

Abstract
Although cellular immunity to acute lymphocytic choriomeningitis virus (LCMV) infection has been well characterized in experimental studies in mice, the T cell response to this virus in humans is incompletely understood. Thus, we analyzed the breadths, magnitudes, and differentiation phenotypes of memory LCMV-specific CD8+ and CD4+ T cells in three human donors displaying a variety of disease outcomes after accidental needle stick injury or exposure to LCMV. Although only a small cohort of donors was analyzed at a single time point postinfection, several interesting observations were made. First, we were able to detect LCMV-specific CD8+ and CD4+ T cell responses directly ex vivo at 4 to 8 years after exposure, demonstrating the longevity of T cell memory in humans. Second, unlike in murine models of LCMV infection, we found that the breadths of memory CD8+ and CD4+ T cell responses were not significantly different from one another. Third, it seemed that the overall CD8+ T cell response was augmented with increasing severity of disease, while the LCMV-specific CD4+ T cell response magnitude was highly variable between the three different donors. Next, we found that LCMV-specific CD8+ T cells in the three donors analyzed seemed to undergo an effector memory differentiation program distinct from that of CD4+ T cells. Finally, the levels of expression of memory, costimulatory, and inhibitory receptors on CD8+ and CD4+ T cell subsets, in some instances, correlated with disease outcome. These data demonstrate for the first time LCMV-specific CD8+ and CD4+ T cells in infected humans and begin to provide new insights into memory T cell responses following an acute virus infection.
INTRODUCTION
Lymphocytic choriomeningitis virus (LCMV), a single-stranded RNA virus and archetypical member of the family Arenaviridae, is a human pathogen maintained in a rodent host reservoir. Infection outcomes can range from subclinical disease to aseptic meningitis, encephalitis, life-threatening infections in immunosuppressed individuals, and severe congenital defects (5, 6, 9, 19). Despite the pathogenic nature of LCMV, there are no U.S. Food and Drug Administration-approved vaccines available. Moreover, antiviral therapy is limited to the use of the guanosine analogue ribavirin, which can lead to adverse side effects such as thrombocytosis, severe anemia, and birth defects (18, 37). Animal models of LCMV infection have clearly demonstrated that virus-specific CD8+ and CD4+ T cells play a pivotal role in viral clearance and protection (17, 20, 26, 27, 58, 59, 61). However, to date, human T cell responses to LCMV have not been characterized in detail.
We have begun to understand human LCMV-specific T cell responses by recent work with HLA transgenic mice, identifying HLA-restricted CD8+ and CD4+ T cell epitopes (10, 31, 32). Immunization of mice with HLA-restricted CD8+ T cell epitopes is sufficient to confer protection against subsequent infection with LCMV in HLA transgenic mice (10). However, the degree of overlap between T cell epitopes identified in HLA transgenic mouse models and humans is incomplete (30, 55). Thus, whether the epitopes defined in the HLA transgenic model are useful to characterize human responses remains to be ascertained experimentally.
Human virus-specific CD8+ T cell responses have been extensively characterized in the context of persistent virus infections, including infections with cytomegalovirus (CMV), Epstein-Barr virus (EBV), hepatitis C virus (HCV), and human immunodeficiency virus type 1 (HIV-1). By examining the expression profiles of surface markers such as CD45RA, CCR7, CD62L, CD28, and CD27, human CD8+ T cells from different persistent virus infections could be categorized into phenotypically distinct memory populations (2, 12, 49). More recently, there have been several investigations examining the differentiation phenotypes of antiviral CD8+ T cells responding to live-attenuated viral vaccines, such as those for smallpox virus (Dryvax) and yellow fever virus (YFV)-17D (1, 13, 38). These studies have shown that memory CD8+ T cells generated after an acute infection with live-attenuated vaccines exhibit phenotypic and functional characteristics that are distinct from those described for human CD8+ T cells specific for persistent viruses (1, 38). Importantly, effector memory CD45RA+ CCR7− CD8+ T cells from persistent infections have been defined as terminally differentiated, while after acute infections, CD45RA-expressing CD8+ T cells exhibit a robust proliferative capacity. Although a great deal of knowledge characterizing Dryvax- and YFD-17D-specific CD8+ T cells has been gained, results from these studies are difficult to generalize to other acute virus infections because of the attenuation state of the virus inoculum and a lack of data in regard to virus-specific CD4+ T cells. Other acute virus infections have also been studied, such as hantavirus and influenza A virus (23, 56); however, these are not ideal experimental systems because exposure to virus and virus reencounter cannot be controlled.
In the present study, we report the identification of LCMV-specific CD8+ and CD4+ T cell epitopes restricted by various HLA molecules. In the case of HLA molecules previously investigated in HLA transgenic mouse models, we report an excellent overlap between epitopes recognized in humans and HLA transgenic mice. Utilizing these T cell epitopes, we characterized the breadths, magnitudes, and differentiation phenotypes of the memory CD8+ and CD4+ T cell responses generated after acute LCMV infection in a small cohort of human donors who displayed a range of disease outcomes.
MATERIALS AND METHODS
Ethics statement.
Research conducted for this study was performed in accordance with approvals from the Institutional Review Board at the La Jolla Institute for Allergy and Immunology. All participants provided written informed consent prior to participation in the study.
Study subjects and PBMC samples.
Healthy volunteers (>18 years of age) with a history of accidental needle stick injury or exposure to LCMV were recruited to the study following informed consent. Seropositivity was confirmed by measuring the serum IgM and IgG antibody titers for LCMV through immunofluorescence assay (Focus Diagnostics, Inc., Cypress, CA). Peripheral blood mononuclear cells (PBMC) were then isolated from heparinized blood or leukapheresis by gradient centrifugation with Histopaque-1077 (Sigma-Aldrich, St. Louis, MO), suspended in fetal bovine serum (FBS) containing 10% dimethyl sulfoxide (DMSO), and cryopreserved in liquid nitrogen. Donor PBMC were typed for HLA-A, -B, and -DR by high-resolution PCR (Blood Systems Laboratories, Tempe, AZ). Approval for all procedures was obtained from the Institutional Review Board (FWA 00000032). Donor AD108 was exposed to LCMV Armstrong clone 53b in 2003, and blood draws were taken in both 2008 and 2009. Donors AD123 and AD124 were exposed to LCMV clone 13 in 2001 and 2002, respectively, and both provided blood donations in 2009.
Bioinformatic analyses.
Candidate epitopes were identified by a consensus epitope prediction approach essentially as described previously (41). For major histocompatibility complex (MHC) class I-restricted peptides, three prediction methods (artificial neural network [ANN], average relative binding [ARB], and the stabilized matrix method [SMM]), as described in reference 62, were applied to select peptides derived from the glycoprotein (GPC) (NCBI accession number P09991) and the nucleoprotein (NP) (NCBI accession number P09992) sequences of LCMV strain Armstrong clone 53b. Nine-mer peptides were ranked according to the their predicted ability to bind to the donor's specific HLA-A and -B molecules. The median rank in the three predictions was used to select the top 3% of class I peptides, which comprised 30 peptides/allele. For HLA-DR-restricted peptides, 15-mer peptides overlapping by 10 residues that covered the entire GPC and NP sequences were ranked using the same median rank consensus approach between ARB, SMM-align, and PROPRED predictions (60). Due to the lower confidence in MHC class II prediction performance, we chose the top 30% of class II peptides, resulting in 60 peptides/DR allele.
Peptides.
