Expression assay of β-galactosidase revealed that pMC-P1(P2) showed low enzyme activity, whereas pMC-P2(P1) showed high enzyme activity (Table 7). This result is quite in contrast to the results obtained with pMC-P1 and pMC-P2. Taken together, these results confirm our above-mentioned hypotheses that P2 mRNA is inactive for translation of the fliA gene and that the 11-bp sequence present at the 5′ end of P1 mRNA but absent from P2 mRNA is important for efficient translation of the fliA gene.
Effect of altered 5′ UTR sequences on fliA translation.
The SD sequence of mRNA is complementary to specific regions of 16S RNA and is known to act as a ribosome-binding site (43). The fliA gene has a very short SD sequence (AGG), which may be a cause of its inefficient translation. We constructed plasmid pMC-P2(SD*), in which the SD sequence together with its flanking sequence was replaced with a sequence identical to the 5′ UTR sequence of the His6-tagged fusion protein gene on the expression vector pQE80L (Fig. 1C). Since this has a good SD sequence (AGGAG), it was expected to act as a stronger ribosome-binding site than the original SD sequence of the fliA gene. As shown in Table 7, this plasmid produced high β-galactosidase activity, though the mRNA was transcribed from the P2 promoter. This result suggests that the attenuated ribosome-binding activity of P2 mRNA is responsible for suppression of the autogenous activation of the fliA gene.
It should be noted that the SD sequence of the fliA gene on P1 mRNA is identical to that on P2 mRNA (Fig. 1B and C). This suggests that the 11-bp sequence of the 5′ end of P1 mRNA may assist its ribosome-binding activity. This sequence is AT rich, with 9 bases being A or T. It is known that, at least in certain genes, an AT-rich sequence upstream of the SD sequence assists mRNA binding to the ribosome through its interaction with the S1 protein (4, 20). In order to test whether the AT-rich region of the 11-bp sequence of the 5′ end of P1 mRNA is important for fliA translation, we constructed plasmid pMC-P1(GC*), in which the sequence UAA is replaced with CGG, making the 11-bp sequence less AT rich (Fig. 1C). As shown in Table 7, this plasmid produced only a low level of β-galactosidase activity, suggesting an important role of the AT-rich region of the 5′ end of P1 mRNA in fliA translation. We anticipate that this region may enhance fliA translation by assisting P1 mRNA with binding to the ribosome through its interaction with S1. However, we cannot exclude an alternative possibility that the alteration of the 5′-terminal sequence affects mRNA stability.
Expression of FliA and FliZ proteins from P2 mRNA.
The above-mentioned results altogether indicate that the fliA gene is not translated efficiently from P2 mRNA, which makes the fliAZ operon unable to be activated autogenously by FliA. In order to confirm this further, we examined the effect of FliA overexpression on intracellular FliA-3×FLAG and FliZ-3×FLAG levels in the absence of FlhD4C2 using the flhD mutant of KK1004AFZF. As shown in Fig. 2B, when the excess amount of FliA was supplied from pQE-fliA, the chromosomally encoded FliA-3×FLAG protein was not observed, whereas the chromosomally encoded FliZ-3×FLAG protein was produced efficiently. This indicates that P2 mRNA has no or only a little, if any, contribution to FliA expression, whereas FliZ is produced efficiently from P2 mRNA.
Contribution of P1 and P2 mRNAs to motility development.
Next, we examined the contribution of P1 and P2 mRNAs to motility development. For this purpose, we constructed two sets of pFZY1-based recombinant plasmids carrying various combinations of the fliA promoter and 5′ UTR sequences: one set has the fliA gene, while the other set has both the fliA and fliZ genes. We compared their ability to restore motility to the ΔfliAZ mutant cells.
The ΔfliAZ mutant cells harboring pF-P1/P2-fliA, pF-P1-fliA, or pF-P2(P1)-fliA formed spreading colonies on MGC motility agar, whereas those harboring pF-P2-fliA or pF-P1(P2)-fliA did not (Fig. 3A). This result conforms to the above-mentioned conclusion that P2 mRNA is inactive for expression of the FliA protein. On the other hand, the ΔfliAZ mutant cells harboring pF-P1/P2-fliAZ or pF-P1-fliAZ formed a larger spreading colony than those harboring pF-P1/P2-fliA or pF-P1-fliA, respectively (Fig. 3A and B). This is consistent with our previous results (46) and indicates that the fliZ gene enhances motility development of the cells. Importantly, the ΔfliAZ mutant cells harboring pF-P1/P2-fliAZ formed a larger spreading colony than those harboring pF-P1-fliAZ (Fig. 3B). This indicates that P2 mRNA must contribute to FliZ expression, leading to enhancement of motility development.
