

Abstract
The transcription factor Sox2 is a key player in the maintenance of pluripotency and “stemness.” We have previously shown that Sox2 maintains self-renewal in the osteoblast lineage while inhibiting differentiation (U. Basu-Roy et al., Cell Death Differ. 17:1345-1353, 2010; A. Mansukhani, D. Ambrosetti, G. Holmes, L. Cornivelli, and C. Basilico, J. Cell Biol. 168:1065-1076, 2005). Sox2 also interferes with Wnt signaling by binding β-catenin, a central mediator of the Wnt pathway. Here we show that these multiple functions of Sox2 are encoded in distinct domains. The self-renewal function of Sox2 is dependent on its transcriptional activity and requires both its DNA-binding and C-terminal activation regions, while only the third C-terminal transactivation (TA) region is required for binding β-catenin and interfering with Wnt-induced transcription. The results of gene expression analysis upon Sox2 deletion strongly support the notion that Sox2 maintains stemness. We show also that Sox2 suppresses differentiation by attenuating Wnt signaling by posttranscriptional and transcriptional mechanisms and that adenomatous polyposis coli (APC) and GSK3β, which are negative regulators of the Wnt pathway, are direct Sox2 targets in osteoblasts. Several genes, such as the FoxP1 and BMI-1 genes, that are associated with stemness are downregulated upon Sox2 inactivation. Constitutive expression of the Polycomb complex member BMI-1 can bypass the Sox2 requirement for self-renewal but does not affect differentiation. Our results establish a connection between Sox2 and BMI-1 in maintaining self-renewal and identify BMI-1 as a key mediator of Sox2 function.
INTRODUCTION
To form bone, fat, muscle, and cartilage, multipotent mesenchymal stem cells (MSCs) must commit to a specific cell lineage and then undergo a sequential program of proliferation and differentiation. Primitive osteoprogenitors are thought to arise from multipotent MSCs that commit to and then differentiate into preosteoblast and mature bone-forming osteoblasts (4, 41). Signaling networks of fibroblast growth factors (FGFs), bone morphogenetic proteins (BMPs), Indian Hedgehog homolog (IHH), Wnt, and Notch orchestrate this complex process (20, 23), and the transcription factors Runx2 and Osterix (OSX) are essential for formation of the osteoblastic lineage (22, 32). However, little is known about other factors that regulate early cell fate decisions and the mechanisms that determine the balance between self-renewal and differentiation of osteoprogenitor cells (20).
We recently reported that the transcription factor Sox2 is essential for the self-renewal and proliferation of osteoblast precursors. Conditional knockout (cko) of the Sox2 gene in the osteoblastic lineage generates mice that, although mosaic with respect to Sox2 inactivation, are strongly osteopenic, and Sox2 inactivation in cultured primary osteoblasts has been shown to cause exhaustion of their proliferative ability and induction of senescence (6). Sox2 expression is highly enriched in a small subset of primary osteoblasts that form nonadherent spheres that are thought to represent a stem-like population (6). These findings suggested that Sox2 marks and maintains a population of multipotent or unipotent osteoblast stem cells that is responsible for self-renewal of the osteoblast lineage.
Sox2, a member of the Sox HMG box family of transcription factors, is required early in embryonic development to maintain pluripotency and self-renewal in embryonic stem (ES) cells (37). Sox2 is a maternally transmitted factor that is first expressed in the cells of the inner cell mass of the blastocyst. In the developing embryo, Sox2 expression is extinguished in most tissues, but it remains strongly expressed in stem cells of the central nervous system (CNS), retina, and the primordial gut (40, 42, 50) and plays an essential role in the maintenance of undifferentiated lineage progenitors such as neural stem cells (15, 40). It is also a critical reprogramming factor for the conversion of somatic cells to induced pluripotent stem cells (iPSC) (48).
In the osteoblast lineage, Sox2 is expressed at relatively low levels in immature cells, both in vitro and in vivo, and is induced by FGF signaling that stimulates proliferation of preosteoblasts and inhibits their differentiation (25). Constitutive expression of Sox2 can increase sphere formation and by itself can inhibit osteoblast differentiation. Sox2 also participates in the inhibitory effect of FGF on the prodifferentiation Wnt pathway by binding to β-catenin, a major effector of canonical Wnt signaling, and inhibiting its transcriptional activity (1, 25).
To identify the mechanisms and pathways by which Sox2 regulates the osteoblast lineage, we have dissected the Sox2 domains required for the functions of Sox2 that are responsible for self-renewal, inhibition of the Wnt pathway, and differentiation of osteoblasts. Using deletion mutants and chimeric fusions of the Sox2 DNA-binding domain with heterologous activation or repression domains, we demonstrate that the self-renewal and proliferation of osteoblastic cells requires both DNA binding and the transcriptional activation function of Sox2. While the C-terminal β-catenin binding domain is sufficient for inhibition of the Wnt pathway, the transcriptional activity of Sox2 also contributes to inhibition of the Wnt response.
An extensive microarray analysis of the gene expression changes in osteoblasts following Sox2 inactivation identified a number of genes whose expression is affected, directly or indirectly, by Sox2. The effects include downregulation of a large number of “stemness” genes and, as expected, of genes regulating cell proliferation. In contrast, expression of many genes in the Wnt pathway was disregulated. Negative regulators of Wnt signaling, such as adenomatous polyposis coli (APC) and GSK3β, are significantly downregulated upon Sox2 deletion and upregulated by its overexpression.
Finally, we show that BMI-1, a Polycomb complex transcriptional repressor which is known to be important for stemness in neural and hematopoietic cells, is an important Sox2 target gene in the osteoblast lineage. It is strongly downregulated following Sox2 inactivation, and constitutive BMI-1 expression can rescue the cell senescence induced by Sox2 deletion. Thus, Sox2 promotes osteoprogenitor self-renewal and expansion by activating transcription of critical stemness genes, and we have identified BMI-1 as a key downstream target in this process.
MATERIALS AND METHODS
Cell culture.
The Sox2flox/flox and Sox2flox/− immortalized osteoblast cell lines have been previously described (6). A stable Sox2flox/− osteoblast line expressing a Wnt-responsive luciferase reporter construct was generated by transfecting Sox2flox/− cells with a pcDNA-TOPflash plasmid (1) and selecting pools in G418 (800 μg/ml). The OB1 cell line is an immature osteoblast cell line that was previously characterized in our laboratory (26). The OB1-TOP cells express stably integrated Wnt-responsive luciferase constructs (1). Primary osteoblasts from P1 calvaria were isolated as previously described (26). All cells were maintained in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal bovine serum, in a humidified incubator with 7% CO2.
Colony assay.
For colony assay experiments, Sox2flox/flox or Sox2flox/− immortalized osteoblast cell lines were transduced with FUCRW lentivirus (29) constructs in the presence of Polybrene (8 μg/ml). Endogenous Sox2 deletion was elicited by infection with either high-titer green fluorescent protein (GFP)-or CRE-GFP-expressing lentivirus. At 72 h after infection, 1,000 cells/well were plated in triplicate in 6-well plates, and colonies were counted after 7 to 10 days by the use of crystal violet staining.
Western blot analysis and antibodies.
Whole-cell lysates were prepared in 0.5% of Triton X-100 lysis buffer and used for Western blotting and immunoprecipitation. Anti-active β-catenin antibody was from Millipore (clone 8E7). Anti-phospho-β-catenin (Thr 41 and Ser 45) (catalog no. 9565) and anti-Sox2 antibody (catalog no. 2748) were from Cell Signaling. For detection of Sox2 deletion mutants, specific Sox2 antibodies from Novus Biologicals (catalog no. NB110-79875) and Activemotif (catalog no. 39823) were used. For immunoprecipitation, the Sox2 antibody used was from Santa Cruz (catalog no. sc-17320). β-Catenin monoclonal and GSK3β antibodies were from BD Transduction Laboratories. Actin monoclonal antibody was from Sigma. LexA antibody (catalog no. sc-7544) was from Santa Cruz. Recombinant mouse Wnt3A was from R&D Systems. CHIR99021 was from Stemgent. BMI-1 and γ-tubulin antibodies were from Sigma.
Expression vectors, cloning, and lentivirus production.