Peptides were synthesized as crude material by Pepscan Systems (Lelystad, The Netherlands). Candidate epitopes were resynthesized by A and A Labs (San Diego, CA) and purified to 95% or greater homogeneity by reverse-phase high-pressure liquid chromatography (HPLC). The IEDB submission identification number for the MHC class I and II epitopes is 1000386.
MHC-peptide binding assay.
Quantitative assays to measure the binding affinity of peptides to purified HLA class I and II molecules were based on the inhibition of binding of a radiolabeled standard peptide and were performed essentially as described elsewhere (11, 51, 52). Briefly, after a 2-day incubation, binding of the radiolabeled peptide to the corresponding MHC class I or II molecule was determined by capturing MHC/peptide complexes on Greiner Lumitrac 600 microplates (Greiner Bio-One, Monroe, NC) coated with the W6/32 antibody (anti-HLA class I) or L243 antibody (anti-DRA) and measuring bound cpm using the Topcount microscintillation counter (Packard Instrument). The concentration of peptide yielding 50% inhibition of the binding of the radiolabeled probe peptide (IC50) was then calculated. Peptides were typically tested at 6 different concentrations covering a 100,000-fold dose range and in 3 or more independent assays. Under the conditions utilized, where [label] < [MHC] and IC50 ≥ [MHC], the measured IC50s are reasonable approximations of the dissociation constant (KD) values.
Ex vivo IFN-γ ELISPOT assay.
Gamma interferon (IFN-γ) enzyme-linked immunospot (ELISPOT) assays were performed as previously described (43). In brief, 2 × 105 PBMC were incubated in the presence of peptide pools (10 μg/ml/peptide) corresponding to the donor's haplotype. Peptide pools yielding positive responses were deconvoluted, and individual peptides were tested at 10 μg/ml. To ensure MHC class I restriction when using MHC class I predicted binding peptides, CD8+ T cells were purified by positive selection using anti-CD8 magnetic beads (Miltenyi Biotec, Auburn, CA). To ensure class II restriction when using MHC class II predicted binding peptides, CD8+ T cells were depleted from the PBMC with anti-CD8 beads (Miltenyi Biotec). After 20 h of incubation at 37°C, plates were developed, and responses were calculated as described previously (43). Criteria for positivity were ≥20 net spot-forming cells (SFC)/106 cells, a stimulation index of ≥2.0, and a P value of ≤0.05 using a Student's t test in at least 2 experiments.
Tetramer staining and magnetic bead enrichment.
MHC class I tetramers conjugated using allophycocyanin (APC)-labeled streptavidin (A*0201, B*0702, B*1501, and B*4402) were provided by the NIH Tetramer Core Facility (Atlanta, GA). MHC class II tetramers conjugated using phycoerythrin (PE)-labeled streptavidin (DRB1*1101 and DRB1*0701) were provided by the Tetramer Core Laboratory at the Benaroya Research Institute at Virginia Mason (Seattle, WA). PBMC (∼50 × 106) were incubated in 0.5 ml phosphate-buffered saline (PBS) containing 0.5% bovine serum albumin (BSA) and 2 mM EDTA (pH 8.0) (magnetically activated cell sorting [MACS] buffer) with a 1:100 dilution of class I tetramer for 30 min at 4°C or a 1:50 dilution of class II tetramer for 1 h at 37°C. The exception was the HLA-A*0201 GPC10-18 tetramer, which was incubated with PBMC at a 1:100 dilution for 1 h at 37°C. Tetramer-specific T cell populations were positively enriched by incubating cells with 50 μl of anti-APC or 50 μl of anti-PE microbeads (Miltenyi Biotech) for 20 min at 4°C. After washing, cells were resuspended in 5 ml MACS buffer and passed through a magnetized LS column (Miltenyi Biotech). The column was washed three times with 3 ml of MACS buffer, and after removal from the magnetic field, cells were collected with 5 ml of MACS buffer. The enriched tetramer-specific CD8+ or CD4+ T cells were resuspended in 0.1 ml MACS buffer and stained for cell surface antigens CD8-peridinin chlorophyll protein (PerCP) Cy5.5/PB (RPA-T8; eBioscience), CD27-fluorescein isothiocyanate (FITC) (M-T271; BD Biosciences), CD28-PE/APC (CD28.2; BD), CD62L-FITC (prey56; eBioscience), CD45RA-PECy7 (hl100; eBioscience), CD127-PE (RDR5; eBioscience), CCR5-FITC (2D7/CCR5; BD), CCR7-PE/APC (3D12; eBioscience), LAG3-FITC (R&D Systems), and PD1-PE/APC (MIH4; BD) and for CD4-PerCP Cy5.5/PB (OKT4 eBioscience; RPA-T4 BD), CD14-PerCP Cy5.5 (HCD14; BioLegend), and CD19-PerCP Cy5.5 (SJ25C1; BD) to exclude non-T cell-containing populations that bound tetramer. Fluorescence minus one (FMO) staining controls were used to set quadrant gates. Samples were acquired on an LSR II flow cytometer (BD Immunocytometry Systems) and analyzed using FlowJo software.
RESULTS
Breadth and magnitude of LCMV-specific memory CD8+ T cell responses in a small cohort of individuals with resolved acute LCMV infection.
We examined responses in three subjects who displayed various degrees of symptoms after accidental needle stick injury or exposure with two different strains of LCMV (Table 1). Subject AD108 was infected with the Armstrong clone 53b strain of LCMV, while subjects AD123 and AD124 were infected with the clone 13 strain. All three subjects displayed LCMV-specific serum IgG antibodies but were negative for LCMV-specific serum IgM (data not shown). The specificity of the serology assay was confirmed by testing sera from a total of 30 subjects with no history of LCMV exposure. All 30 subjects were negative for both LCMV-specific IgG and IgM antibodies. AD108 was clinically unaware of his infection, thus allowing us to study an asymptomatic acute LCMV infection. AD123 and AD124 were both symptomatic, and infection resulted in either a flu-like illness or aseptic meningitis, respectively. To investigate the breadth and magnitude of LCMV-specific memory CD8+ and CD4+ T cell responses following an acute LCMV infection, peptides derived from the GPC and NP predicted to bind to the donors' specific haplotypes and an HLA-A*0201-restricted epitope, Z49-58, previously identified in HLA transgenic animals, were synthesized. PBMC isolated from the three donors were stimulated with the candidate peptides, and positive peptides were identified on the basis of their ability to stimulate IFN-γ production from CD8+ T cells in an ELISPOT assay.
Table 1.
Characteristics of LCMV-seropositive donors
| Donor | Sex | HLA class I | HLA class II (DRB1) | LCMV strain | Yr of infection | Route of infection | Symptoms and duration |
|---|---|---|---|---|---|---|---|
| AD108 | Male | A*2402 A*3201 B*0702 B*1501 | *1101 *1401 | Armstrong clone 53b | 2003 | Unknown | None |
| AD123 | Male | A*0201 B*1401 B*4402 | *0701 *0801 | Clone 13 | 2001 | Leg needle stick | Flu-like illness and bacterial infection, 60 days |
| AD124 | Male | A*0201 A*2402 B*4801 B*5401 | *0803 *0901 | Clone 13 | 2002 | Eye | Aseptic meningitis, 4 to 5 days |
In total, nine unique CD8+ T cell epitopes were defined from the three individuals. CD8+ T cells specific for the GPC, NP, and Z proteins were detected, indicating that all three viral proteins are immunogenic in humans (Table 2 and Fig. 1). As a specificity control, CD8+ T cells isolated from three LCMV-seronegative individuals who expressed the same HLA class I molecules as our donor subjects (representative data are shown in Fig. 1) were also examined. Because the defined epitopes were restricted by 6 different HLA class I alleles, a total of 18 HLA-matched, LCMV seronegative PBMC (3 donors/allele) were tested to ensure that the epitope-specific responses were truly LCMV specific. None of the epitopes were recognized by CD8+ T cells isolated from the seronegative donors, thus confirming the specificity of the defined epitopes.