Fig. 3.
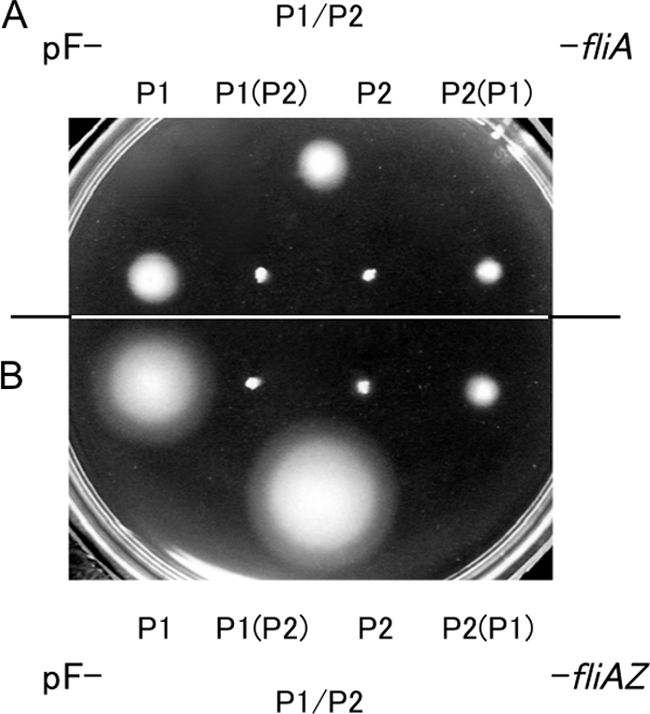
Contribution of the P1 and P2 promoters to motility development. Single colonies of strain KK1004iAZ harboring the indicated plasmid formed in MGC agar plates containing ampicillin were stabbed onto an MGC motility agar plate containing ampicillin and incubated at 37°C for 8 h. Plasmids used included pF-P1/P2-fliA, pF-P1-fliA, pF-P1(P2)-fliA, pF-P2-fliA, and pF-P2(P1)-fliA (A) and pF-P1/P2-fliAZ, pF-P1-fliAZ, pF-P1(P2)-fliAZ, pF-P2-fliAZ, and pF-P2(P1)-fliAZ (B).
DISCUSSION
In a previous report (14), we showed both in vivo and in vitro that the fliAZ operon is transcribed from the class 3 promoter (P2) as well as from the class 2 promoter (P1) in Salmonella. Because the fliA gene encodes σ28, responsible for transcription from the class 3 promoter (39), this operon should be activated autogenously by transcription from the P2 promoter. However, our earlier work (27) and recently published work by Wozniak et al. (48) indicated that transcription from the P1 promoter is a prerequisite to transcription from the P2 promoter in the expression of the fliAZ operon. This enigma has been solved by the present study, showing that the transcript from the P2 promoter is translationally inert in the expression of the fliA gene.
According to our findings reported here, the control of fliAZ operon expression is summarized as follows. The fliAZ operon is expressed first from the P1 promoter, which is transcribed by σ70 RNA polymerase in the presence of the class 1 gene product FlhD4C2. From this transcript, the fliA and fliZ genes are both translated. The expressed FliZ protein activates autogenously the transcription from the P1 promoter by repressing the anti-FlhD4C2 gene ydiV, while the expressed FliA protein (σ28) associates with RNA polymerase core enzyme and then transcribes the fliAZ operon from the P2 promoter. The FliZ protein is also expressed from the P2 transcript, which results in further enhancement of transcription from the P1 promoter. On the other hand, the fliA gene is not expressed from the P2 transcript, which ensures suppression of the autogenous activation of the fliA gene.