Sox2 and Sox2 deletion mutants and chimeric constructs were cloned in the FUCRW lentiviral vector (29). BMI-1 and Foxp1 were cloned from murine ES cells and osteoblast cDNA, respectively, into the XbaI and EcoRI sites of FUCRW vector. The FUGW (GFP control) and CRE-internal ribosome entry site-GFP (CRE-IRES-GFP)-expressing lentivirus constructs used were a kind gift from Richard Huganir (49). All lentiviruses used were produced using 293T cells and pVSV-G, pLP1, and pLP2 as helper plasmids. To obtain Sox2-expressing lentiviral constructs, Sox2 was amplified from pCEP4-Sox2 (3) by using XbaI linkers. Sox2 PCR products were digested with XbaI and subcloned into XbaI sites of FUCRW. To construct lentiviral constructs expressing Sox2 1-310, 1-278,1-255,1-248,1-230, 1-129, 31-319, 31-129, 121-319, and LexA, pCEP4 deletion mutants (3) were digested with NheI and BamHI and then blunt-end cloned in the XbaI and EcoRI sites of FUCRW. LexA 121-319 DNA fragments were amplified by using pCEP4-LexA 121-319 plasmid with primers containing XbaI linkers (25). LexA 121-319 PCR products were digested with XbaI and cloned into FUCRW. The HMG (amino acids [aa] 1 to 129)-VP16 chimeric molecule was cloned in FUCRW. The region comprising aa 1 to 129 of Sox2 was amplified by PCR using a forward primer (F1), 5′-GCT CTA GAA TGT ATA ACA TGA TGG AGA CG, containing an XbaI site and a reverse primer (R1), 5′-ACG CGC GCG GCT CGT CTT GGT TTT CCG CCG CGG, containing the 5′ forward sequences of VP16. The VP16 activation domain was generated by PCR using a forward primer (F2), 5′-AGC CGC GCG CGT ACG AAA AAC, and a reverse primer (R2), 5′-CGG AAT TCC TAC CCA CCG TAC TCG TCA A, containing EcoRI sites. The HMG and VP16 PCR products were combined for use as a template for PCR with primer F1 and R2. The resulting PCR product was digested with XbaI and EcoRI and inserted into XbaI-EcoRI sites of FUCRW. The HMG (aa 1 to 129)-Engrailed repressor, LexA VP16 chimeric construct, and Sox2 Δ129-254 were constructed by the same strategy as that used for the HMG VP16 product. An Engrailed repressor domain (kindly provided by A. Sharrocks) was amplified by PCR (52). The forward primers and reverse primers for each chimeric protein were as follows: for the HMG Engrailed repressor, the F1 forward primer was the same as the forward primer to Sox2, the R1 reverse primer was 5′-CTC CAG GGC CGT CTT GGT TTT CCG CCG CGG, the F2 forward primer was 5′-GCC CTG GAG GAT CGC TGC AGC, and the R2 reverse primer was 5′-CGG AAT TCG GAT CCC AGA GCA GAT TTC TC; for LexA VP16AD, the F1 forward primer was 5′-GCT CTA GAA TGA AAG CGT TAA CGG CCA GG, the R1 reverse primer was 5′-CGT ACG CGC GCG GCT AGC GGC CGC CGG GGC CTC CAT, and the F2 forward and R2 reverse primers were the same as those used for VP16 AD; and for Sox2 Δ129-254, the R1 reverse primer was AAC CAC GGG CTG GTA CTT ATC CTT CTT CAT, the F2 forward primer was AAG TAC ACG CCC GTG GTT ACC TCT TCC TCC, and the F1 forward and R2 reverse primers were the same as those used for Sox2.
Microarray hybridization.
After infection with enhanced GFP (EGFP) (control)- or Cre (linked to IRES-GFP to detect GFP expression)-expressing lentivirus for 24, 48, or 72 h, total cellular RNA was extracted from Sox2flox/− and Sox2flox/flox cells as previously described (6) and purified (RNeasy; Qiagen). Three replicate experiments were performed for each set of conditions. RNA quality was assessed using an Agilent 2100 Bioanalyzer and a Nanodrop ND-1000 spectrophotometer. cRNA was prepared using 100 ng of RNA and an Affymetrix 3′ IVT Express labeling kit and array processing protocol (Affymetrix). cRNAs were hybridized to mouse genome 430A 2.0 arrays (Affymetrix), which contain 22,690 gene probes. Arrays were processed and scanned at the New York University Medical Center Genome Technology Center according to procedures recommended by the manufacturer (Affymetrix).
Analysis of gene expression data.
Raw expression data (CEL files) were generated using GCOS 1.4 software (Affymetrix) and normalized using GeneSpring GX11 software (Agilent, Santa Clara, CA), the Robust Multichip Average algorithm (RMA) for probe level normalization, and principal component analysis (PCA) for examining variances in the data. Genes expressed differentially between GFP and CRE samples at each time point were identified on the basis of statistically significant differences (t test; P < 0.05) in relative transcript intensity signal values and a change of ≥1.66-fold by the use of GeneSpring software. Significantly enriched gene sets were determined by (i) the Gene Set Enrichment Analysis (GSEA) method (http://www.broadinstitute.org/gsea) using the entire data set at 48 and 72 h and (ii) differential expression analysis (change > 1.66-fold; P ≤ 0.05) of differences between GFP- and CRE-expressing lentivirus-infected cells at 48 or 72 h after infection.
In sublists of genes differentially regulated in the two cell lines, enrichment scores were determined for gene sets containing differentially expressed genes by the functional annotation feature of the DAVID bioinformatics database (http://david.abcc.ncifcrf.gov/).
Wnt-responsive luciferase activity.
OB1-TOP cells were transduced with Sox2 expression constructs and treated with recombinant Wnt3A protein (100 ng/ml) for 24 h, after which luciferase activity was measured using Promega luciferase assay reagents as described by the manufacturer. Wnt activation in Sox2 null cells in a Sox2flox/− osteoblast line expressing a Wnt-responsive luciferase reporter construct was measured after 72 h of CRE-expressing lentivirus infection and 24 h of Wnt3A stimulation.
ChIP assay.
The chromatin immunoprecipitation (ChIP) assay was performed by established protocols (34, 35). A sonicator (Misonix 3000) was used to shear chromatin. For immunoprecipitation, chromatin samples were precleared with IgG and 15 μg of precleared samples was incubated overnight with polyclonal anti-sox2 goat antibody (catalog no. AF2018; R&D Systems). A 5-μg volume of each sample was used as the input. Protein A beads were added, and samples were washed at least 5 times. Primers (see Fig. 6E) to A included forward (F) primer AAC ATC AAC CCC CAT CTG AA and reverse (R) primer GCA GGC CAG AAA TAC AGA GC; primers to B included forward (F) primer AGG GCA TCC ACC TTA ACC TT and reverse (R) primer CCA GAG TTC ATC CCT ACC ACA; and primers to C included forward (F) primer AGT CAG GAT TGC CAG TGT CC and reverse (R) primer AAT TGC TCG CAG CAG TTT TT.
Fig. 6.

Deregulation of the Wnt signaling pathway in Sox2-depleted cells. (A) Heat map showing expression of Wnt pathway genes in triplicate samples of GFP- and CRE-infected Sox2flox/flox cells at 24, 48, and 72 h. The most strongly upregulated (orange) and downregulated (blue) gene clusters are shown in an expanded view on the right. (B) Gene expression analysis of Wnt target genes in osteoblasts following Sox2 deletion. Sox2flox/flox cells were transduced with either GFP- or CRE-expressing lentivirus for 72 h. Expression levels of the indicated genes were analyzed using qRT-PCR and specific primers. All values are normalized to actin and are expressed as fold changes in comparison to vector-infected control results. (C) APC and GSK3β are downregulated in Sox2-depleted osteoblasts. Sox2flox/flox cells were transduced with either GFP- or CRE-expressing lentivirus for 72 h. Expression levels of APC and GSK3β were analyzed using qRT-PCR and specific primers. All values are normalized to actin and are expressed as fold changes in comparison to vector-infected control results. *, P < 0.05. (D) Wnt reporter activity in Sox2-depleted osteoblasts. Sox2flox/− cells containing stably transfected pTOPflash vector were infected with either GFP- or CRE-expressing lentivirus for 48 h and treated with control or Wnt3A-conditioned media. Luciferase activity was measured as described in Materials and Methods. *, P < 0.05. (E) APC and GSK3β are direct targets of Sox2 in osteoblasts. Cells were infected with empty vector or Sox2 and used for ChIP analysis using anti-Sox2 antibody or IgG as a negative control. PCR was performed using immunoprecipitated chromatin and primers specific for APC or GSK3β promoter regions (see Fig. S4 at http://www.med.nyu.edu/sites/all/files/Seo_et_al_MCB_Supplementary_figures.pdf).
Quantitative RT-PCR (qRT-PCR) analysis.
Total cellular RNA extracted from cells after infection with EGFP (control) or Cre (linked to IRES-GFP to detect GFP expression) virus for 24, 48, or 72 h was purified as described previously (6). For expression analysis of Sox2-regulated genes in bones of osteoblast-specific Sox2 knockout mice, total RNA from calvaria (p1 pups) or femurs (8-week-old mice) was extracted after the bone was cleared of mesenchyme and surrounding connective tissue. Reverse transcription (RT) and real-time PCR analysis were carried out using specific primers. Actin or 18S rRNA was used as a normalization control.
RESULTS
Sox-mediated self-renewal of osteoblasts requires it to be transcriptionally active.
Sox2 is a member of the HMG domain, SRY-related transcription factor family that acts by binding to the DNA consensus sequence T/AT/ACAAAGA (17, 51, 54). It spans 319 amino acids and contains a 79-amino-acid HMG DNA binding domain and several C-terminal transcriptional activation domains (54). So far, Sox2 has been shown to directly regulate transcription, generally in combination with other DNA binding partners of the POU domain family, the best known of which is Oct4 (2, 3, 54). In osteoblasts, however, Sox2 can perform other functions, such as the inhibition of Wnt signaling, that do not involve its transcriptional activity, as it does not require the Sox2 DNA binding region. We had previously shown that Cre-mediated Sox2 excision in Sox2 floxed cultured osteoblasts resulted in abolishing their colony-forming ability and that the ability to form colonies could be rescued by the introduction of a transgenic copy of wild-type Sox2 (6). To determine which of the Sox2 domains was critical for the self-renewal of the osteoblast lineage, we introduced into Sox2flox/− immortalized osteoblasts (in which one of the Sox2 alleles is a null and the other is flanked by LoxP sequences) a transgenic copy of the wild type or of deletion mutants of Sox2 by the use of lentivirus vectors, which typically results in >90% cell transduction. Cells were then infected with a GFP (control)- or Cre-expressing lentivirus to induce Sox2 excision, and the colony-forming ability was measured as described previously (6).