Table 2.
Properties of LCMV-derived HLA class I-restricted CD8+ T cell epitopes
| Donor | Epitopea | Peptide sequence | ELISPOT assay result (avg no. of SFC/106 CD8+ T cells)b | Predicted HLA restriction | Measured binding affinity (nM)c | Restriction (tetramer staining)d |
|---|---|---|---|---|---|---|
| AD108 | GPC38-46 | FATCGIFAL | 63 | B*0702 | 6,230 | ND |
| GPC447-455 | YLVSIFLHL | 46 | B*1501 | 6,920 | ND | |
| NP246-254 | AAVKAGAAL | 46 | B*0702 | 9.8 | B*0702 | |
| NP414-422 | KQFKQDSKY | 73 | B*1501 | 13 | B*1501 | |
| AD123 | GPC447-455 | YLVSIFLHL | 172 | A*0201 | 35 | A*0201 |
| NP45-53 | SEVSNVQRI | 84 | B*4402 | 12 | B*4402 | |
| NP69-77 | SLNQTVHSL | 348 | A*0201 | 53 | A*0201 | |
| Z49-58 | YLCRHCLNLL | 130 | A*0201 | 3.9 | A*0201 | |
| AD124 | GPC10-18 | ALPHIIDEV | 413 | A*0201 | 57 | A*0201 |
| GPC11-19 | LPHIIDEVI | 151 | B*5401 | 1,400 | ND | |
| NP246-254 | AAVKAGAAL | 299 | B*4802 | ND | ND |
Peptide position within LCMV strain Armstrong clone 53b GPC, NP, or Z protein.
Number of net SFC per 106 purified CD8+ T cells. Data are averaged from two independent experiments.
Peptides were tested for binding to their predicted HLA class I allele. ND, binding was not determined because the purified HLA was not available.
Restriction was determined by HLA class I tetramer staining. ND, tetramer staining was not performed.
Fig. 1.

Direct ex vivo detection of LCMV-specific memory CD8+ T cell responses. Purified CD8+ T cells from either the LCMV-seropositive donors (black bars) or a representative HLA-matched, LCMV-seronegative donor (white bars) were incubated with 10 μg/ml peptide, after which the number of IFN-γ-producing cells was enumerated in an ELISPOT assay. Shown are the average net SFC/106 CD8+ T cells for AD108 (A), AD123 (B), and AD124 (C) in response to the defined epitopes from at least two independent experiments. The dashed lines indicate the threshold for peptide positivity (≥20 net SFC/106 cells). For each individual experiment, samples were tested in triplicate. Error bars indicate standard errors of the means (SEM).
Since three or four CD8+ T cell epitopes were identified in each individual, disease severity and/or the infecting virus strain did not appear to affect the overall breadth of the memory CD8+ T cell response. However, the magnitude of the total CD8+ T cell response seemed to correlate with increased clinical severity, with the lowest responses detected in AD108 (total of 228 net SFC/106 cells) and significantly higher responses observed in AD123 and AD124 (totals of 734 and 863 net SFC/106 cells, respectively) (P < 0.05).
As shown in Table 2, the HLA restriction of the newly defined LCMV-specific CD8+ T cell populations, inferred by the bioinformatic predictions, was further supported by experiments measuring epitope binding to purified HLA molecules in vitro. In 7 out of 10 cases, the epitope bound the relevant HLA molecule with high affinity (l to 100 nM range), and in the 3 remaining cases it did so with weak to intermediate affinity (100 to 10,000 nM range). Two CD8+ T cell epitopes (GPC447-455 and NP246-254) were recognized in more than one donor and were restricted by distinct MHC class I types. An overlapping epitope sequence was also defined in AD124, with GPC10-18 binding to HLA-A*0201 and GPC11-19 to HLA-B*5401.
LCMV-specific memory CD4+ T cell responses in same three individuals with resolved acute infection.
CD4+ T cells targeted both the GPC and NP, with three unique epitopes derived from the GPC and nine from the NP (Table 3 and Fig. 2). A single epitope (NP521-535), originally defined in HLA-DRB1*0101 transgenic mice (33), was also recognized by AD123 (Table 4). Epitope-specific responses for AD108 and AD123 were not detectable in HLA-matched, LCMV-seronegative donors (representative data are given in Fig. 2). The overall breadth of the LCMV-specific memory CD4+ T cell response did not appear to correlate with disease severity and/or the infecting virus strain. Unlike the case for the LCMV-specific CD8+ T cell response, the magnitude of the total CD4+ T cell response was highly variable between the three different donors examined. The total net SFC/106 CD4+ T cells for AD108, AD123, and AD124 were 121, 1,279, and 100, respectively.
Table 3.
Properties of LCMV-derived HLA class II-restricted CD4+ T cell epitopes
| Donor | Epitopea | Peptide sequence | ELISPOT assay result (avg no. of SFC/106 CD4+ T cells)b | Predicted HLA restriction | Measured binding affinity (nM)c | Restriction (tetramer staining)d |
|---|---|---|---|---|---|---|
| AD108 | NP86-100 | KNVLKVGRLSAEELM | 121 | *1101 | 23 | *1101 |
| AD123 | GPC66-80 | DIYKGVYQFKSVEFD | 239 | *0701 | 16 | *0701 |
| GPC71-85 | VYQFKSVEFDMSHLN | 190 | *0701 | 373 | ND | |
| GPC341-355 | HLFKTTVNSLISDQL | 71 | *0701 | 2.0 | ND | |
| NP106-120 | LEKLKAKIMRSERPQ | 33 | *0801 | 403 | ND | |
| NP236-250 | NISGYNFSLGAAVKA | 328 | *0701 | 13 | *0701 | |
| NP261-275 | LESILIKPSNSEDLL | 134 | *0701 | 60 | *0701 | |
| NP281-295 | AKRKLNMFVSDQVGD | 53 | *0701 | 228 | ND | |
| NP311-325 | EGWPYIACRTSIVGR | 45 | *0701 | 480 | ND | |
| NP356-370 | VGLSYSQTMLLKDLM | 26 | *0701 | 3.8 | ND | |
| NP411-425 | VDQKQFKQDSKYSHG | 128 | *0701 | 339 | ND | |
| NP521-535 | MDCIIFESASKARLP | 32 | *0801 | 75 | ND | |
| AD124 | GPC66-80 | DIYKGVYQFKSVEFD | 62 | *0901 | 791 | ND |
| GPC71-85 | VYQFKSVEFDMSHLN | 38 | *0901 | 1,200 | ND |
Peptide position within LCMV strain Armstrong clone 53b GPC or NP.
Number of net SFC per 106 purified CD4+ T cells. Data are averaged from two independent experiments.
Peptides were tested for binding to their predicted HLA class II allele.
Restriction was determined by HLA class II tetramer staining. ND, tetramer staining was not determined.
Fig. 2.

