

Abstract
Here we show that the ubiquitin-proteasome system is required for the efficient replication of rotavirus RRV in MA104 cells. The proteasome inhibitor MG132 decreased the yield of infectious virus under conditions where it severely reduces the synthesis of not only viral but also cellular proteins. Addition of nonessential amino acids to the cell medium restored both viral protein synthesis and cellular protein synthesis, but the production of progeny viruses was still inhibited. In medium supplemented with nonessential amino acids, we showed that MG132 does not affect rotavirus entry but inhibits the replication of the viral genome. It was also shown that it prevents the efficient incorporation into viroplasms of viral polymerase VP1 and the capsid proteins VP2 and VP6, which could explain the inhibitory effect of MG132 on genome replication and infectious virus yield. We also showed that ubiquitination is relevant for rotavirus replication since the yield of rotavirus progeny in cells carrying a temperature-sensitive mutation in the E1 ubiquitin-activating enzyme was reduced at the restrictive temperature. In addition, overexpression of ubiquitin in MG132-treated MA104 cells partially reversed the effect of the inhibitor on virus yield. Altogether, these data suggest that the ubiquitin-proteasome (UP) system has a very complex interaction with the rotavirus life cycle, with both the ubiquitination and proteolytic activities of the system being relevant for virus replication.
INTRODUCTION
Group A rotaviruses are a major cause of severe gastroenteritis in young children. The rotavirus infectious particles are formed by a triple-layer protein capsid that encloses a segmented double-stranded RNA (dsRNA) genome. The outermost layer is composed by proteins VP4 and VP7, which are important for the first interactions of the virus with the cell surface (14). Rotavirus enters cells by endocytosis; some strains use the classical clathrin-mediated endocytosis, while others enter through a not fully characterized endocytic pathway (21, 49). During or shortly after cell entry, the infecting triple-layer particle (TLP) loses the external protein layer and is converted to a double-layer particle (DLP). Once in the cytoplasm, the DLP, which is transcriptionally active, starts transcribing the viral genome (29). The viral mRNAs direct the synthesis of six structural proteins (VP1 to VP4, VP6, and VP7) and six nonstructural proteins (NSP1 to NSP6). In addition to their function as mRNAs, the viral transcripts also serve as RNA templates for the synthesis of negative-strand RNAs to form the double-stranded RNA (dsRNA) genomic segments. The newly synthesized viral proteins are recruited to viroplasms, electrodense cytoplasmic structures where the viral genome replicates and double-layer intermediate replication particles assemble (42). The DLPs newly formed in the viroplasms bud through the membrane of the endoplasmic reticulum into the lumen of this organelle. During this process, the DLPs acquire a transient lipid envelope that is subsequently lost to yield mature infectious TLPs (43). Finally, in MA104 cells, the virus is released into the medium by cell lysis.
The ubiquitin-proteasome (UP) system is the major nonlysosomal protein degradation system in eukaryotic cells (11, 46). Ubiquitin is a small 76-amino-acid protein that is covalently attached to cellular proteins in a three-step reaction: the final product is an isopeptide bond between the carboxy terminus of ubiquitin and the epsilon amino of a lysine residue of the target protein, although ubiquitination has also been reported to occur at histidine, cysteine, serine, threonine, and the N-terminal methionine of some proteins (4, 38, 57). As ubiquitin possesses seven lysines, polyubiquitin chains can be formed. Ubiquitination is the signal to direct proteins to the proteasome (54). The type (poly- versus monoubiquitination) and the site of linkage determine if the target protein is directed to the proteasome or if ubiquitination is involved in the control of the protein activity. Ubiquitination of particular targets is involved in a variety of cellular processes, such as control of cell division, signal transduction, transcriptional regulation, development, immune response, endocytosis, cellular trafficking, and cell survival control (22, 24, 25, 36).
Many viruses manipulate the UP system to favor their replication. For example, proteasome inhibitors affect the replication of herpesvirus (13), vaccinia virus (50), influenza virus (59), human immunodeficiency virus (52), and cytomegalovirus (55), among other viruses. Many viruses encode proteins that can modify the host's ubiquitin machinery, and some viruses even encode their own ubiquitinating or deubiquitinating enzymes (26). Recently, the nonstructural protein NSP1 of rotavirus was described as a ubiquitin ligase that controls the turnover of several factors implicated in the cellular immune response (18, 19). In this work, we explored the participation of the UP system during a single replication cycle of rotavirus. It was found to be required for the efficient translation of cellular and viral proteins and to activate cellular or viral factors that facilitate the replication of the virus genome.