The Sox2 deletion mutants and data revealing their ability to rescue colony formation are shown in Fig. 1. Cre-expressing virus infection of Sox2flox/− cells caused an approximately 90% loss of colony-forming ability; the surviving colonies represented cells which had escaped Cre-expressing virus infection. In contrast, as previously shown, expression of a transgenic copy of wild-type Sox2 rescued essentially the entire cell population from the lethality induced by endogenous Sox2 excision (Fig. 1B). Deletion of the 31 N-terminal amino acids of Sox2 upstream of the DNA binding domain (aa 31 to 319) did not affect rescue, but deletions of the C-terminal 71 amino acids, which contain the strongest Sox2 TA domain (R1), abolished the rescue ability of Sox2. Importantly, deletion of the HMG DNA binding domain also abolished rescue, and this result was not affected by the replacement of the HMG domain by the DNA binding domain of LexA, which provides nuclear location signals (NLS) that are contained within the Sox2 HMG domain (Fig. 1B). These results therefore indicate that Sox2 maintains osteoblast self-renewal by regulating transcription, as both the DNA binding and transcriptional activation domains are required. Most transcription factors, however, can function as transcriptional activators or repressors, depending on the cellular context. To determine whether the ability of Sox2 to maintain osteoblast proliferation requires activation or repression of gene transcription, we created “chimeric” transcription factors in which the Sox2 DNA binding domain was fused to the herpesvirus VP16 activation domain (HMG-VP16) or the Engrailed repressor domain (HMG-Eng) (52). Both the HMG-VP16 and HMG-Eng domains behave as potent activators and repressors, respectively, in other systems (52, 53). The results obtained show that the Sox2-VP16 chimeric factor could rescue the colony-forming ability in Sox2-depleted cells as efficiently as wild-type Sox2, while the Sox2-Engrailed chimera exhibited no activity, despite being expressed at levels higher than that of the Sox2-VP16 fusion (Fig. 1C and D). Thus, maintenance of osteoblast self-renewal by Sox2 requires its function as a transcriptional activator.
Fig. 1.

Identification of Sox2 domains required for self-renewal and the inhibition of Wnt signaling. (A) Schematic representation of Sox2 constructs and summary of their activity in rescue of colony formation and Wnt inhibition. +++, complete effect; ++ and +, partial effect; +/−, negligible effect; −, no effect; HMG, DNA binding domain of Sox2; LEXA, DNA binding domain of LexA; RI, R2, and R3, transactivation domains of Sox2; VP16, activation domain of VP16; Eng, repressor domain of Engrailed. The results of a colony assay and a Wnt reporter assay are presented in panel B and Fig. 2A. (B) Colony assay with Sox2 deletion mutants. Sox2-floxed (Sox2flox/flox) osteoblasts were transduced with vector, Sox2, or the indicated mutants for 36 h and then reinfected with either GFP or CRE lentivirus for 72 h. A total of 1,000 cells were plated in triplicate in six-well plates. Colonies were stained and counted after 10 days. Numbers of colonies obtained in the CRE infection are plotted as percentages of the number of colonies obtained in the corresponding GFP infection. *, P < 0.05. (C) Colony assay with Sox2 chimeric proteins. Sox2flox/flox osteoblasts were transduced with vector, Sox2, HMG-VP16, or HMG-Eng, and a colony assay was performed as described for panel B. *, P < 0.05. (D) Western analysis of Sox2 mutants and chimeric proteins. Sox2flox/flox osteoblasts were infected with the indicated constructs as described above. Expression of viral constructs was verified using antibodies against the C-terminal or HMG domain of Sox2 or LexA. Error bars, standard deviations (SD).
Inhibition of Wnt signaling by Sox2 involves independent Sox2 functions.
As discussed above, Sox2, which is strongly induced by FGF in osteoblasts, also plays a major role in the inhibition of Wnt signaling mediated by FGF in these cells (25). This function had been previously mapped to the C-terminal half of Sox2, which can bind β-catenin, and did not require the DNA binding HMG domain (25). These experiments, however, had been conducted using 293 cells transfected with a Wnt reporter plasmid and a plasmid encoding a constitutively activated β-catenin lacking the N-terminal domain that is targeted for phosphorylation and β-catenin degradation. As the roles of Wnt signaling differ in different cell types (13, 38), we therefore considered it necessary to repeat those experiments in the proper cellular context of Wnt-treated osteoblasts expressing only endogenous β-catenin. We used OBI-TOP, an immature osteoblast cell line that we had previously described (1) that carries a stably integrated Wnt-responsive TOPflash plasmid. In these cells, Wnt3A-induced luciferase activity is repressed by FGF (1). OBI-TOP cells were transduced with lentivirus vectors expressing wild-type Sox2 or the deletion mutants shown in Fig. 1 and were then treated with Wnt3A, and the luciferase activity was measured (Fig. 1 and 2A).
Fig. 2.

Independent domains of Sox2 inhibit the Wnt-β-catenin pathway. (A and B) Wnt-responsive luciferase activity in osteoblasts expressing Sox2 mutants. OB1-TOP cells (osteoprogenitor cells containing a stably integrated Wnt-responsive luciferase reporter construct) were infected with empty vector or the indicated constructs for 36 h and plated in triplicate. The next day, cells were treated with Wnt3A (100 ng/ml) for 10 h (A) or with CHIR 99021 (4.5 μM) for 18 h (B), and luciferase activity was measured. *, P < 0.05. (C and D) Wnt-responsive luciferase activity in osteoblasts expressing Sox2-chimeric fusion proteins. OB1-TOP cells were infected with empty vector, Sox2, HMG-VP16, or HMG-Eng and treated with Wnt3A (100 ng/ml) for 10 h (C) or with CHIR 99021 (4.5 μM) for 18 h (D), and luciferase activity was measured as described in Materials and Methods. *, P < 0.05. Error bars, SD.
Progressive Sox2 deletions from the protein C terminus showed that amino acids 255 to 319 were essential for inhibition of Wnt-mediated luciferase activity. Deletion of this protein segment abolished the ability of Sox2 to interfere with the Wnt response of the TOPflash reporter (Fig. 2A). In contrast to the results of the rescue experiments, the DNA binding domain of Sox2 is not required for inhibiting Wnt signaling, as it can be replaced by the LexA DNA binding domain, which has no recognition sequences in mammalian DNA but contains nuclear localization signals (NLS) that substitute for the NLS encoded in the Sox2 HMG domain. These results therefore map the Sox2 region required for Wnt inhibition to the last 71 C-terminal amino acids (248 to 319). Additionally, to determine the effects of Sox2 on the Wnt pathway downstream of the region corresponding to β-catenin activation and stabilization, we stimulated the canonical Wnt pathway by the use of Chir99021, a potent GSK3β inhibitor (Fig. 2B). The Chir99021 results with were similar to those obtained with Wnt3A treatment, in line with the hypothesis that the major effect of Sox2 on the Wnt pathway is inhibition of the β-catenin function. An unexpected result was that the chimeric HMG-VPI6 protein was also able to inhibit the luciferase response to Wnt3A stimulation and, to a lesser extent, to Chir99021 (Fig. 2C and D). This was not a result of the intrinsic ability of the VP16 TA domain to inhibit Wnt signaling or to bind β-catenin, as a chimeric protein consisting of the LexA DNA binding domain fused to the VP16 TA domain exhibited no activity (not shown).
We therefore considered the possibility that the transcriptional activity of Sox2 would contribute to inhibition of Wnt signaling by directly regulating expression of genes in the Wnt pathway. In line with this hypothesis, we were able to show that Sox2 overexpression strongly induces expression of APC and GSK3β, two genes that play a key role in limiting the activity of the Wnt pathway, and that this induction was also promoted by the HMG-VP16 chimera but not by the LexA-Sox2 chimeric protein (Fig. 3A). Additionally, both wild-type Sox2 and the HMG-VP16 chimera induced a higher level of β-catenin phosphorylation (Fig. 3B). Further evidence that Sox2 regulates expression of APC, GSKβ, and other genes in the Wnt pathway is presented in the next section.
Fig. 3.