Direct ex vivo detection of LCMV-specific memory CD4+ T cell responses. PBMC enriched for CD4+ T cells from either the LCMV-seropositive donors (black bars) or a representative HLA-matched, LCMV-seronegative donor (white bars) were incubated with 10 μg/ml peptide. The number of IFN-γ-producing cells was enumerated in an ELISPOT assay. Shown are the average net SFC/106 CD4+ T cells for AD108 (A), AD123 (B), and AD124 (C) in response to the defined epitopes from at least two independent experiments. An HLA-matched, LCMV-seronegative donor for AD124 was not available. The dashed lines indicate the threshold for peptide positivity (≥20 net SFC/106 cells). For each individual experiment, samples were tested in triplicate. Error bars indicate SEM.
Table 4.
Summary of overlap between HLA class I- and II-restricted LCMV T cell epitope recognition in human and HLA transgenic mouse systems
| Epitope | Sequence | Identificationa in: |
Reference(s)b | |
|---|---|---|---|---|
| Human system | Transgenic mouse system | |||
| GPC10-18 | ALPHIIDEV | Y | Y | 10 |
| GPC11-19 | LPHIIDEVI | Y | NT | |
| GPC34-43 | AVYNFATCGI | NT | Y | 10, 31 |
| GPC38-46 | FATCGIFAL | Y | N | |
| GPC112-120 | FTNDSIISH | N | Y | 31 |
| GPC46-55 | LVSFLLLAGR | NT | Y | 31 |
| GPC447-455 | YLVSIFLHL | Y | Y | 10 |
| NP45-53 | SEVSNVQRI | Y | NT | |
| NP69-77 | SLNQTVHSL | Y | Y | 10 |
| NP246-254 | AAVKAGAAL | Y | N | |
| NP414-422 | KQFKQDSKY | Y | NT | |
| NP521-535 | MDCIIFESASKARLP | Y | Y | 33 |
| Z24-33 | TTYLGPLSCK | NT | Y | 31 |
| Z49-58 | YLCRHCLNLL | Y | Y | 10, 31 |
Y, epitope was identified; N, epitope was not identified. Class I epitope recognition in HLA transgenic mice was examined following inoculation of mice with the LCMV strain Armstrong (10) or recombinant VACV (rVACV) expressing LCMV GPC, NP, or Z (31). Class II epitope recognition was examined after priming mice with an rVACV expressing one of the LCMV proteins followed by boosting with LCMV-specific peptide pools (33). Epitope-specific T cell responses were measured by IFN-γ ELISPOT assays. NT, not tested because the epitope sequence was not predicted to bind to the donor's HLA molecules or because HLA transgenic mice were not available.
Reference(s) for epitope identification in HLA transgenic mice.
The restricting alleles of the LCMV-specific CD4+ T cell populations inferred by the predicted binding affinities were further supported by the measured binding affinities (Table 3). In 7 out of 14 cases, the epitope bound the relevant HLA molecule with high affinity (l to 100 nM range), and in the 7 remaining cases it did so with weak to intermediate affinity (100 to 10,000 nM range). Two GPC-specific epitopes (GPC66-80 and GPC71-85) were recognized in more than one donor and were restricted by different HLA-DRB1 molecules. GPC66-80 and GPC71-85 both bound to HLA-DRB1-*0701 in AD123 and to HLA-DRB1-*0901 in AD124.
HLA restriction of LCMV-specific T cell populations determined by MHC-peptide tetramer staining.
To further characterize the dynamics of the LCMV-specific T cell response and the differentiation phenotypes of LCMV-specific CD8+ and CD4+ T cells, MHC-peptide tetramer reagents were prepared for selected epitopes with binding affinities to the predicted HLA allele of <100 nM (Tables 2 and 3). Because the frequencies of LCMV-specific CD8+ and CD4+ T cell populations detected by ELISPOT assay in the three individuals were relatively low, we employed tetramer staining followed by a magnetic bead enrichment technique (see Materials and Methods) to increase the sensitivity of detection (4, 45).
The tetramer staining experiments confirmed HLA restriction of seven CD8+ and four CD4+ T cell responses (representative data are given in Fig. 3). We were able to detect LCMV-specific T cell responses at frequencies 30- to 600-fold above background. For some CD8+ and CD4+ T cell populations, we were unable to confirm the HLA restriction inferred on the basis of the HLA predictions (Tables 2 and 3), because purified HLAs for binding assays and/or tetramer preparations were not available. Only a small number of background-level tetramer-positive cells were detected with the LCMV-specific tetramers in the control HLA-matched, LCMV-seronegative individuals (Fig. 3), thus confirming that tetramer specificity was derived from the LCMV peptide and HLA molecule combination.
Fig. 3.

HLA restriction of LCMV-specific CD8+ and CD4+ T cell responses using MHC tetramers. PBMC from AD108, AD123, or HLA-matched, seronegative individuals were stained with MHC class I tetramers (A) or class II tetramers (B), and tetramer-positive cells were isolated following magnetic bead enrichment. Plots are gated on either CD3+ CD8+ (A) or CD3+ CD4+ (B) lymphocytes, and the numbers indicate the total number of tetramer-positive cells isolated from each donor's PBMC.
Phenotypic characterization of the LCMV-specific memory CD8+ T cells.
Next, we assessed the differences between effector and central memory T cells using CD45RA, a transmembrane tyrosine phosphatase reexpressed on late-differentiated CD8+ T cells (12), and CCR7, a chemokine receptor that controls homing to secondary lymphoid organs (49). As shown in Fig. 4, we found that LCMV-specific tetramer-positive CD8+ T cells consisted predominantly of CD45RA+ CCR7− effector memory cells in the three different donors analyzed. We also analyzed the expression of CD62L, an L-selectin lymph node homing molecule. LCMV-specific CD8+ T cell subsets consisted mainly of CD62Llow effector memory cells but also contained CD62Lhigh central memory cells (Fig. 4), particularly in subjects AD108 and AD124. Interestingly, the majority of tetramer-positive CD8+ T cells isolated from AD123 consistently displayed CD45RAlow and CD62Llow expression compared with the other two donors (medians of 34% CD45RA+ and 12% CD62L+). In most instances, we found similar expression patterns of CD45RA, CCR7, and CD62L in both the tetramer-positive CD8+ T cells and the total CD8+ T cell population within the same donor.
Fig. 4.

Memory phenotype of LCMV-specific CD8+ T cells. PBMC from donors AD108, AD123, and AD124 were stained with the MHC class I tetramers HLA-B*1501 NP414-422, HLA-A*0201 Z49-58, and HLA-A*0201 GPC10-18, respectively. Tetramer-positive cells were enriched following incubation with magnetic beads. There was a 10-fold reduction in the number of tetramer-positive CD8+ T cells isolated from AD124 when examining CD127, CCR5, CD27, CD28, LAG3, and PD1 expression, as fewer PBMC were initially used. Plots are gated on either total CD3+ CD8+ T cells (black background) or LCMV-specific CD3+ CD8+ T cells (red dots). The numbers represent the percentages of tetramer-positive CD3+ CD8+ T cells in the gate.
To further determine the phenotypic nature of the LCMV-specific memory CD8+ T cells, we examined the coexpression of CD127 (interleukin-7 receptor alpha [IL-7Rα]), which marks precursors destined to differentiate into long-lived memory cells (25), and the chemokine receptor CCR5, which is associated with activated memory cells (46). We found that the majority of tetramer-positive CD8+ T cells reexpressed CD127 in subject AD108 (median of 69% CD127+), whereas CD127 was expressed at progressively lower levels in AD123 and AD124 (medians of 22% and 10% CD127+, respectively). Bimodal expression patterns were observed for CCR5 in all three donors.
Characterization of the LCMV-specific CD8+ T cells in terms of expression of costimulation and inhibitory receptors.