We reported previously that cells defective in the anti-σ28 gene flgM showed impaired cell growth and this growth inhibition was relieved by a coexisting mutation in the flagellin gene fliC, which is transcribed by σ28 RNA polymerase (27). This indicates that flagellin overexpression is detrimental to cell growth. If the fliA gene was activated autogenously, FliA should be produced in much excess, leading to flagellin overproduction, which should cause severe growth inhibition. In fact, when the fliA gene was fully induced from pQE-fliA in the flgM mutant, cell growth was completely inhibited (our unpublished result). Therefore, it is reasonable that the cell has an ability to suppress the autogenous activation of the fliA gene. Usually, FliA also activates the flgM gene (10, 24), and the expressed FlgM protein can inactivate FliA to prohibit flagellin overexpression. However, under the condition where the intracellular FlgM concentration becomes low, translational silencing of the fliA gene in the P2 transcript could ensure normal cell growth by suppressing flagellin overproduction. For example, such a condition can occur upon completion of hook-basal body assembly, where FlgM is exported efficiently out of the cell through the flagellum-specific secretion pathway (12, 24) and the intracellular concentration of FlgM becomes low, leading to activation of the σ28 activity of the FliA protein (19). We anticipate that, under the condition where the FlgM level is low, the σ28 activity of the FliA protein expressed from the P1 transcript suffices for production of large enough amounts of flagellin molecules for filament assembly.
As shown in Fig. 2A, the intracellular level of FliZ increased in the flgM mutant, probably owing to the increased level of P2 transcript. This result is consistent with our previous results showing that the flgM mutation caused enhanced expression of the class 2 operons (27) and this enhancement was cancelled by the coexisting fliZ mutation (28). Curiously, however, the flgM mutation did not increase the intracellular level of FliA (Fig. 2A). This is apparently inconsistent with the above-mentioned observation that the intracellular level of FliZ was increased by the flgM mutation, because the FliZ protein expressed should increase the P1 transcript of the fliAZ operon and thus enhance FliA expression. Recently, however, we showed that FliZ overproduction had only a moderate effect on class 2 transcription, though the effect of the fliZ mutation was remarkable (46). This suggests that the enhanced FliZ level in the flgM mutant does not lead to upregulation of FliA production, which conforms to our observation in Fig. 2A. However, at present, we cannot rule out the possibility that the Salmonella cell has some unknown mechanism for FliA homeostasis.
Next, we would like to consider the biological implication of the P2 promoter in the fliAZ operon. FliZ is expressed from both the P1 and P2 transcripts, whereas FliA is expressed only from the P1 transcript. This situation may cause differential expression of the two genes fliA and fliZ within the same operon. We guess that FliZ is required at a higher level than FliA for efficient production of the flagellar structure. During the flagellar assembly process, transcription from the P2 promoter should be enhanced upon completion of hook-basal body assembly. At this step, FliZ should be expressed at the maximal rate, leading to enhanced expression of the class 2 operons, which can trigger another cycle of flagellar biogenesis. This may be responsible for the FliZ-mediated induction of the kinetic switch in flagellar gene expression proposed by Saini et al. (41). Alternatively, the cells may require FliZ at a much higher level than FliA, because FliZ is involved in regulation of many cellular processes, in addition to flagellar biogenesis. They include transcriptional control of SPI1 genes (6, 16, 18) and that of the type 1 fimbrial genes (8, 42).
In this study, we showed several lines of evidence suggesting that the translational silencing of the fliA gene in the P2 transcript is attributable to its attenuated ribosome-binding activity. The AU-rich sequence in the 5′ end of the P1 transcript has a significant effect on translation of the fliA gene (Table 7). It is known that the AU-rich sequence upstream of the SD sequence is a target of ribosome protein S1 (4) and plays an important role in mRNA stabilization and efficient translation in E. coli (20). Although we have not analyzed further the role of the AU-rich sequence on fliA translation, we believe that it assists P1 mRNA in its binding to the ribosome through interaction with S1, since the modified P2 transcript, in which the SD sequence was replaced with a stronger one, showed much enhanced translation activity of the fliA gene (Table 7).
In addition to the fliAZ operon, the fliDST, flgMN, and flgKL operons are known to be transcribed from both class 2 and class 3 promoters in the flagellar regulon of Salmonella (10, 24, 26, 49). In all cases, the transcripts from the class 3 promoters have shorter 5′ UTR sequences than those from the class 2 promoters. However, unlike in the fliAZ operon, the class 2 and class 3 promoters do not overlap with each other and the class 3 transcripts have longer 5′ UTR sequences than the P2 transcript of the fliAZ operon. Furthermore, the class 3 transcripts have AU-rich 5′ UTRs and better SD sequences than P2 mRNA of the fliAZ operon. Consistent with these structural features, the class 3 transcripts have activities to efficiently translate the first genes in these three operons (our unpublished results). Therefore, the fliAZ operon is a special case among the flagellar operons whose transcription is controlled by multiple promoters. This may reflect the deleterious effect of FliA overproduction on cell growth.