Sox2 inhibits Wnt signaling by induction of APC and GSK3β mRNA and by binding to β-catenin. (A) Gene expression analysis of APC and GSK3β in osteoblasts expressing Sox2 and chimeric proteins. OB1 cells were infected with vector, Sox2, LexA-121-319, or HMG-VP16 for 48 h. Expression levels of APC and GSK3β were analyzed using qRT-PCR and specific primers. All values are normalized to actin and are expressed as severalfold increases compared to vector-infected control results. *, P < 0.05. (B) Western analysis of phosphorylated β-catenin. Empty vector, Sox2, LexA 121-319, and HMG-VP16 viral vectors were expressed in OB1 cells, and phospho-β-catenin (serine 33 and threonine 45) was detected after 48 h. (C) Interaction of Sox2 (R1) with β-catenin. OB1 cells were infected with empty vector, Sox2, Sox2-Δ129-254 (containing R1 only), LexA 121-319, or Sox2 1-255 for 36 h. Cells were treated with control media or Wnt3A-conditioned media for 24 h. Immunoprecipitation (IP) was performed with anti-Sox2 antibody against the C terminus of Sox2 or against the HMG domain, and blot analysis was performed with anti-β-catenin antibody, anti-Sox2 HMG antibody, or anti-Sox2 C-terminal antibody. The lower panel shows expression levels of proteins in the whole-cell lysate used in IP. Error bars, SD.
We also further verified that the ability of the Sox2 C-terminal domain to inhibit Wnt signaling corresponded to the domain that interacted with β-catenin. As shown in Fig. 3C, mutant Sox2 plasmids that encoded the C-terminal portion of the Sox2 protein linked to the LexA DNA binding domain, or just to the C-terminal 64 amino acids (Sox2 Δ129-254), were able to interact with β-catenin, whereas a Sox2 mutant lacking this region (Sox2 1-255) was not. This interaction was enhanced by Wnt3A treatment, which increased β-catenin nuclear translocation (Fig. 3C).
In summary, we interpret these experiments as indicating that the ability of Sox2 to interfere with the Wnt pathway results from two mechanisms: (i) binding of the Sox2 C-terminal domain to β-catenin, which impairs β-catenin transcriptional function, and (ii) regulation of expression of critical genes in the Wnt pathway, which requires the transcriptional activity of Sox2, as further discussed in the next section.
Sox2 overexpression inhibits osteoblast differentiation (25). To identify the Sox2 domains required for this inhibition, we transduced the Sox2 mutants described in Fig. 1 into primary calvarial osteoblasts and determined their ability to undergo differentiation. Only wild-type Sox2, the 31-319 mutant, and the LexA-121-319 chimeric protein could inhibit differentiation (see Fig. S1 in the supplemental material). Importantly, deletion of the C-terminal 41 amino acids (mutant 1-278) abolished the ability of Sox2 to inhibit differentiation. Thus, these results suggest that the inhibition of osteoblast differentiation induced by Sox2 was mainly due to Sox2's ability to interfere with Wnt signaling.
Changes in gene expression profiles upon Sox2 deletion.
To investigate the mechanisms by which Sox2 inactivation causes loss of self-renewal and senescence and to identify potential Sox2 downstream targets, we examined the global gene expression profiles of the Sox2-floxed osteoprogenitor cells in which the Sox2 gene was deleted by CRE-mediated excision. Excision, growth arrest, and depletion of Sox2 were verified in accordance with our previous method (6).
Sox2flox/flox or Sox2flox/− cells were infected with GFP (control)- or CRE-expressing virus, and gene expression profiles were examined at 24, 48, and 72 h postinfection using Affymetrix mouse 430A 2.0 arrays. An overall profile of the data indicated that very few significant changes in gene expression were detected in GFP- versus CRE-infected cells at 24 h and that significant changes were detected at 48 h, but the most striking and extensive alterations were observed at 72 h (Fig. 4A) (see also Fig. S2A at http://www.med.nyu.edu/sites/all/files/seo_et_al_MCB_Supplementary_figures.pdf). By differential expression analysis (change > 1.66-fold by t test; P < 0.05) of Sox2flox/flox cells, 37, 421, and 6,342 entities were found to have changed at 24, 48, and 72 h, respectively. A similar pattern but with many more and earlier changes was seen with Sox2flox/− cells, possibly because only one Sox2 allele needs to be deleted in those cells (see Fig. S2 at the URL listed above). As seen in Fig. 4A, most (4,713 [74%] of 6,342 entities) of the changes resulted in downregulation, in line with the notion that Sox2 acts mostly as a transcriptional activator in these cells (Fig. 4A). The minimal change at 24 h was most likely due to the time required for Sox2 excision and the decay of the remaining endogenous Sox2 protein (Fig. 4A). At 72 h, many gene expression changes may have been an indirect effect of the inhibition of proliferation upon Sox2 excision. We therefore verified that the changes we focused on were also evident at earlier times in both cell lines. The changes in the expression levels of several Sox2-regulated genes are shown in Table S1 in the supplemental data).
Fig. 4.

Gene expression changes following Sox2 deletion. (A) Expression profiles of GFP- and CRE-infected Sox2flox/flox cells, showing entities upregulated >1.66-fold at 72 h. Normalized expression values plotted on a log2 scale were determined relative to expression in GFP-infected samples. Expression changes upregulated more than 2-fold are shown in red, and those downregulated over 2-fold are shown in blue. The right panel shows Sox2 and CRE protein expression as determined by Western analysis of GFP- and CRE-infected Sox2flox/flox cells at the indicated time points. Tubulin was used as a loading control. (B) Cell cycle genes are downregulated upon Sox2 deletion. A GSEA enrichment plot showing expression enrichment of a set of cell cycle-related genes is presented. A negative enrichment score indicates that expression of the majority of these genes in the GFP-infected cells was enriched. Bars represent individual genes in a ranked data set list. A heat map of the genes in the leading edge (most highly ranked) with the greatest significant differences revealed in experiments performed using triplicate samples is shown.
Genes significantly affected by Sox2 knockout (upregulated and downregulated more than 1.66-fold) were analyzed by functional annotation showing clustering of genes based on pathways and gene ontology keywords (see Table S2 at http://www.med.nyu.edu/sites/all/files/Seo_et_al_MCB_Supplementary_figures.pdf). The microarray data were also analyzed directly using the Gene Set Enrichment Analysis software (GSEA), a tool that evaluates the data in an unbiased manner using curated gene sets, based on prior knowledge (31, 46). Gene sets significantly enriched in GFP (downregulated upon Sox2 deletion)-infected cells or CRE (upregulated upon Sox2 deletion)-infected cells yielded insights into the biological functions regulated by Sox2 in osteoprogenitors.
Both types of analysis revealed that cell cycle- and mitosis-related genes were significantly downregulated upon Sox2 deletion, a predictable pattern consistent with the arrest of proliferation of the cells (Fig. 4B) (see Table S2 at http://www.med.nyu.edu/sites/all/files/Seo_et_al_MCB_Supplementary_figures.pdf). Several genes (Rasa1, KitL, Foxp1, Bmi-1, Tnrc6A, and SmarcaD1) related to stem cell maintenance in neural, hematopoietic, and embryonic stem cells were also significantly downregulated upon Sox2 deletion, further implicating Sox2 in the maintenance of stemness features in the osteoprogenitor cells (Fig. 5C). The downregulation of BMI-1, Foxp1, and KitL was confirmed by qRT-PCR (Fig. 5B). There was a significant downregulation of the genes (encoding Igf1, Igf1r, GSK3β, Pten, and Pdk2) in the MTOR mitogen/nutrient/energy/sensing pathway that are responsible for translational control and ATP sensing (not shown). Among the genes upregulated by the Sox2 deletion, there was also a significant enrichment of genes related to mitochondria and mitochondrial function and oxidation-reduction, suggesting that Sox2 depletion affects these processes (see Table S2 at http://www.med.nyu.edu/sites/all/files/Seo_et_al_MCB_Supplementary_figures.pdf). These findings may provide clues helping to identify the mechanism by which Sox2 deletion leads to osteoprogenitor senescence, as changes in oxidation are known to lead to senescence. In line with previous results, expression of many genes (e.g., osteocalcin, ALP, osteopontin, MEPE genes) characteristic of osteoblast differentiation was unchanged by Sox2 inactivation (not shown).
Fig. 5.

Stem cell genes are downregulated upon Sox2 deletion. (A) A GSEA enrichment plot showing expression enrichment of a set of common stem cell-related genes at 48 h is presented. Bars represent individual genes in a ranked data set list. Expression of the majority of these genes was enriched in the GFP-infected cells. A heat map of the genes in the leading edge showing the strongest downregulation in CRE-infected cells is present. (B) Gene expression analysis of stemness genes in osteoblasts following Sox2 deletion. Sox2flox/flox cells were transduced with either GFP or CRE lentivirus for 72 h. Expression levels of indicated genes were analyzed using qRT-PCR and specific primers. All values are normalized to actin and are expressed as fold changes in comparison to vector-infected control results. (C) Heat map of the indicated stem cell gene sets in GFP- or CRE-infected Sox2flox/− cells at 24, 48, and 72 h after infection. The expression level of all 22,690 entities is shown as a control. Significant downregulation upon Sox2 deletion is indicated in blue. Expression of BMI-1 from two independent BMI-1 probes in the same samples is shown in the bottom panel. Error bars, SD.
Gene expression changes in the Wnt pathway.