To assess the ability of the memory CD8+ T cells to respond to costimulatory signals, we studied the expression of CD27 and CD28. These costimulatory receptors have also been used to phenotypically differentiate distinct memory CD8+ T cell subsets following persistent virus infections (2). We found that tetramer-positive CD8+ T cells from AD108 and AD123 showed uniformly high expression of CD27 and heterogeneous expression of CD28, while only ∼30% of tetramer-positive CD8+ T cells from AD124 expressed both CD27 and CD28.
We then examined the expression of two inhibitory receptors, programmed death 1 (PD1) and lymphocyte activation gene 3 (LAG3), that both negatively regulate T cell activation and proliferation when upregulated transiently in acute LCMV Armstrong infection and long term in clone 13 infection of mice (3, 7). Recently, it was shown that blockade of both PD1 and LAG3 improved CD8+ T cell responses and reduced the viral load in LCMV clone 13-infected mice (7). We found that tetramer-positive CD8+ T cells from all three donors did not express PD1, which is consistent with a resolved acute infection and clearance of virus. In contrast, we observed upregulated expression of LAG3 in the three donors (medians of 6% for AD108, 19% for AD123, and 4% for AD124). Interestingly, a high degree of variability was observed when examining the expression of LAG3 in other tetramer-positive CD8+ T cell populations from AD123. Ten percent of HLA-A*0201 NP69-77, 23% of HLA-A*0201 GPC447-455, and 42% of HLA-B*4402 NP45-53 were positive for LAG3 (median of 19% LAG3+) (data not shown).
Phenotypic characterization of the LCMV-specific memory CD4+ T cells.
Our understanding of CD4+ T cell differentiation following acute virus infection in humans remains unclear. To begin to fill this knowledge gap, we applied the tetramer enrichment technique to phenotypically characterize LCMV-specific CD4+ T cell populations isolated from AD108 and AD123. We were unable to phenotype CD4+ T cells from AD124, as the MHC class II molecules DRB1*0803 and DRB1*0901 were not available for tetramer preparation. The expression of markers used to dissect the CD8+ T cell populations was also used to characterize the LCMV-specific CD4+ T cells.
Overall, we observed two key differences between the tetramer-positive CD8+ and CD4+ T cell populations analyzed. First, we found that tetramer-positive CD4+ T cells consisted predominantly of CD45RA− CCR7− CD62L− effector memory cells (Fig. 5). Second, when examining CD27 and CD28 expression, we found that the tetramer-positive CD4+ T cells uniformly expressed high levels of both costimulatory receptors. Expression of CD127 was divergent between the two donors analyzed. Greater than 70% of tetramer-positive CD4+ T cells isolated from AD108 reexpressed high levels of CD127, whereas the majority of tetramer-positive CD4+ T cells from AD123 expressed CD127 at only low to moderate levels (median of 16% CD127+). Interestingly, similar to the case for LCMV-specific CD8+ T cells from AD123, LAG3 expression was also upregulated on tetramer-positive CD4+ T cells in this donor. This phenotype was seen on other tetramer-positive CD4+ T cell populations isolated from AD123 (median of 27% LAG3+) (data not shown). Both CCR5 and PD1 were not expressed on any of the CD4+ T cell populations analyzed.
Fig. 5.
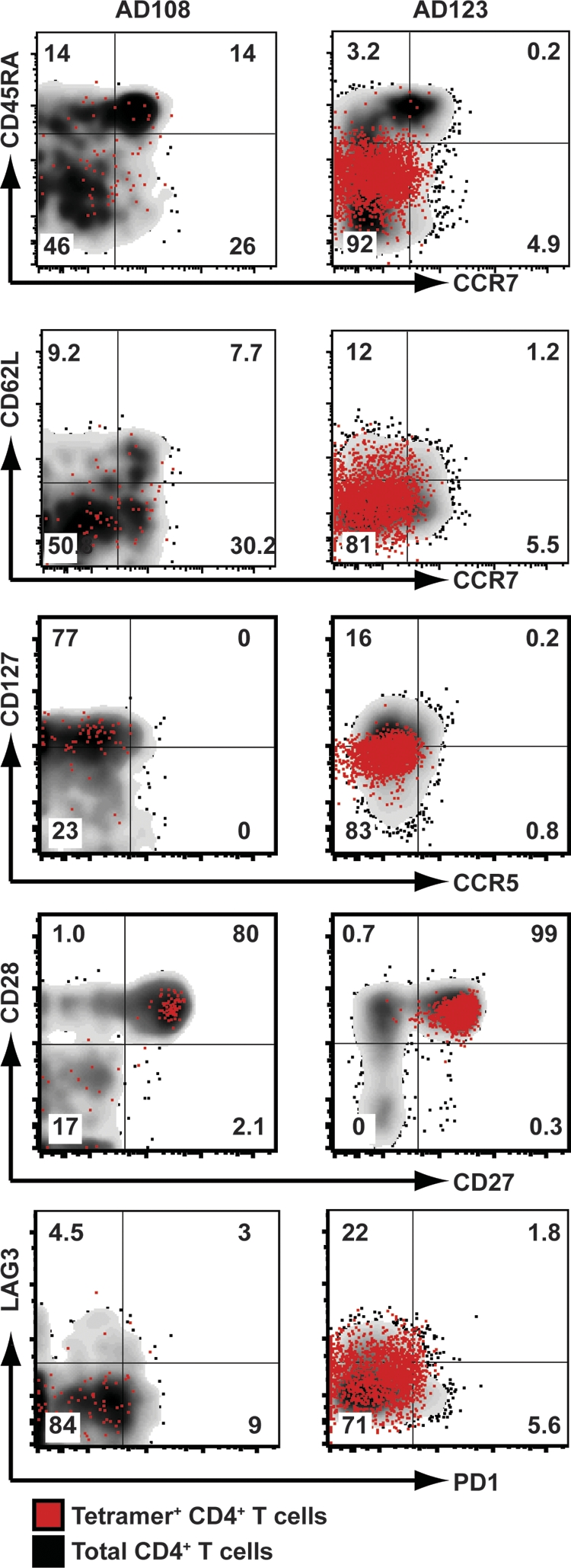
Memory phenotype of LCMV-specific CD4+ T cells. PBMC from donors AD108 and AD123 were stained with the MHC class II tetramers HLA-DRB1*1101 NP86-100 and HLA-DRB1*0701 NP236-250, respectively. Tetramer-positive cells were enriched following incubation with magnetic beads. Plots are gated on either total CD3+ CD4+ T cells (black background) or LCMV-specific CD3+ CD4+ T cells (red dots). The numbers represent the percentages of tetramer-positive CD3+ CD4+ T cells in the gate.
DISCUSSION
The present study represents the first characterization of human memory T cell responses to LCMV in the setting of accidental needle stick injury or exposure with different strains of LCMV in three individuals who displayed distinct disease outcomes. Responses against a total of 9 unique CD8+ and 12 CD4+ T cell epitopes were detected directly ex vivo at 4 to 8 years after LCMV exposure. We were able to rapidly identify these T cell epitopes, as HLA binding predictions are increasingly more accurate and IFN-γ ELISPOT assays allow quick screening of a large number of peptides. Most importantly, the integration of bioinformatic approaches with cellular immune assays might lead to the development of optimized vaccines and drugs tailored to personalized prevention and treatment of virus infection. Thus, with the advent of epitope identification in humans, personalized medicine is becoming a reality.