MATERIALS AND METHODS
Cells and viruses.
The green monkey epithelial cell line MA104 was grown in Advanced Dulbecco's modified Eagle's medium (Advanced-DMEM) (Invitrogen, Carlsbad, CA) supplemented with 4% fetal bovine serum (FBS). Murine fibroblasts ts20b and H38.5 were provided by H. L. Ozer, University of Medicine and Dentistry of New Jersey, New Jersey Medical School. Fibroblasts were grown in high-glucose Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated FBS and nonessential amino acids (NEAA; obtained from Gibco as a 100× solution. The amino acids were used at a 1× concentration of 100 μM each. Rhesus rotavirus strain RRV was obtained from H. B. Greenberg, Stanford University, Stanford, CA, and was propagated in MA104 cells as described previously (39). Rotavirus lysates were activated with trypsin (10 μg/ml; Gibco BRL) for 20 min at 37°C.
Plasmids, antibodies, and inhibitors.
The plasmid construct (pBJ5-HA-Ub), expressing a hemagglutinin (HA)-tagged ubiquitin, was kindly provided by S. Das, National Institute of Allergy and Infectious Diseases, Bethesda, MD. Antipeptide polyclonal antibody SPA-812 to heat shock protein 70 (Hsp70) was from StressGen; mouse monoclonal antibodies (MAbs) to VP2 (3A8) and VP6 (255/60) were kindly provided by H. B. Greenberg (Stanford University, Stanford, CA). Rabbit polyclonal sera to purified RRV TLPs, VP1, NSP2, NSP5, and vimentin have been described previously (1, 17, 32). The secondary antibodies used were goat anti-rabbit or anti-mouse coupled to Alexa 647, Alexa 568, or Alexa 488 (Molecular Probes, Eugene, OR). MG132, lactacystin, and proteasome inhibitor I were from Merck Biosciences (Darmstadt, Germany).
Immunofluorescence.
MA104 cells were grown on coverslips at 80% confluence, infected with RRV at a multiplication of infection (MOI) of 1, and then incubated in MEM containing nonessential amino acids in the presence or absence of 10 μM MG132. At 8 hpi, the cells were fixed with 2.5% formaldehyde (freshly prepared from paraformaldehyde) for 20 min at room temperature. Fixed cells were permeabilized by incubation with blocking buffer (1% bovine serum albumin [BSA] diluted in phosphate-buffered saline [PBS]-50 mM NH4Cl) plus 0.5% Triton X-100 for 15 min. Primary and secondary antibodies were incubated for 1 h in blocking buffer at room temperature. Coverslips were mounted on glass slides using Fluokeep (Argene, Varilhes, France), and the samples were observed under a fluorescence microscope (Zeiss Axioskop 2) coupled to a digital camera (Photometrics Cool Snap HQ). The images were then digitally captured and prepared in Adobe Photoshop 6.0. The estimation of the viroplasms' size was carried out as reported previously (7). All images were acquired with a 60× objective with a real-time charge-coupled device (CCD) camera in 256 gray scales, and the size of the images was 1,392 by 1,040 pixels, with 8 bits. The estimation of viroplasm size was done using the Analyze particle function of the ImageJ 1.32j program (Wayne Rasband, NIH).
Real-time PCR.
For detection of the plus and minus strands of rotavirus gene 10, the viral RNA was amplified as described previously (1, 32), and the threshold cycle (CT) for each amplified sample was calculated (31). Data are expressed as the relative difference in either negative- or positive-strand RNA with respect to the total amount of RNA present at time zero postinfection, assigning this sample the arbitrary value of 1. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA was used as an internal control for all samples analyzed (1, 32).
Gel electrophoresis for virus genome detection.
MA104 cells were infected with rotavirus RRV at an MOI of 3, NEAA were added, and the cells were treated or not with MG132, proteasome inhibitor I, or lactacystin. At 9 h postinfection (hpi), total RNA was extracted with TRIzol (Invitrogen, Carlsbad, CA) according to the manufacturer's instructions. One microgram of RNA was loaded per well in a 1.5% agarose gel in Tris-acetate-EDTA (TAE) buffer and, after electrophoresis, the RNA was visualized by ethidium bromide staining using a Typhoon Trio scanner (General Electric).
Determination of virus yield.