The Wnt signaling pathway has an overall anabolic effect on bone, and the canonical Wnt pathway promotes differentiation in osteoprogenitor cells (5, 8). We found that expression of several genes involved in Wnt signaling is altered upon Sox2 deletion. Known Wnt targets (Ctgf, CcnD1, Axin2, Timp3, and Wisp2) were upregulated along with Wnt ligands Wnt2 and Wnt5A and Wnt receptors Fzd1 and Fzd2, whereas negative Wnt regulators such as APC and GSK3β were downregulated, providing compelling evidence that the Wnt signaling pathway is derepressed upon Sox2 deletion (Fig. 6). qRT-PCR analysis was used to verify the increase in expression levels of osteoblastic Wnt target genes Axin2, Ctgf, and Timp3 (Fig. 6B) and the decrease in APC and GSK3β expression (Fig. 6C). To further verify the increased activity of the Wnt pathway after Sox2 inactivation, we introduced into our Sox2flox/− osteoblasts the TOPflash reporter plasmid driven by a Wnt-responsive promoter and tested the activity of this plasmid following Sox2 excision. Basal luciferase activity was strongly increased by Sox2 inactivation, and the response to Wnt3A treatment was also increased (Fig. 6D). This was also reflected in increased expression of active β-catenin (not shown). These findings are consistent with the inhibitory function of Sox2 in the canonical Wnt pathway in osteoblasts. The downregulation of APC and GSK3β suggests that, in addition to binding β-catenin and thereby inhibiting Wnt target gene transcription, Sox2 also regulates directly gene expression of negative Wnt regulators as well as of Wnt ligands and receptors.
To verify this hypothesis, we conducted a ChIP experiment in which the binding of Sox2 to putative DNA binding sites in the promoters of APC and GSK3β was measured. The results shown in Fig. 6E demonstrate a clear binding of Sox2 to fragments of the APC and GSK3 promoters that contain a Sox2 consensus binding site. Furthermore, binding was increased in chromatin extracted from cells overexpressing Sox2 (Fig. 6E). Thus, Sox2 can interfere with Wnt signaling in osteoblasts through its previously demonstrated ability to bind β-catenin and by direct transcriptional regulation of genes in the Wnt pathway.
BMI-1 is a downstream effector of Sox2 in osteoblast self-renewal.
As seen in the microarray analysis, several stemness genes were downregulated in osteoblasts in which the endogenous Sox2 gene was inactivated by CRE-mediated excision. Many of those genes, such as the BMI-1 and Foxp1 genes, have been implicated in the self-renewal of cells from other tissues such as the neural and hematopoietic stem cells (30, 39). Additionally, recent reports have demonstrated that these two genes are necessary for the maintenance of the mesenchymal lineage. FoxP1 is a transcription factor of the FOX/winged-helix DNA binding family that has been identified as a direct target of Sox2 in ES cells (12), and BMI-1 is a Polycomb group protein that is generally involved in chromatin remodeling, repression of gene expression, and stem cell maintenance (39, 47). In addition, BMI-1 knockout mice exhibit defective bone density, suggesting a role in maintaining osteoblast proliferation or commitment (55). We tested whether these two genes could rescue the failure to self-renew in Sox2 null osteoblasts. We introduced, via lentivirus-mediated gene transduction, transgenic copies of BMI-1 and Foxp1 and tested whether they could restore the ability to self-renew in Sox2 null osteoblasts. As seen in Fig. 7A, CRE-mediated deletion of Sox2 led to decreases in the endogenous levels of BMI-1 and Foxp1 proteins, confirming our microarray results. Introduction of a transgenic copy of BMI-1 substantially reduced the lethality caused by Sox2 excision, as seen by rescue of the colony-forming ability of Sox2-depleted osteoblasts. However, introduction of Foxp1 did not promote the self-renewal of Sox2 null osteoblasts (Fig. 7B). To confirm that the presence of BMI-1 was sufficient to rescue the lethality caused by Cre-mediated Sox2 excision, we propagated individual clones that overexpressed BMI-1 and lacked Sox2. Figure 7C shows that these clones had undetectable levels of Sox2 and high BMI-1 protein levels compared to the control cells. When the BMI-1-positive, Sox2-negative clones were tested for the ability to form colonies (Fig. 7D), it proved similar to that of control BMI-1-positive, Sox2-positive cells. However, the colonies formed by these cells were smaller than those formed by the control cells, and they proliferated more slowly than the BMI-1-positive, Sox2-positive cells, as demonstrated by a small but statistically significant decrease in BrdU incorporation (not shown). We tested whether BMI-1 overexpression, whether in a Sox2-positive or a Sox2 null background, could affect osteoblast differentiation by determining the ability of BMI-1-overexpressing cells to differentiate spontaneously or in response to BMP. Osteoblast differentiation was not inhibited by BMI-1 (Fig. 7E). These results indicate that BMI-1 is a critical downstream effector of the self-renewal machinery regulated by Sox2 but that it cannot by itself influence osteoblast differentiation. In line with the finding that BMI-1 knockout mice have reduced levels of osteoprogenitors (55), we found that depletion of BMI-1 by short hairpin RNA (shRNA) decreased colony formation in osteoblasts that could not be rescued by Sox2 overexpression (Fig. 7F), further supporting the notion that BMI-1 is downstream of Sox2.
Fig. 7.

BMI-1 is a critical regulator of Sox2-dependent self-renewal. (A) Western analysis of Sox2, Foxp1, and BMI-1 expression. Sox2flox/− cells were infected with empty vector or Sox2-, Foxp1-, or BMI-1-expressing lentivirus, and then endogenous Sox2 was deleted using CRE-expressing virus-based excision as described in Materials and Methods. Protein expression was analyzed by immunoblotting with the indicated antibodies. (B) Colony assay of Sox2-depleted cells expressing transgenic Sox2, Foxp1, or BMI-1. Cells were infected as described for panel A, and 1,000 cells were plated in triplicate. Colonies were stained and counted after 10 days. Numbers of colonies obtained after CRE infection are plotted as percentages of the numbers of colonies in the corresponding GFP infection. Each experiment was repeated at least twice. Results from a representative experiment are shown. *, P < 0.05. (C and D) High-BMI-1, Sox2-null cells can be propagated in culture. Colonies obtained from BMI-overexpressing and Sox2-deleted osteoblasts were analyzed by Western blotting (C) and colony assay (D) to determine the levels of the indicated proteins. The colony assay was performed as described above. (E) Differentiation of Sox2-positive, high-Bmi-1 osteoblasts and Sox2-negative, high-Bmi-1 osteoblasts. Sox2-positive, high-Bmi-1 cells and Sox2-negative, high-Bmi-1 cells were differentiated in the presence of BMP2 (100 ng/ml) and stained for alkaline phosphatase activity at the indicated times. (F) Sox2 overexpression does not restore colony formation by Bmi-1-depleted osteoblasts. OB1 cells were transduced with either an empty vector or Sox2-expressing virus and then infected with either scrambled or Bmi-1-specific shRNA. Western blot analysis confirmed expression of Sox2 and a decrease in Bmi-1 protein levels. For the colony assay, 1,000 cells were plated in triplicate. Numbers of colonies were counted after 10 days and are plotted as a percentage of control numbers. Each experiment was repeated at least twice. Results from a representative experiment are shown. *, P < 0.05. Error bars, SD.
Misregulation of Sox2-regulated genes in vivo.
To determine whether the changes in gene expression induced by Sox2 inactivation in osteoblast cell culture were also observed in an in vivo model of Sox2 inactivation, we measured the mRNA levels of a few Sox2-regulated genes in the bones (femurs and calvaria) of mice with an osteoblast-specific Sox2 conditional knockout (cko) that we have previously described (6) (Fig. 8). Although these mice are mosaic with respect to Sox2 inactivation, a significant decrease in expression of BMI-1 and APC mRNA was observed whereas the levels of CTGF mRNA were increased, in line with the changes we observed in cell culture. However, GSK3β expression was unchanged. The discrepancy observed in the case of GSK3β could be due to compensatory mechanisms that occur in vivo that would not be operational in our experiments involving acute Sox2 inactivation in culture.
Fig. 8.
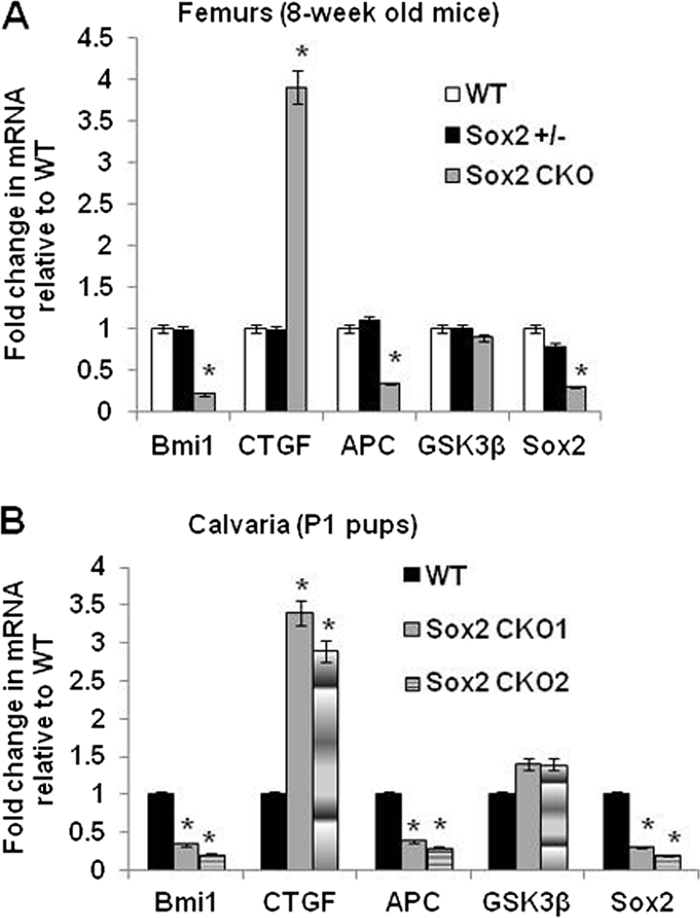
Expression of Sox2-regulated genes in the bones of mice with osteoblast-specific conditional knockout of Sox2 (6). Total RNA was extracted from femurs of 8-week old mice (A) and calvaria of P1 pups (B). Expression of indicated genes was analyzed by qRT-PCR using specific primers. All values are normalized to 18S rRNA and are expressed as fold change compared to wild-type (WT) results. Sox2 CKO, Sox2 conditional knockout. *, P < 0.05. Error bars, SD.