Despite our findings, the small number of individuals available to us for study and the significant time elapsed from infection dictate that these evaluations should be interpreted with caution. The data trends and conclusions suggested in this study would be strengthened by increasing the number of subjects and/or by analysis of the same three subjects at multiple time points to enable documentation of differentiation changes of the defined T cell populations. Unfortunately, we have been constrained by the wide geographic distribution of our donors and thus are unable to obtain repeat PBMC donations from the same three subjects. Further studies with additional donors and time points postinfection would be required to more firmly establish the observations reported here. These are areas of future research.
Although the donor cohort size was small, we detected a high degree of overlap in the epitopes recognized in infected humans and in previous studies utilizing HLA transgenic mice. Of the 5 HLA-A*0201-restricted LCMV-specific CD8+ T cell epitopes originally defined in HLA-A*0201 transgenic mice, 4 were also recognized by HLA-A*0201-positive donors in this study, thus demonstrating an 80% concordance rate between humans and mice. The facts that multiple HLA-A*0201-positive donors recognized the HLA-A*0201-restricted CD8+ T cell epitopes and that immunization with these epitopes protected mice against LCMV challenge further validate their potential use as vaccine candidates. The high rate of concordance between the two systems underscores the importance of HLA transgenic mice as a suitable model for identifying human T cell epitopes from small RNA viruses, such as LCMV. HLA transgenic mice also serve as a preliminary probe for identifying T cell epitopes from viruses that infrequently infect humans and that could be viewed as potential bioterrorist threats.
In the three donors analyzed, we found that the breadths of the memory CD8+ and CD4+ T cell responses did not significantly differ from one another and were not influenced by disease severity and/or the infecting LCMV strain. In contrast, the overall magnitudes of memory CD8+ and CD4+ T cell responses varied between the different donors. Compared to the total CD8+ T cell response magnitude, the total CD4+ T cell response was similar in AD108, higher in AD123, and lower in AD124. These findings were interesting, as they differ from what was previously observed in murine models of LCMV infection and also in the case of other acute viral infections in humans. Thus, it appears that different viruses might induce qualitatively different T cell responses. In murine models of LCMV, studies have demonstrated that a broader repertoire of CD8+ T cell responses exists after an acute LCMV infection (16, 32). Murine models have also demonstrated major differences in the magnitudes of memory CD8+ and CD4+ T cell responses. Memory is stable in CD8+ T cell responses, while the levels of memory CD4+ T cells are 20-fold lower and gradually decline (15, 24). Our findings also differ from those for other acute viral infections in humans, including severe acute respiratory syndrome (SARS) coronavirus (35) and vaccinia virus (VACV) (38), where memory CD8+ T cell responses are more frequent and greater in magnitude than CD4+ T cell responses (10, 31).
We also found that the total LCMV-specific CD8+ T cell response was augmented with increasing severity of disease, while the total CD4+ T cell response was highly variable between the different donors. This variability could be due to the small size of the donor cohort analyzed. These findings are in agreement with studies on other acute virus infections in humans, including Sin Nombre virus and dengue virus, which have demonstrated that elevated levels of virus-specific CD8+ T cells correlate with heightened disease severity (29). Infection with Sin Nombre virus, a hantavirus that causes fulminant life-threatening disease, results in high frequencies of tetramer-specific CD8+ T cells in patients with severe hantavirus pulmonary syndrome compared to those in patients with moderate disease (29, 39, 40). Similarly, higher frequencies of circulating dengue virus-specific CD8+ T cells are seen in patients with acute secondary dengue virus infection (40). Thus, whether disease outcome correlates with the virus-specific T cell response seems to depend on the infecting virus, and the fine balance between protective immunity and immunopathology. However, in our case, we have been able to study only infections resolved some time previously, so it is difficult to determine the importance of these cells in the development of acute infection and pathogenesis.
To our knowledge, this is one of the first studies where the differentiation phenotypes of human LCMV-specific CD8+ and CD4+ T cells have been compared following an acute, live nonattenuated virus infection in the absence of antigenic reencounter (14, 28, 36, 50). When examining the differences between effector memory and central memory on the basis of CD45RA, CCR7, and CD62L expression patterns, we found that LCMV-specific CD8+ T cells had a predominantly effector memory phenotype, with the majority of tetramer-positive CD8+ T cells reexpressing CD45RA. This CD45RA+ effector memory phenotype confirms what was previously observed in vaccinees acutely infected with either Dryvax or YFV-17D (1, 13, 38, 44) and expands these observations to LCMV infection. Interestingly, this general observation held true for the three different donors, despite dramatic differences in the severity of disease associated with the infection. Interestingly, we noted that in some instances MHC-peptide tetramer staining detected a 5-fold-greater number of LCMV-specific T cells than was demonstrated to produce IFN-γ (HLA-B*1501-restricted NP414-422 and HLA-A*0201-restricted Z49-58). It has been previously reported that in some cases there is a 1:1 correspondence between the number of antigen-specific T cells detected by IFN-γ ELISPOT and tetramer staining assays, while in other cases this correspondence is not observed. As noted by Murali-Krishna et al. (42), in BALB/c mice infected with LCMV, both IFN-γ ELISPOT and tetramer staining detected approximately 50% of CD8+ T cells recognizing the immunodominant epitope NP118-126. However, in LCMV-infected C57BL/6 mice, 33% of CD8+ T cells recognizing the immunodominant H-2Db-restricted NP396-404 epitope were detected by IFN-γ ELISPOT, while only 23% of CD8+ T cells bound the H-2Db NP396-404 tetramer. In humans, there is also a correlation between the number of antigen-specific T cells detected by IFN-γ ELISPOT and tetramer staining assays; however, the ratio is usually not 1:1. In agreement with our data, in HIV-infected individuals, tetramer staining detected approximately a 5- to 10-fold-greater number of CD8+ T cells recognizing HLA-A02-restricted Gag and Pol-specific epitopes than were demonstrated to produce IFN-γ (22, 54). A larger number of antigen-specific T cells could be detected by tetramer staining assays, because tetramer-positive T cells might be producing other cytokines or no cytokines at all. Another possibility is that a portion of tetramer-positive T cells are no longer able to proliferate and are undergoing apoptosis.
The availability of novel MHC class II tetramers allowed us for the first time to evaluate the memory phenotype of virus-specific CD4+ T cells resulting from an acute viral infection in humans. Importantly, we demonstrated that LCMV-specific CD4+ T cells had an effector memory phenotype different from that of CD8+ T cells. The majority of tetramer-positive CD4+ T cells lacked CD45RA expression. The lack of CD45RA expression has been suggested to reflect a possibly less mature or differentiated effector memory phenotype (48, 53). Thus, it seems that following acute LCMV infection in humans, CD4+ T cells have a less mature memory phenotype than CD8+ T cells. Considering the phenotypic differences between CD8+ and CD4+ T cell subsets unearthed in the present study, this suggests that a more thorough examination of virus-specific CD4+ T cells is needed in future studies.
The majority of LCMV-specific CD8+ and CD4+ T cells expressed high levels of both CD28 and CD27, which is consistent with the early differentiation phenotype also seen in influenza virus-infected individuals and Dryvax and YFD-17D vaccinees (1, 23, 38). We expanded on these findings by demonstrating that the level of CD28 and CD27 expression appears to correlate with disease outcome. Specifically, we found that the tetramer-positive CD8+ T cells from AD124 were predominantly CD28− CD27−, suggesting a late differentiated phenotype also seen in CMV infections (2). Interestingly, early CD28+ CD27+ differentiated antiviral T cells have been associated with protective immunity (47), suggesting that a more severe disease outcome, as seen in AD124, might be related to the quality of the antiviral CD8+ T cell response.