Cell monolayers in 48- or 96-well plates were infected with virus at an MOI of 3 focus-forming units (FFU) per cell and then incubated for 8 to 12 h at 37°C in the presence of the indicated drugs. At this time, the cells were lysed by two freeze-thaw cycles, and the lysates were treated with 10 μg of trypsin/ml for 20 min at 37°C. The yield of infectious virus was determined as follows. Confluent MA104 cells in 96-well plates were washed twice with phosphate-buffered saline (PBS), and serial 2-fold dilutions of the above-mentioned viral lysate were adsorbed to cells for 60 min at 37°C. After the adsorption period, the virus inoculum was removed, the cells were washed once with PBS, MEM was added, and the infection was left to proceed for 14 h at 37°C. RRV-infected cells were detected by an immunoperoxidase focus detection assay using a rabbit hyperimmune serum to rotavirus as described previously (39). The FFU were counted with the help of a Visiolab 1000 station (Biocom, France), as previously reported (20).
Plasmid transfection.
Plasmids were introduced into cells by reverse transfection. Two hundred nanograms of each plasmid was mixed with 3 μl of Lipofectamine LTX (Invitrogen, Carlsbad, CA) in 50 μl of MEM directly in a well of a 96-well plate. Twenty minutes later, 10,000 cells were added per well in DMEM, and the cells were incubated for 48 h and then infected as described above (21).
Virus entry assay.
MA104 cells in 96-well plates were infected with about 1,400 FFU/well. The cells were incubated with MG132 1 h before and during the virus adsorption period (1 h at 37°C) and washed twice with PBS to remove the virus inoculum, MEM containing MG132 and MAb 159 to neutralize the extracellular membrane-attached virus that did not enter the cell during the adsorption period was replaced, and the cells were further incubated for 14 h at 37°C. NEAA were added or not to the cell medium as indicated. At 14 h postinfection, the cell monolayers were fixed, and the infected cells were detected by an immunoperoxidase focus detection assay (39).
Western blot.
Cells were infected with rotavirus RRV as described above. Eight hours postinfection, the cells were lysed with Laemmli sample buffer, and the proteins were separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to nitrocellulose membranes (Millipore, Bedford, MA). Membranes were blocked with 5% nonfat dried milk in PBS and incubated at 4°C with the indicated primary antibodies in PBS, followed by incubation with secondary, species-specific, Alexa 647-conjugated antibodies. The immunofluorescence signal was detected in a Thypoon Trio scanner (General Electric). For detection of viral proteins, we used a mix of antibodies to detect all proteins in a single incubation step, with exception of Fig. 3E, where VP1 was detected in parallel lanes (32). For quantification of the signal, the ImageQuant software was used. For detection of free ubiquitin, Tris-Tricine gels were used (51).
Fig. 3.

The effect of the proteasome inhibitor MG132 on protein synthesis is reversed by addition of nonessential amino acids, but the effect on the yield of progeny virus is not. MA104 cells infected with rotavirus RRV (A) or uninfected (B) were incubated for 12 h in the presence or absence of 10 μM MG132 and 100 μM each NEAA, as indicated. In panel A, the viral proteins were detected by Western blotting, and vimentin (Vim) was used as a loading control. In panel B, during the last hour of incubation, 25 μCi/ml of Easy Tag EXPRESS-35S labeling mix was added; the cells were then washed with PBS and lysed with Laemmli sample buffer. The proteins were resolved by SDS-PAGE and subjected to autoradiography. (C) Cells in 96-well plates were infected at an MOI of 3, and at 12 hpi, the cells were lysed by two freeze-thaw cycles, and the viral titer was determined by an immunoperoxidase assay. Data represent the arithmetic means ± SEM of 3 independent experiments performed in duplicate. (D) MA104 cells were infected with rotavirus RRV at an MOI of 3 and incubated in MEM plus NEAA (NE), MEM alone as control (C), MEM plus 100 μM serine and asparagine (SA), or NEAA without serine and asparagine (−SA) in the presence (+) or absence (−) of 10 μM MG132. At 12 hpi, the viral proteins were detected by Western blotting. Vimentin (Vim) was used as loading control. (E) MA104 cells were infected at an MOI of 3 and incubated by 8 h in MEM plus NEAA in the absence (lane 1) or presence (lane 2) of 10 μM MG132. The cells were lysed in Laemmli sample buffer, and the proteins were analyzed by Western blotting using polyclonal antibodies to VP1 (upper panel) or a mixture of sera containing antibodies to purified rotavirus particles, vimentin, NSP2, and NSP5 (lower panel).