DISCUSSION
The results presented in this report show that Sox2 maintains self-renewal in the osteoblast lineage by activating transcription and by inhibiting the Wnt prodifferentiation pathway. The multiple functions of this transcription factor are both independent and overlapping. While inhibition of Wnt-induced transcription requires only the C-terminal, β-catenin binding domain of Sox2, Sox2 also transcriptionally regulates a number of genes in the Wnt pathway (Fig. 9).
Fig. 9.

Model for the multiple mechanisms by which Sox2 regulates the osteoblast lineage. Sox2 maintains stemness and self-renewal by transcriptional mechanisms, fostering expression of genes such as Bmi-1 that are necessary for the self-renewal of osteoprogenitor cells. Sox2 also regulates transcription of genes in the Wnt pathway (e.g., APC, GSK3β, and Fzd receptor genes) whose upregulation or downregulation would inhibit Wnt signaling. Additionally, Sox2 downregulates canonical Wnt signaling by interacting through its C-terminal activation domain (R1) with β-catenin, thus inhibiting the prodifferentiation Wnt pathway.
Extensive analysis of the changes in osteoblast gene expression that follow Sox2 inactivation confirm the hypothesis that Sox2 maintains osteoblasts in a stem-like state. While the changes in gene expression are multiple and have novel implications for the functions of this transcription factor, the most striking downregulation is observed with genes that have been considered diagnostic of stemness in embryonic, hematopoietic, and neural stem cells. Constitutive expression of one of those genes, the BMI-1 gene, rescues osteoblasts from the failure to self-renew following Sox2 inactivation.
Sox2 maintains osteoblast self-renewal by acting as a transcriptional activator.
Although Sox2 has a well-established canonical transcription factor function, its ability to interact with β-catenin and inhibit Wnt-induced transcription does not require the DNA binding domain and thus does not involve direct transcriptional regulation. We conducted a mutational analysis of the Sox2 domains required for self-renewal and inhibition of the Wnt pathway in osteoblasts. The results of these experiments clearly show that self-renewal requires Sox2 to act as a transcriptional activator. It requires the Sox2 DNA binding domain and the strongest C-terminal activation domain. Furthermore, the HSV-1/VP16 transcription-activating domain, an acidic sequence that has been shown to behave as a strong activation domain in other systems, could substitute for the Sox2 TA domain to restore colony-forming ability in Sox2 null cells.
The 79-aa HMG DNA binding domain of Sox2 closely resembles other HMG domains of Sox proteins with respect to DNA target recognition as well as the ability to bend DNA (19); therefore, it is unlikely that it alone can determine Sox2 target specificity. Several POU domain-containing partner factors for Sox2 have been identified, the best studied of which are Oct3-Oc4 (POU5F1) in embryonal stem cells (36). These partners confer target specificity by interacting with the Sox2-HMG domain and at adjacent sites in DNA. The specific Sox2 partner is thought to be an essential feature of the tissue-specific functions of Sox2 (10, 14, 33). The interaction of Sox2 with Oct4 that leads to activation of transcription of FGF4 is exquisitely specific (54). The POU family protein Oct1 cannot replace Oct4 in FGF4 transactivation, although it can bind to the same DNA element recognized by Oct4 (54). However, a Sox2 partner has not yet been identified in osteoblasts.
In our previous studies of the Sox2 domains involved in transcriptional regulation of the FGF4 genes in ES and embryonic carcinoma (EC) cells, we had identified 3 TA domains in the C-terminal portion of the Sox2 protein (3, 54). The two domains located closest to the 5′ end (R3 and R2) appeared to function only in conjunction with Oct4 to activate FGF4 transcription, while the most C-terminal domain (R1) was also capable of activating transcription by itself, using a reporter plasmid driven by multi-merized Sox2 binding sites (3, 14). In the present system, the finding that the strongest Sox2 activation domain is required and that it can be replaced by the VP16 TA domain suggests that in osteoblasts, Sox2 acts alone or in combination with a different partner than Oct4.
The ability of Sox2 to interfere with Wnt signaling mapped to the C-terminal R1 domain. Indeed, the same C-terminal domain can bind β-catenin. An unexpected result, however, was the ability of the HMG-VP16 chimera to depress the Wnt response. As discussed in detail in the next section, that appears to be have been due to the fact that Sox2 transcriptionally regulates several genes in the Wnt pathway whose up- or downregulation following Sox2 overexpression would negatively influence Wnt signaling.
Thus, although the inhibition of the Wnt pathway cannot by itself account for the ability of Sox2 to promote osteoblast self-renewal, it may contribute to it by keeping in check signals that would promote cell differentiation. In line with this notion, the Sox2 mutant constructs whose overexpression can inhibit osteoblast differentiation are the same as those that interfere with the Wnt response, suggesting that inhibition of osteoblast differentiation does not require the self-renewal function of Sox2.
Genes regulated by Sox2 in osteoblasts.
The microarray expression analysis showed that the majority of genes with expression profiles that are altered upon Sox2 deletion are downregulated, corroborating its role as a primarily transcriptional activator. The significant alterations reveal predictable and unexpected gene sets regulated by Sox2 in osteoblasts. Predictably, genes highly expressed in proliferating cells exhibit reduced expression in Sox2-deleted cells. Several genes known to regulate stemness in other systems (44) are also downregulated. Many of those genes, such as the FoxP1, Huwei1, Zcchc14, and Rasa1 genes, have been identified as direct targets of Sox2 in ES cells (9). To assess which of the regulated genes may be direct targets of Sox2, we analyzed expression of Sox2 target genes previously defined in studies of embryonic stem cells (9, 12, 27) On the basis of ChIP analyses and gene regulation, Boyer et al. demonstrated that transcription factors SmarcaD1 (chromatin remodeling), Myst3 (histone acetylation), and SkiL (transforming growth factor beta [TGF-β] signaling inhibitor) are direct targets of Sox2 in embryonic stem cells (12). All three genes were significantly downregulated upon Sox2 deletion in the osteoprogenitor cells (see Table S1 in the supplemental material). An analysis of genes in a large (734-gene) set of Sox2 targets previously defined by ChIP in studies of embryonic stem cells (9, 12) reveals an overlap of 214 genes with differentially regulated genes in control versus Sox2-deleted osteoprogenitor RNAs (see Fig. S3A at http://www.med.nyu.edu/sites/all/files/Seo_et_al_MCB_Supplementary_figures.pdf). The most upregulated of these genes include Fzd2, Ctgf, and Tead2, while the downregulated genes include stem cell transcription factors and factors involved in chromatin remodeling (Rasa1, Smarcad1, Nusap1, Huwei1, Foxp1, and Jarid2). The severalfold changes in regulation of several of these genes are shown in Table S1 in the supplemental material).
Our analysis of significant pathways and gene ontology terms associated with 173 of these potential targets significantly regulated in both cell lines includes Wnt signaling and RNA processing (see Fig. S3B at http://www.med.nyu.edu/sites/all/files/Seo_et_al_MCB_Supplementary_figures.pdf). These data further support a role for Sox2 in regulating Wnt signaling in these cells and strongly implicate it in processes related to RNA processing. Interestingly, some of the regulated genes (such as Huwei1, Kif11, and Foxp1) do not appear to contain binding sites for Oct4 or Nanog, the other two pluripotency factors in ES cells, which further suggests that Sox2 may function alone or with an unidentified partner at several of the target genes. Accordingly, we did not find evidence of either Oct4 or Nanog expression in osteoblasts. Analysis of upregulated gene sets after Sox2 deletion suggests that Sox2 plays some unexpected roles in suppressing mitochondrial redox-related functions, fatty acid oxidation, and metalloprotease activity as well as in RNA splicing and micro-RNA processing. The increase in mitochondrial redox activity could contribute to the senescent phenotype in Sox2 null osteoblasts. Sox2 is known to bind DNA in the minor groove, a common feature in RNA-binding proteins. These potential functions of Sox2 remain to be explored.
Regulation of Wnt signaling by FGF and Sox2.