Finally, CD127 and LAG3 expression also correlated with clinical manifestation. CD127 is reexpressed on antiviral CD8+ T cells in resolved infections (8, 25) but is not present at significant levels on CMV- and EBV-specific CD8+ T cells, demonstrating that persistent viral antigen suppresses CD127 expression (21, 34, 57). In our study, CD127 was expressed at high levels on LCMV-specific CD8+ and CD4+ T cells in the asymptomatic AD108 donor, while CD127 expression rapidly declined with increased disease severity in AD123 and AD124. LAG3 expression was also upregulated on all tetramer-positive T cell subsets examined, especially CD8+ and CD4+ T cells in donor AD123. Similarly, the upregulation of CD127 on HBV-specific CD8+ T cells correlated with a loss of PD1 expression (8). These results suggest a reciprocal relationship between CD127 and inhibitory receptor expression.
In conclusion, these data collectively demonstrate for the first time that memory CD8+ and CD4+ T cells can be identified in infected humans. Most notably, the rapid identification of T cell epitopes from infected humans suggests that personalized prevention and treatment of virus infection are possible. In addition, this study not only confirms previous findings with live attenuated viral vaccines but also begins to show that disease outcome correlates with both the magnitude of the memory CD8+ T cell response and the phenotypes of both LCMV-specific CD8+ and CD4+ T cells. These data suggest that a closer examination of LCMV-specific CD8+ and CD4+ T cells with a larger donor cohort is needed in future studies.
ACKNOWLEDGEMENTS
This work was supported by National Institute of Allergy and Infectious Diseases grant U54AI065359 (M.J.B.) and National Institutes of Health contract NO1-AI-40023 (A.S.). M.J.B. and B.P. were also supported in part by funds from the Pacific Southwest RCE (AI065359).
We thank Carrie Moore, Sandy Ngo, and Amiyah Steen for performing the MHC binding assays.
This is La Jolla Institute for Allergy and Immunology and Kyowa Hakko Kirin California publication number 1317.
Footnotes
Published ahead of print on 7 September 2011.
REFERENCES
- 1. Akondy R. S., et al. 2009. The yellow fever virus vaccine induces a broad and polyfunctional human memory CD8+ T cell response. J. Immunol. 183: 7919–7930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Appay V., et al. 2002. Memory CD8+ T cells vary in differentiation phenotype in different persistent virus infections. Nat. Med. 8: 379–385 [DOI] [PubMed] [Google Scholar]
- 3. Barber D. L., et al. 2006. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature 439: 682–687 [DOI] [PubMed] [Google Scholar]
- 4. Barnes E., et al. 2004. Ultra-sensitive class I tetramer analysis reveals previously undetectable populations of antiviral CD8+ T cells. Eur. J. Immunol. 34: 1570–1577 [DOI] [PubMed] [Google Scholar]
- 5. Barton L. L., Hyndman N. J. 2000. Lymphocytic choriomeningitis virus: reemerging central nervous system pathogen. Pediatrics 105: E35. [DOI] [PubMed] [Google Scholar]
- 6. Barton L. L., Mets M. B., Beauchamp C. L. 2002. Lymphocytic choriomeningitis virus: emerging fetal teratogen. Am. J. Obstet. Gynecol. 187: 1715–1716 [DOI] [PubMed] [Google Scholar]
- 7. Blackburn S. D., et al. 2009. Coregulation of CD8+ T cell exhaustion by multiple inhibitory receptors during chronic viral infection. Nat. Immunol. 10: 29–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boettler T., et al. 2006. Expression of the interleukin-7 receptor alpha chain (CD127) on virus-specific CD8+ T cells identifies functionally and phenotypically defined memory T cells during acute resolving hepatitis B virus infection. J. Virol. 80: 3532–3540 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bonthius D. J., et al. 2007. Congenital lymphocytic choriomeningitis virus infection: spectrum of disease. Ann. Neurol. 62: 347–355 [DOI] [PubMed] [Google Scholar]
- 10. Botten J., et al. 2007. HLA-A2-restricted protection against lethal lymphocytic choriomeningitis. J. Virol. 81: 2307–2317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Calvo-Calle J. M., Strug I., Nastke M. D., Baker S. P., Stern L. J. 2007. Human CD4+ T cell epitopes from vaccinia virus induced by vaccination or infection. PLoS Pathog. 3: 1511–1529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Champagne P., et al. 2001. Skewed maturation of memory HIV-specific CD8 T lymphocytes. Nature 410: 106–111 [DOI] [PubMed] [Google Scholar]
- 13. Co M. D., Kilpatrick E. D., Rothman A. L. 2009. Dynamics of the CD8 T-cell response following yellow fever virus 17D immunization. Immunology 128: e718–e727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Day C. L., et al. 2003. Ex vivo analysis of human memory CD4 T cells specific for hepatitis C virus using MHC class II tetramers. J. Clin. Invest. 112: 831–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Boer R. J., Homann D., Perelson A. S. 2003. Different dynamics of CD4+ and CD8+ T cell responses during and after acute lymphocytic choriomeningitis virus infection. J. Immunol. 171: 3928–3935 [DOI] [PubMed] [Google Scholar]
- 16. Dow C., et al. 2008. Lymphocytic choriomeningitis virus infection yields overlapping CD4+ and CD8+ T-cell responses. J. Virol. 82: 11734–11741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elsaesser H., Sauer K., Brooks D. G. 2009. IL-21 is required to control chronic viral infection. Science 324: 1569–1572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Enria D. A., et al. 1987. Tolerance and antiviral effect of ribavirin in patients with Argentine hemorrhagic fever. Antiviral Res. 7: 353–359 [DOI] [PubMed] [Google Scholar]
- 19. Fischer S. A., et al. 2006. Transmission of lymphocytic choriomeningitis virus by organ transplantation. N. Engl. J. Med. 354: 2235–2249 [DOI] [PubMed] [Google Scholar]
- 20. Frohlich A., et al. 2009. IL-21R on T cells is critical for sustained functionality and control of chronic viral infection. Science 324: 1576–1580 [DOI] [PubMed] [Google Scholar]
- 21. Fuller M. J., et al. 2005. Emergence of CD127high functionally competent memory T cells is compromised by high viral loads and inadequate T cell help. J. Immunol. 174: 5926–5930 [DOI] [PubMed] [Google Scholar]
- 22. Goepfert P. A., et al. 2000. A significant number of human immunodeficiency virus epitope-specific cytotoxic T lymphocytes detected by tetramer binding do not produce gamma interferon. J. Virol. 74: 10249–10255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hoji A., Rinaldo C. R., Jr 2005. Human CD8+ T cells specific for influenza A virus M1 display broad expression of maturation-associated phenotypic markers and chemokine receptors. Immunology 115: 239–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Homann D., Teyton L., Oldstone M. B. 2001. Differential regulation of antiviral T-cell immunity results in stable CD8+ but declining CD4+ T-cell memory. Nat. Med. 7: 913–919 [DOI] [PubMed] [Google Scholar]
- 25. Kaech S. M., et al. 2003. Selective expression of the interleukin 7 receptor identifies effector CD8 T cells that give rise to long-lived memory cells. Nat. Immunol. 4: 1191–1198 [DOI] [PubMed] [Google Scholar]
- 26. Kagi D., et al. 1994. Cytotoxicity mediated by T cells and natural killer cells is greatly impaired in perforin-deficient mice. Nature 369: 31–37 [DOI] [PubMed] [Google Scholar]