The promoter region of the fliAZ operon of E. coli has a structure similar to that of S. enterica (38, 53), suggesting identical expression control in these two bacteria. In contrast, as reported by Lanois et al. (34), the fliAZ operons from other gammaproteobacteria such as Proteus mirabilis, Serratia marcescens, Xenorhabdus nematophila, and Yersinia enterocolitica have a class 2 promoter-like sequence but lack a class 3 promoter-like sequence. This suggests that the regulatory mechanism of the fliAZ operon in these organisms may be distinct from that in S. enterica and E. coli.
ACKNOWLEDGMENTS
We thank the National BioResource Project, E. coli, at the National Institute of Genetics, Japan, for plasmid vector pBAD18kan used in this study.
Footnotes
Published ahead of print on 9 September 2011.
REFERENCES
- 1. Aldridge P., Hughes K. T. 2002. Regulation of flagellar assembly. Curr. Opin. Microbiol. 5: 160–165 [DOI] [PubMed] [Google Scholar]
- 2. Amann E., Ochs B., Abel K. J. 1988. Tightly regulated tac promoter vectors useful for the expression of unfused and fused proteins in Escherichia coli. Gene 69: 301–315 [DOI] [PubMed] [Google Scholar]
- 3. Barembruch C., Hengge R. 2007. Cellular levels and activity of the flagellar sigma factor FliA of Escherichia coli are controlled by FlgM-modulated proteolysis. Mol. Microbiol. 65: 76–89 [DOI] [PubMed] [Google Scholar]
- 4. Boni I. V., Isaeva D. M., Musychenko M. L., Tzareva N. V. 1991. Ribosome-messenger recognition: mRNA target sites for ribosomal protein S1. Nucleic Acids Res. 19: 155–162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Casadaban M. J., Chou J., Cohen S. N. 1980. In vitro gene fusions that join an enzymatically active β-galactosidase segment to amino-terminal fragments of exogenous proteins: Escherichia coli plasmid vectors for the detection and cloning of translational initiation signals. J. Bacteriol. 143: 971–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Chubiz J. E., Golubeva Y. A., Lin D., Miller L. D., Slauch J. M. 2010. FliZ regulates expression of the Salmonella pathogenicity island 1 invasion locus by controlling HilD protein activity in Salmonella enterica serovar Typhimurium. J. Bacteriol. 192: 6261–6270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Claret L., Hughes C. 2002. Interaction of the atypical prokaryotic transcription activator FlhD2C2 with early promoters of the flagellar gene hierarchy. J. Mol. Biol. 321: 185–199 [DOI] [PubMed] [Google Scholar]
- 8. Clegg S., Hughes K. T. 2002. FimZ is a molecular link between sticking and swimming in Salmonella enterica serovar Typhimurium. J. Bacteriol. 184: 1209–1213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Datsenko K. A., Wanner B. L. 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc. Natl. Acad. Sci. U. S. A. 97: 6640–6655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gillen K. L., Hughes K. T. 1993. Transcription from two promoters and autoregulation contribute to the control of expression of the Salmonella typhimurium flagellar regulatory gene flgM. J. Bacteriol. 175: 7006–7015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Guzman L.-M., Belin D., Carson M. J., Beckwith J. 1995. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J. Bacteriol. 177: 4121–4130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hughes K. T., Gillen K. L., Semon M. J., Karlinsey J. E. 1993. Sensing structural intermediates in bacterial flagellar assembly by export of a negative regulator. Science 262: 1277–1280 [DOI] [PubMed] [Google Scholar]
- 13. Ide N., Ikebe T., Kutsukake K. 1999. Reevaluation of the promoter structure of the class 3 flagellar operons of Escherichia coli and Salmonella. Genes Genet. Syst. 74: 113–116 [DOI] [PubMed] [Google Scholar]
- 14. Ikebe T., Iyoda S., Kutsukake K. 1999. Structure and expression of the fliA operon of Salmonella typhimurium. Microbiology 145: 1389–1396 [DOI] [PubMed] [Google Scholar]
- 15. Ikebe T., Iyoda S., Kutsukake K. 1999. Promoter analysis of the class 2 flagellar operons of Salmonella. Genes Genet. Syst. 74: 179–183 [DOI] [PubMed] [Google Scholar]