Metabolic labeling.
MG132 was added to confluent MA104 cells grown in 96-well plates. Seven hours posttreatment, the medium was replaced by MEM without methionine, supplemented with 25 μCi/ml of Easy Tag EXPRESS-35S labeling mix (Dupont, NEN), maintaining the same concentration of MG132, and incubated for 1 h. Cells were then washed and lysed with Laemmli sample buffer. The proteins were separated by SDS-PAGE and subjected to autoradiography.
Statistical analysis.
Statistical analysis was performed using a paired, two-tailed t test and the Prism 4.0 software for Macintosh (GraphPad Software, Inc.).
RESULTS
Proteasome inhibitors reduce virus replication.
To determine if proteasome activity was required for rotavirus replication, we tested the effect of proteasome inhibitors MG132 and lactacystin and proteasome inhibitor I on virus yield. For this, MA104 cells were infected with the simian rotavirus strain RRV, and after the adsorption period, the cell medium was replaced by medium containing the drugs at the concentrations indicated in Fig. 1. At 12 hpi, the cells were lysed and the virus titer was determined by an immunoperoxidase focus-forming assay. The inhibitors were tested for cell toxicity; at the incubation times and concentrations assayed, the inhibitors did not cause an increase in lactate dehydrogenase (LDH) release (data not shown).
Fig. 1.
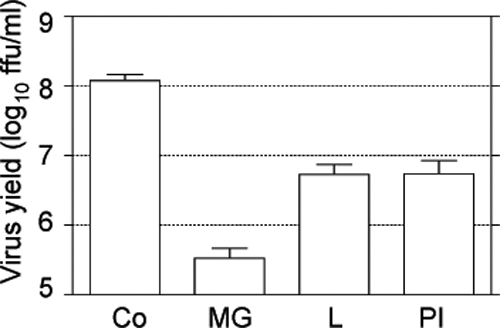
Proteasome inhibitors reduce the yield of rotavirus progeny. MA104 cells grown to confluence in 96-well plates were infected with rotavirus RRV at an MOI of 3. After the adsorption period, the virus inoculum was removed and cell medium containing the indicated drugs was added. Virus was collected at 12 hpi, and the viral titer was determined by an immunoperoxidase focus-forming assay. The data are shown as FFU/ml and represent the arithmetic means ± standard errors of the means (SEM) of 3 independent experiments performed in duplicate. The drugs used were MG132 (MG) at 10 μM, lactacystin (L) at 7.5 μM, and proteasome inhibitor I (PI) at 20 μM. Co, control cells.
All of the inhibitors tested caused a significant reduction in the yield of virus progeny (Fig. 1), suggesting that the UP system is important for rotavirus replication. MG132 had the most pronounced effect since it decreased virus production by more than 2 logs; thus, we used this inhibitor to further characterize the role of proteasome activity on the virus life cycle. To generate additional evidence to link the proteasome activity to rotavirus replication, we used RNA interference to knockdown the expression of several subunits of the proteasome (PSMD2, PSMD8, and PSMD14). When the synthesis of the proteasome subunits was downregulated by the corresponding small interfering RNA (siRNA), the yield of virus progeny was reduced; however, under these conditions, cell survival was compromised, as determined by an LDH release assay (data not shown).
MG132 inhibits protein translation.
To determine the level at which the proteasome activity is required for virus replication, we first tested the effect of the inhibitor on viral protein synthesis. A concentration-dependent reduction in the synthesis of both structural and nonstructural (as judged by the presence of NSP5) viral proteins was caused by MG132 at 8 hpi, while the stress protein Hsp70 was induced (Fig. 2A). Previous reports have shown that this drug induces a translational arrest of cellular protein synthesis (27, 35, 37). Thus, to determine if this was the case for MA104 cells, we analyzed the effect of MG132 on the incorporation of radioactive methionine and cysteine into cellular proteins. After incubation of cells for 8 h in the presence of the drug, a reduction in cellular protein synthesis was found at concentrations of 1 μM or higher (Fig. 2B), indicating that MG132 inhibits cellular protein translation, as well as viral protein synthesis.
Fig. 2.