The Wnt pathway plays important roles in osteoblast and bone development and is generally considered to be a prodifferentiation signal in osteoblasts and an overall anabolic signal in bone tissue (16, 21, 28, 43). Our previous studies of the antagonistic effect of FGF on osteoblast differentiation and on the Wnt prodifferentiation pathway had identified multiple mechanisms by which FGF signaling inhibited Wnt-induced transcription in osteoblasts. We showed that Sox2 induction by FGF played a role in this process via the ability of Sox2 to bind β-catenin and inhibit Wnt-induced transcription (1, 25). Those reports also showed that Wnt induction of several osteoblast-specific Wnt target genes was inhibited by FGF but that other genes in the Wnt pathway (most notably the Fzd Wnt receptors) were also downregulated by FGF by mechanisms apparently unrelated to the inhibition of canonical Wnt signaling.
Our current findings reveal new aspects of the role of Sox2 in inhibiting Wnt signaling in osteoblasts. In this report, we show that the Sox2 interaction with β-catenin occurs via its C-terminal domain (aa 255 to 319) and identify another mechanism of Wnt-β-catenin inhibition by Sox2. Sox2 deletion results in greatly reduced expression of GSK3β and APC, which negatively regulate the Wnt pathway by their participation in the β-catenin destruction complex. In contrast, we found that Sox2 overexpression strongly upregulated GSK3β and APC and increased phosphorylation of β-catenin (Thr-Ser). Sox2 binds to specific regions upstream of the transcription initiation sites of both GSK3β and APC promoters in osteoblasts. Significantly, the Sox2 binding sequences in APC and GSK3β are conserved across different species, suggesting a general mechanism by which Wnt-β-catenin signaling is regulated by Sox2.
The Sox2 HMG-VP16 fusion protein was also able to induce APC and GSK3β expression, which could account for the inhibitory effect of the VP16 fusion on the TOPflash Wnt reporter. Thus, the transcriptional function of Sox2 also contributes to negative regulation of Wnt-β-catenin signaling by maintaining expression of negative Wnt regulators. In line with this, we also demonstrated that basal Wnt activity is derepressed by Cre-mediated deletion of Sox2 in osteoblasts. Although that would be expected to lead to increased differentiation, expression of osteoblast differentiation-related genes such as the Runx2, OSX, ALP, Col1, and OPN genes is not increased upon Sox2 inactivation. It is possible that differentiation requires some degree of proliferation and that the Sox2-deleted cells were unable to sustain proliferation. In fact, depletion of Sox2 by the activity of shRNA in undifferentiated osteosarcoma cells, where low levels of Sox2 expression are maintained, leads to upregulation of Wnt signaling and robust osteoblastic differentiation (6a).
Among the genes upregulated upon Sox2 deletion are Ctgf, Wisp2, Axin2, and Timp3, which are known Wnt targets in other systems and are downregulated by FGF (1). However, the upregulated genes also include the genes encoding Wnt receptors Fzd1 and Fzd2 and Wnt ligands Wnt2, -3, -5, and -10a, which have not been recognized as Wnt targets. Thus, it is likely that Sox2 regulates expression of these genes at the transcriptional level. Indeed, we have found that Sox2 overexpression downregulates the level of Fzd1 RNA (not shown). While confirmation would require further studies, that result implies that the transcription of these genes would be negatively regulated by Sox2, which would act as a transcriptional repressor in this case. The interaction of Sox2 with β-catenin and the inhibitory effect of the association are likely to vary at target promoters according to the specific cofactors and complexes assembled. In ES cells, Sox2 signaling and Wnt signaling are both required to maintain stemness and pluripotency and thus do not appear to have overall antagonistic functions. Taken together, our results suggest that Sox2 is a major mediator of the antagonistic effect of FGF on the Wnt pathway in osteoblasts. Sox2 levels could act as a rheostat to determine the extent of proliferation or differentiation, with high Sox2 levels leading to Wnt repression and maintenance of the undifferentiated state.
BMI-1 is a critical downstream target of Sox2.
The stem cell gene Bmi-1 was strongly downregulated upon Sox2 deletion and upregulated by Sox2 overexpression. BMI-1 belongs to the Polycomb group of transcriptional repressors, is part of the Polycomb PRC1 repressor complex, and has several important functions ascribed to it (39). Importantly, BMI-1 is required for the self-renewal of neural and hematopoietic stem cells (30). PRC1 and -2 are essential for chromatin silencing and maintaining pluripotency, and BMI-1 is thought to maintain self-renewal by repressing genes involved in senescence. In ES cells, PRC1 and -2 act as chromatin modifiers to ensure proper lineage commitment by silencing expression of key regulator genes that specify lineage commitment and differentiation (45, 47). In addition to other defects, mice deficient in BMI-1 exhibit low osteoblast numbers and bone density, suggesting a role in osteoblastogenesis (55).
The rescue of Sox2-deficient osteoprogenitors by a single gene, Bmi-1, was somewhat surprising, given the large number of genes regulated by Sox2. Although BMI-1-rescued colonies were smaller, the cells were able to bypass senescence and regain the capacity for self-renewal. Thus, the complementation of Sox2 function by BMI-1 may be only partial. In osteoprogenitor cells, Polycomb group genes Bmi-1, Suz12, and Ezh2 are all downregulated upon Sox2 deletion, suggesting that Sox2 may maintain stemness in osteoprogenitors by maintenance of Polycomb complexes for self-renewal and repression of lineage commitment. Although their function is unclear, Polycomb complexes are also associated with several noncoding RNAs (ncRNAs) (7). We found that many ncRNAs (e.g., Xist and Tugb1, ncRNAs) are also downregulated in Sox2-deficient cells. Recently, BMI-1 has been implicated in other important cellular processes such as DNA repair and mitochondrial redox homeostasis (18, 24). We found that several mitochondrial oxidation genes are upregulated upon Sox2 deletion, suggesting an alteration in redox homeostasis that could lead to cell senescence. Thus, BMI-1 may substitute for Sox2 in osteoprogenitor self-renewal by preventing senescence due to misregulation of oxidation homeostasis.
Importantly, Sox2-positive or null clones overexpressing BMI-1 are able to undergo osteoblast differentiation. Thus, BMI-1, unlike Sox2, cannot block osteoblast differentiation, which implies that BMI-1 cannot entirely substitute for Sox2 for all Sox2 functions. Bmi-1 has not been described as a direct Sox2 target gene in embryonic stem cells, and examination of the promoter region does not reveal putative Sox2 binding sites. Currently, the mechanism by which Sox2 maintains Bmi-1 expression for self-renewal is not known. Micro-RNAs that repress Bmi-1 expression have been previously described (11), and our preliminary analysis of micro-RNAs regulated by Sox2 suggests that such regulation may represent a potential mechanism by which Sox2 helps to maintain Bmi-1 expression. Future experiments are planned to explore this hypothesis and the mechanisms by which BMI-1 rescues Sox2 deficiency in the osteoblast lineage.
Supplementary Material
ACKNOWLEDGMENTS
This investigation was supported by grants ARO51358 from the NIAMS and DE 013765 from the NIDCR. U.B.-R. is a recipient of a fellowship from the Children's Cancer Research Fund.
We thank Angus Wilson for the gift of VP16 plasmid, A. D. Sharrocks for the Engrailed plasmid, and Sally Temple for the BMI-1 shRNA plasmid. We thank the NYULMC Genome Technology Center for expert assistance with microarray gene expression profiling.
Footnotes
Supplemental material for this article may be found at http://mcb.asm.org/.
Published ahead of print on 19 September 2011.