- 16. Iyoda S., Kamidoi T., Hirose K., Kutsukake K., Watanabe H. 2001. A flagellar gene fliZ regulates the expression of invasion genes and virulence phenotype in Salmonella enterica serovar Typhimurium. Microb. Pathog. 30: 81–90 [DOI] [PubMed] [Google Scholar]
- 17. Iyoda S., Kutsukake K. 1995. Molecular dissection of the flagellum-specific anti-sigma factor, FlgM, of Salmonella typhimurium. Mol. Gen. Genet. 249: 417–424 [DOI] [PubMed] [Google Scholar]
- 18. Kage H., Takaya A., Ohya M., Yamamoto T. 2008. Coordinated regulation of expression of Salmonella pathogenicity island 1 and flagellar type III secretion systems by ATP-dependent ClpXP protease. J. Bacteriol. 190: 2470–2478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Karlinsey J. E., et al. 2000. Completion of the hook-basal body complex of the Salmonella typhimurium flagellum is coupled to FlgM secretion and fliC transcription. Mol. Microbiol. 37: 1220–1231 [DOI] [PubMed] [Google Scholar]
- 20. Komarova A. V., Tchufistova L. S., Dreyfus M., Boni I. V. 2005. AU-rich sequences within 5′ untranslated leaders enhance translation and stabilize mRNA in Escherichia coli. J. Bacteriol. 187: 1344–1349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Komeda Y., Kutsukake K., Iino T. 1980. Definition of additional flagellar genes in Escherichia coli K12. Genetics 94: 277–290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Komeda Y. 1982. Fusions of flagellar operons to lactose genes on a Mu lac bacteriophage. J. Bacteriol. 150: 16–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koop A. H., Hartley M. E., Bourgeois S. 1987. A low-copy-number vector utilizing β-galactosidase for the analysis of gene control elements. Gene 52: 245–256 [DOI] [PubMed] [Google Scholar]
- 24. Kutsukake K. 1994. Excretion of the anti-sigma factor through a flagellar substructure couples the flagellar gene expression with flagellar assembly in Salmonella typhimurium. Mol. Gen. Genet. 243: 605–612 [DOI] [PubMed] [Google Scholar]
- 25. Kutsukake K. 1997. Autogenous and global control of the flagellar master operon, flhD, in Salmonella typhimurium. Mol. Gen. Genet. 254: 440–448 [DOI] [PubMed] [Google Scholar]
- 26. Kutsukake K., Ide N. 1995. Transcriptional analysis of the flgK and fliD operons of Salmonella typhimurium which encode flagellar hook-associated proteins. Mol. Gen. Genet. 247: 275–281 [DOI] [PubMed] [Google Scholar]
- 27. Kutsukake K., Iino T. 1994. Role of the FliA-FlgM regulatory system on the transcriptional control of the flagellar regulon and flagellar formation in Salmonella typhimurium. J. Bacteriol. 176: 3598–3605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kutsukake K., Ikebe T., Yamamoto S. 1999. Two novel regulatory genes, fliT and fliZ, in the flagellar regulon of Salmonella. Genes Genet. Syst. 74: 287–292 [DOI] [PubMed] [Google Scholar]
- 29. Kutsukake K., Nakashima H., Tominaga A., Abo T. 2006. Two DNA invertases contribute to flagellar phase variation in Salmonella enterica serovar Typhimurium strain LT2. J. Bacteriol. 188: 950–957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kutsukake K., Nambu T. 2000. Bacterial flagellum: a paradigm for biogenesis of transenvelope supramolecular structures. Recent Res. Dev. Microbiol. 4: 607–615 [Google Scholar]
- 31. Kutsukake K., Iyoda S., Ohnishi K., Iino T. 1994. Genetic and molecular analyses of the interaction between the flagellum-specific sigma and anti-sigma factors in Salmonella typhimurium. EMBO J. 13: 4568–4576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kutsukake K., Ohya Y., Iino T. 1990. Transcriptional analysis of the flagellar regulon of Salmonella typhimurium. J. Bacteriol. 172: 741–747 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kutsukake K., Ohya Y., Yamaguchi S., Iino T. 1988. Operon structure of flagellar genes in Salmonella typhimurium. Mol. Gen. Genet. 214: 11–15 [DOI] [PubMed] [Google Scholar]
- 34. Lanois A., Jubelin G., Givaudan A. 2008. FliZ, a flagellar regulator, is at the crossroads between motility, haemolysin expression and virulence in the insect pathogenic bacterium Xenorhabdus. Mol. Microbiol. 68: 516–533 [DOI] [PubMed] [Google Scholar]