- 27. Kalams S. A., Walker B. D. 1998. The critical need for CD4 help in maintaining effective cytotoxic T lymphocyte responses. J. Exp. Med. 188: 2199–2204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kasprowicz V., et al. 2006. Tracking of peptide-specific CD4+ T-cell responses after an acute resolving viral infection: a study of parvovirus B19. J. Virol. 80: 11209–11217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kilpatrick E. D., et al. 2004. Role of specific CD8+ T cells in the severity of a fulminant zoonotic viral hemorrhagic fever, hantavirus pulmonary syndrome. J. Immunol. 172: 3297–3304 [DOI] [PubMed] [Google Scholar]
- 30. Kotturi M. F., et al. 2009. Of mice and humans: how good are HLA transgenic mice as a model of human immune responses? Immunome Res. 5: 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kotturi M. F., et al. 2009. A multivalent and cross-protective vaccine strategy against arenaviruses associated with human disease. PLoS Pathog. 5: e1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kotturi M. F., et al. 2007. The CD8+ T-cell response to lymphocytic choriomeningitis virus involves the L antigen: uncovering new tricks for an old virus. J. Virol. 81: 4928–4940 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kotturi M. F., et al. 2008. Naive precursor frequencies and MHC binding rather than the degree of epitope diversity shape CD8+ T cell immunodominance. J. Immunol. 181: 2124–2133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lang K. S., et al. 2005. Inverse correlation between IL-7 receptor expression and CD8 T cell exhaustion during persistent antigen stimulation. Eur. J. Immunol. 35: 738–745 [DOI] [PubMed] [Google Scholar]
- 35. Li C. K., et al. 2008. T cell responses to whole SARS coronavirus in humans. J. Immunol. 181: 5490–5500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Lucas M., et al. 2004. Ex vivo phenotype and frequency of influenza virus-specific CD4 memory T cells. J. Virol. 78: 7284–7287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. McKee K. T., Jr., Huggins J. W., Trahan C. J., Mahlandt B. G. 1988. Ribavirin prophylaxis and therapy for experimental argentine hemorrhagic fever. Antimicrob. Agents Chemother. 32: 1304–1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Miller J. D., et al. 2008. Human effector and memory CD8+ T cell responses to smallpox and yellow fever vaccines. Immunity 28: 710–722 [DOI] [PubMed] [Google Scholar]
- 39. Mongkolsapaya J., et al. 2003. Original antigenic sin and apoptosis in the pathogenesis of dengue hemorrhagic fever. Nat. Med. 9: 921–927 [DOI] [PubMed] [Google Scholar]
- 40. Mongkolsapaya J., et al. 2006. T cell responses in dengue hemorrhagic fever: are cross-reactive T cells suboptimal? J. Immunol. 176: 3821–3829 [DOI] [PubMed] [Google Scholar]
- 41. Moutaftsi M., et al. 2006. A consensus epitope prediction approach identifies the breadth of murine T(CD8+)-cell responses to vaccinia virus. Nat. Biotechnol. 24: 817–819 [DOI] [PubMed] [Google Scholar]
- 42. Murali-Krishna K., et al. 1998. Counting antigen-specific CD8 T cells: a reevaluation of bystander activation during viral infection. Immunity 8: 177–187 [DOI] [PubMed] [Google Scholar]
- 43. Oseroff C., et al. 2005. HLA class I-restricted responses to vaccinia recognize a broad array of proteins mainly involved in virulence and viral gene regulation. Proc. Natl. Acad. Sci. U. S. A. 102: 13980–13985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Precopio M. L., et al. 2007. Immunization with vaccinia virus induces polyfunctional and phenotypically distinctive CD8(+) T cell responses. J. Exp. Med. 204: 1405–1416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Psurek A., Neususs C., Pelzing M., Scriba G. K. 2005. Analysis of the lipophilic peptaibol alamethicin by nonaqueous capillary electrophoresis-electrospray ionization-mass spectrometry. Electrophoresis 26: 4368–4378 [DOI] [PubMed] [Google Scholar]
- 46. Rabin R. L., et al. 1999. Chemokine receptor responses on T cells are achieved through regulation of both receptor expression and signaling. J. Immunol. 162: 3840–3850 [PubMed] [Google Scholar]
- 47. Sacre K., et al. 2005. Repertoire, diversity, and differentiation of specific CD8 T cells are associated with immune protection against human cytomegalovirus disease. J. Exp. Med. 201: 1999–2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sallusto F., Geginat J., Lanzavecchia A. 2004. Central memory and effector memory T cell subsets: function, generation, and maintenance. Annu. Rev. Immunol. 22: 745–763 [DOI] [PubMed] [Google Scholar]
- 49. Sallusto F., Lenig D., Forster R., Lipp M., Lanzavecchia A. 1999. Two subsets of memory T lymphocytes with distinct homing potentials and effector functions. Nature 401: 708–712 [DOI] [PubMed] [Google Scholar]
- 50. Scriba T. J., et al. 2005. Ultrasensitive detection and phenotyping of CD4+ T cells with optimized HLA class II tetramer staining. J. Immunol. 175: 6334–6343 [DOI] [PubMed] [Google Scholar]
- 51. Sidney J., et al. 2001. Majority of peptides binding HLA-A*0201 with high affinity crossreact with other A2-supertype molecules. Hum. Immunol. 62: 1200–1216 [DOI] [PubMed] [Google Scholar]
- 52. Sidney J., et al. 1998. Measurement of MHC/peptide interactions by gel filtration, p. 18.13.11-18.13.19. In Coligan J. E., Kruisbeek A. M., Margulies D. H., Shevach E. M., Strober W.(ed.), Current protocols in immunology. John Wiley & Sons, New York, NY [Google Scholar]
- 53. Speiser D. E., et al. 2002. In vivo activation of melanoma-specific CD8(+) T cells by endogenous tumor antigen and peptide vaccines. A comparison to virus-specific T cells. Eur. J. Immunol. 32: 731–741 [DOI] [PubMed] [Google Scholar]
- 54. Sun Y., et al. 2003. A systematic comparison of methods to measure HIV-1 specific CD8 T cells. J. Immunol. Methods 272: 23–34 [DOI] [PubMed] [Google Scholar]
- 55. Terajima M., Ennis F. A. 2006. Using HLA-transgenic mice to identify immunodominant human CD8+ T cell epitopes—does (genome) size matter? Immunol. Lett. 105: 97–98 [DOI] [PubMed] [Google Scholar]
- 56. Van Epps H. L., et al. 2002. Long-lived memory T lymphocyte responses after hantavirus infection. J. Exp. Med. 196: 579–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. van Leeuwen E. M., et al. 2005. IL-7 receptor alpha chain expression distinguishes functional subsets of virus-specific human CD8+ T cells. Blood 106: 2091–2098 [DOI] [PubMed] [Google Scholar]
- 58. von Herrath M. G., Yokoyama M., Dockter J., Oldstone M. B., Whitton J. L. 1996. CD4-deficient mice have reduced levels of memory cytotoxic T lymphocytes after immunization and show diminished resistance to subsequent virus challenge. J. Virol. 70: 1072–1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Walsh C. M., et al. 1994. Immune function in mice lacking the perforin gene. Proc. Natl. Acad. Sci. U. S. A. 91: 10854–10858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Wang P., et al. 2008. A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput. Biol. 4: e1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yi J. S., Du M., Zajac A. J. 2009. A vital role for interleukin-21 in the control of a chronic viral infection. Science 324: 1572–1576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang Q., et al. 2008. Immune epitope database analysis resource (IEDB-AR). Nucleic Acids Res. 36: W513–W518 [DOI] [PMC free article] [PubMed] [Google Scholar]