REFERENCES
- 1. Ambrosetti D., Holmes G., Mansukhani A., Basilico C. 2008. Fibroblast growth factor signaling uses multiple mechanisms to inhibit Wnt-induced transcription in osteoblasts. Mol. Cell. Biol. 28:4759–4771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Ambrosetti D. C., Basilico C., Dailey L. 1997. Synergistic activation of the fibroblast growth factor 4 enhancer by Sox2 and Oct-3 depends on protein-protein interactions facilitated by a specific spatial arrangement of factor binding sites. Mol. Cell. Biol. 17:6321–6329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ambrosetti D. C., Scholer H. R., Dailey L., Basilico C. 2000. Modulation of the activity of multiple transcriptional activation domains by the DNA binding domains mediates the synergistic action of Sox2 and Oct-3 on the fibroblast growth factor-4 enhancer. J. Biol. Chem. 275:23387–23397 [DOI] [PubMed] [Google Scholar]
- 4. Aubin J., Triffit J. 2002. Mesenchymal cells and osteoblast differentiation, p. 59–82 In Bilezikian J., Raisz L. G., Rodan G. A.(ed.), Principles of bone biology, vol. 1 Academic Press, New York, NY [Google Scholar]
- 5. Baron R., Rawadi G. 2007. Targeting the Wnt/beta-catenin pathway to regulate bone formation in the adult skeleton. Endocrinology 148:2635–2643 [DOI] [PubMed] [Google Scholar]
- 6. Basu-Roy U., et al. 2010. The transcription factor Sox2 is required for osteoblast self-renewal. Cell Death Differ. 17:1345–1353 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6a. Basu-Roy U., et al. 19 September 2011. Sox2 maintains self renewal of tumor-initiating cells in osteosarcomas. Oncogene doi:10.1038/onc.2011.405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beisel C., Paro R. 2011. Silencing chromatin: comparing modes and mechanisms. Nat. Rev. Genet. 12:123–135 [DOI] [PubMed] [Google Scholar]
- 8. Bennett C. N., et al. 2005. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl. Acad. Sci. U. S. A. 102:3324–3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ben-Porath I., et al. 2008. An embryonic stem cell-like gene expression signature in poorly differentiated aggressive human tumors. Nat. Genet. 40:499–507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bernard P., Harley V. R. 2010. Acquisition of SOX transcription factor specificity through protein-protein interaction, modulation of Wnt signalling and post-translational modification. Int. J. Biochem. Cell Biol. 42:400–410 [DOI] [PubMed] [Google Scholar]
- 11. Bhattacharya R., et al. 2009. MiR-15a and MiR-16 control Bmi-1 expression in ovarian cancer. Cancer Res. 69:9090–9095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Boyer L. A., et al. 2005. Core transcriptional regulatory circuitry in human embryonic stem cells. Cell 122:947–956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Clevers H. 2006. Wnt/beta-catenin signaling in development and disease. Cell 127:469–480 [DOI] [PubMed] [Google Scholar]
- 14. Dailey L., Basilico C. 2001. Coevolution of HMG domains and homeodomains and the generation of transcriptional regulation by Sox/POU complexes. J. Cell. Physiol. 186:315–328 [DOI] [PubMed] [Google Scholar]
- 15. Ellis P., et al. 2004. SOX2, a persistent marker for multipotential neural stem cells derived from embryonic stem cells, the embryo or the adult. Dev. Neurosci. 26:148–165 [DOI] [PubMed] [Google Scholar]
- 16. Gaur T., et al. 2005. Canonical WNT signaling promotes osteogenesis by directly stimulating Runx2 gene expression. J. Biol. Chem. 280:33132–33140 [DOI] [PubMed] [Google Scholar]
- 17. Harley V. R., Lovell-Badge R., Goodfellow P. N. 1994. Definition of a consensus DNA binding site for SRY. Nucleic Acids Res. 22:1500–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ismail I. H., Andrin C., McDonald D., Hendzel M. J. 2010. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J. Cell Biol. 191:45–60 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kamachi Y., Uchikawa M., Kondoh H. 2000. Pairing SOX off: with partners in the regulation of embryonic development. Trends Genet. 16:182–187 [DOI] [PubMed] [Google Scholar]
- 20. Karsenty G., Kronenberg H. M., Settembre C. 2009. Genetic control of bone formation. Annu. Rev. Cell Dev. Biol. 25:629–648 [DOI] [PubMed] [Google Scholar]
- 21. Krishnan V., Bryant H. U., Macdougald O. A. 2006. Regulation of bone mass by Wnt signaling. J. Clin. Invest. 116:1202–1209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lee K. S., et al. 2000. Runx2 is a common target of transforming growth factor beta1 and bone morphogenetic protein 2, and cooperation between Runx2 and Smad5 induces osteoblast-specific gene expression in the pluripotent mesenchymal precursor cell line C2C12. Mol. Cell. Biol. 20:8783–8792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lian J. B., et al. 2006. Networks and hubs for the transcriptional control of osteoblastogenesis. Rev. Endocr Metab. Disord. 7:1–16 [DOI] [PubMed] [Google Scholar]
- 24. Liu J., et al. 2009. Bmi1 regulates mitochondrial function and the DNA damage response pathway. Nature 459:387–392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mansukhani A., Ambrosetti D., Holmes G., Cornivelli L., Basilico C. 2005. Sox2 induction by FGF and FGFR2 activating mutations inhibits Wnt signaling and osteoblast differentiation. J. Cell Biol. 168:1065–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mansukhani A., Bellosta P., Sahni M., Basilico C. 2000. Signaling by fibroblast growth factors (FGF) and fibroblast growth factor receptor 2 (FGFR2)-activating mutations blocks mineralization and induces apoptosis in osteoblasts. J. Cell Biol. 149:1297–1308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Marson A., et al. 2008. Connecting microRNA genes to the core transcriptional regulatory circuitry of embryonic stem cells. Cell 134:521–533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Maruyama T., Mirando A. J., Deng C. X., Hsu W. 2010. The balance of WNT and FGF signaling influences mesenchymal stem cell fate during skeletal development. Sci. Signal. 3:ra40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Memarzadeh S., et al. 2007. Enhanced paracrine FGF10 expression promotes formation of multifocal prostate adenocarcinoma and an increase in epithelial androgen receptor. Cancer Cell 12:572–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Molofsky A. V., He S., Bydon M., Morrison S. J., Pardal R. 2005. Bmi-1 promotes neural stem cell self-renewal and neural development but not mouse growth and survival by repressing the p16Ink4a and p19Arf senescence pathways. Genes Dev. 19:1432–1437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Mootha V. K., et al. 2003. PGC-1alpha-responsive genes involved in oxidative phosphorylation are coordinately downregulated in human diabetes. Nat. Genet. 34:267–273 [DOI] [PubMed] [Google Scholar]
- 32. Nakashima K., et al. 2002. The novel zinc finger-containing transcription factor Osterix is required for osteoblast differentiation and bone formation. Cell 108:17–29 [DOI] [PubMed] [Google Scholar]
- 33. Nakatake Y., et al. 2006. Klf4 cooperates with Oct3/4 and Sox2 to activate the Lefty1 core promoter in embryonic stem cells. Mol. Cell. Biol. 26:7772–7782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Nelson J. D., Denisenko O., Bomsztyk K. 2006. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat. Protoc. 1:179–185 [DOI] [PubMed] [Google Scholar]
- 35. Nelson J. D., Denisenko O., Sova P., Bomsztyk K. 2006. Fast chromatin immunoprecipitation assay. Nucleic Acids Res. 34:e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Nichols J., et al. 1998. Formation of pluripotent stem cells in the mammalian embryo depends on the POU transcription factor Oct4. Cell 95:379–391 [DOI] [PubMed] [Google Scholar]
- 37. Niwa H. 2007. How is pluripotency determined and maintained? Development 134:635–646 [DOI] [PubMed] [Google Scholar]
- 38. Nusse R. 2005. Wnt signaling in disease and in development. Cell Res. 15:28–32 [DOI] [PubMed] [Google Scholar]
- 39. Park I. K., Morrison S. J., Clarke M. F. 2004. Bmi1, stem cells, and senescence regulation. J. Clin. Invest. 113:175–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pevny L. H., Nicolis S. K. 2010. Sox2 roles in neural stem cells. Int. J. Biochem. Cell Biol. 42:421–424 [DOI] [PubMed] [Google Scholar]
- 41. Pittenger M. F., et al. 1999. Multilineage potential of adult human mesenchymal stem cells. Science 284:143–147 [DOI] [PubMed] [Google Scholar]
- 42. Que J., et al. 2007. Multiple dose-dependent roles for Sox2 in the patterning and differentiation of anterior foregut endoderm. Development 134:2521–2531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rachner T. D., Khosla S., Hofbauer L. C. 2011. Osteoporosis: now and the future. Lancet 377:1276–1287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ramalho-Santos M., Yoon S., Matsuzaki Y., Mulligan R. C., Melton D. A. 2002. “Stemness”: transcriptional profiling of embryonic and adult stem cells. Science 298:597–600 [DOI] [PubMed] [Google Scholar]
- 45. Sauvageau M., Sauvageau G. 2010. Polycomb group proteins: multi-faceted regulators of somatic stem cells and cancer. Cell Stem Cell 7:299–313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Subramanian A., et al. 2005. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U. S. A. 102:15545–15550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Surface L. E., Thornton S. R., Boyer L. A. 2010. Polycomb group proteins set the stage for early lineage commitment. Cell Stem Cell 7:288–298 [DOI] [PubMed] [Google Scholar]
- 48. Takahashi K., Yamanaka S. 2006. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126:663–676 [DOI] [PubMed] [Google Scholar]
- 49. Takamiya K., Mao L., Huganir R. L., Linden D. J. 2008. The glutamate receptor-interacting protein family of GluR2-binding proteins is required for long-term synaptic depression expression in cerebellar Purkinje cells. J. Neurosci. 28:5752–5755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Taranova O. V., et al. 2006. SOX2 is a dose-dependent regulator of retinal neural progenitor competence. Genes Dev. 20:1187–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. van de Wetering M., Clevers H. 1992. Sequence-specific interaction of the HMG box proteins TCF-1 and SRY occurs within the minor groove of a Watson-Crick double helix. EMBO J. 11:3039–3044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vickers E. R., Sharrocks A. D. 2002. The use of inducible engrailed fusion proteins to study the cellular functions of eukaryotic transcription factors. Methods 26:270–280 [DOI] [PubMed] [Google Scholar]
- 53. Wegner M. 2010. All purpose Sox: the many roles of Sox proteins in gene expression. Int. J. Biochem. Cell Biol. 42:381–390 [DOI] [PubMed] [Google Scholar]
- 54. Yuan H., Corbi N., Basilico C., Dailey L. 1995. Developmental-specific activity of the FGF-4 enhancer requires the synergistic action of Sox2 and Oct-3. Genes Dev. 9:2635–2645 [DOI] [PubMed] [Google Scholar]
- 55. Zhang H. W., et al. 2010. Defects in mesenchymal stem cell self-renewal and cell fate determination lead to an osteopenic phenotype in Bmi-1 null mice. J. Bone Miner. Res. 25:640–652 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.