- 35. Lee Y.-Y., Barker C. S., Matsumura P., Belas R. 2011. Refining the binding of the Escherichia coli flagellar master regulator, FlhD4C2, on a base-specific level. J. Bacteriol. 193: 4057–4068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Liu X., Matsumura P. 1994. The FlhD/FlhC complex, a transcriptional activator of the Escherichia coli flagellar class II operons. J. Bacteriol. 176: 7345–7351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Miller J. 1972. Experiments in molecular genetics, p. 431–433 Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 38. Mytelka D. S., Chamberlin M. J. 1996. Escherichia coli fliAZY operon. J. Bacteriol. 178: 24–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Ohnishi K., Kutsukake K., Suzuki H., Iino T. 1990. Gene fliA encodes an alternative sigma factor specific for flagellar operons in Salmonella typhimurium. Mol. Gen. Genet. 221: 139–147 [DOI] [PubMed] [Google Scholar]
- 40. Ohnishi K., Kutsukake K., Suzuki H., Iino T. 1992. A novel transcriptional regulation mechanism in the flagellar regulon of Salmonella typhimurium: an anti-sigma factor inhibits the activity of the flagellum-specific sigma factor, σF. Mol. Microbiol. 6: 3149–3157 [DOI] [PubMed] [Google Scholar]
- 41. Saini S., et al. 2010. FliZ induces a kinetic switch in flagellar gene expression. J. Bacteriol. 192: 6477–6481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saini S., Slauch J. M., Aldridge P. D., Rao C. V. 2010. Role of cross talk in regulating the dynamic expression of the flagellar Salmonella pathogenicity island 1 and type 1 fimbrial genes. J. Bacteriol. 192: 5767–5777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Shine J., Dalgarno L. 1975. Determinant of cistron specificity in bacterial ribosomes. Nature 254: 34–38 [DOI] [PubMed] [Google Scholar]
- 44. Uzzau S., Figueroa-Bossi N., Rubino S., Bossi L. 2001. Epitope tagging of chromosomal genes in Salmonella. Proc. Natl. Acad. Sci. U. S. A. 98: 15264–15269 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Wada T., et al. 2011. EAL domain protein YdiV acts as an anti-FlhD4C2 factor responsible for nutritional control of the flagellar regulon in Salmonella enterica serovar Typhimurium. J. Bacteriol. 193: 1600–1611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Wada T., Tanabe Y., Kutsukake K. 2011. FliZ acts as a repressor of the ydiV gene, which encodes an anti-FlhD4C2 factor of the flagellar regulon in Salmonella enterica serovar Typhimurium. J. Bacteriol. 193: 5191–5198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wang S., Fleming R. T., Westbrook E. M., Matsumura P., McKay D. B. 2006. Structure of the Escherichia coli FlhDC complex, a prokaryotic heteromeric regulator of transcription. J. Mol. Biol. 355: 798–808 [DOI] [PubMed] [Google Scholar]
- 48. Wozniak C. E., Chevance F. F., Hughes K. T. 2010. Multiple promoters contribute to swarming and the coordination of transcription with flagellar assembly in Salmonella. J. Bacteriol. 192: 4752–4762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wozniak C. E., Hughes K. T. 2008. Genetic dissection of the consensus sequence for the class 2 and class 3 flagellar promoters. J. Mol. Biol. 379: 936–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wozniak C. E., Lee C., Hughes K. T. 2009. T-POP array identifies EcnR and PefI-SrgD as novel regulators of flagellar gene expression. J. Bacteriol. 191: 1498–1508 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Yamamoto S., Kutsukake K. 2006. FliT acts as an anti-FlhD2C2 factor in the transcriptional control of the flagellar regulon in Salmonella enterica serovar Typhimurium. J. Bacteriol. 188: 6703–6708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yanagihara S., Iyoda S., Ohnishi K., Iino T., Kutsukake K. 1999. Structure and transcriptional control of the flagellar master operon of Salmonella typhimurium. Genes Genet. Syst. 74: 105–111 [DOI] [PubMed] [Google Scholar]
- 53. Zaslaver A., et al. 2006. A comprehensive library of fluorescent transcriptional reporters for Escherichia coli. Nat. Methods 3: 623–628 [DOI] [PubMed] [Google Scholar]