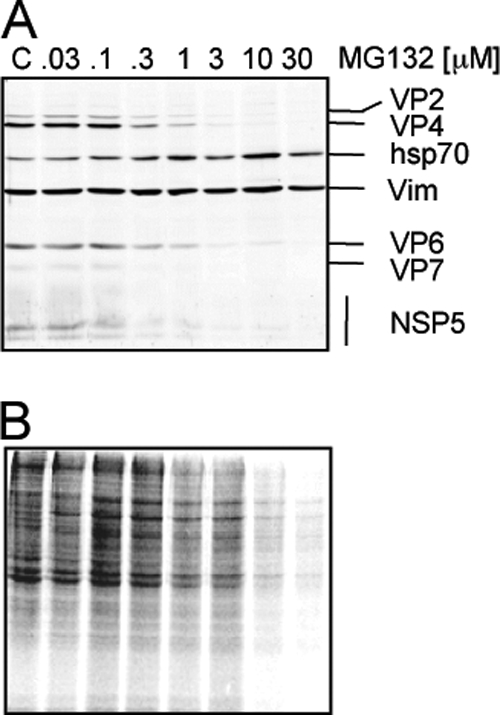
Inhibition of proteasome activity reduces viral and cellular protein synthesis. Cells grown to confluence in 96-well plates were infected (A) or not (B) with rotavirus RRV at an MOI of 3 and then were incubated with the indicated concentrations of MG132 for 8 h, and the cell monolayer was solubilized in Laemmli sample buffer. (A) The proteins were analyzed by Western blotting for detection of rotavirus structural proteins and the virus nonstructural protein NSP5 using specific antibodies. Vimentin (Vim) was used as a loading control, and Hsp70 was detected as a stress indicator. (B) Uninfected MA104 cells were incubated for 8 h with the indicated concentration of MG132. During the last hour of incubation 25 μCi/ml of Easy Tag EXPRESS-35S labeling mix was added; after this period, the cells were washed and lysed with Laemmli sample buffer. Proteins were collected and resolved by SDS-PAGE and then subjected to autoradiography.
The addition of nonessential amino acids reverses the effect of MG132 on cellular and viral protein synthesis, but the virus yield recovers only partially.
Since MG132 was found to inhibit cellular protein synthesis, a specific role of proteasomes in the replication of rotaviruses was difficult to establish. One of the roles assigned to the proteasome is to maintain a pool of free amino acids for protein synthesis (56). We hypothesized that the arrest in cellular and viral protein translation produced by MG132 in MA104 cells could be due to a reduction in the pool level of some limiting amino acids. To test this hypothesis, we incubated MA104 cells in the presence of nonessential amino acids (NEAA), which are not present in MEM. The addition of NEAA to the cell medium reversed the effect of MG132 on both viral protein synthesis (Fig. 3A) and cellular protein synthesis (Fig. 3B); however, under these conditions, the rotavirus progeny yield remained about 10-fold below that obtained in control cells (Fig. 3C). The effect of MG132 on protein synthesis was also reversed by addition of serine and asparagine (Fig. 3D), but not by the mix of NEAA lacking these two amino acids, suggesting that Ser and Asn are key to overcome the protein synthesis effect of the proteasome inhibitor. Altogether, these results indicate that proteasome activity is required for efficient viral and cellular protein synthesis and for efficient rotavirus replication.
Proteasome activity is needed for replication of the virus genome.
Since the addition of NEAA restored viral protein synthesis, and yet there was a 10-fold reduction in virus yield, we decided to evaluate if replication of the viral genome was affected by MG132. To achieve this goal, we quantified the levels of positive and negative RNA strands of RRV gene 10 by real-time PCR. As shown in Fig. 4A, treatment with MG132 reduced the level of viral positive strand by about 50% even in the presence of NEAA, while under these conditions, the level of the negative strand was decreased by about 15-fold. As the negative strand is only present in viral genomic double-stranded RNA (dsRNA), while the positive strand is also present as mRNA, these results suggest that proteasome inhibition reduces viral genome replication. Similar results were obtained when the synthesis of viral genomic dsRNA was evaluated by gel electrophoresis in cells treated with MG132, proteasome inhibitor I, or lactacystin (Fig. 4B). Under these conditions, a clear reduction in the level of the rotavirus genomic dsRNA segments was observed. These results confirm the observations obtained by real-time PCR for rotavirus segment 10 and extend them to all 11 viral dsRNA segments.
Fig. 4.

Proteasome inhibitors block the replication of the viral genome. (A) The levels of RRV gene 10-negative (closed bars) and -positive (open bars) strands were detected by real-time PCR. Cells were grown in 6-well plates, infected in the presence or absence of 10 μM MG132, and at 9 hpi RNA was purified by TRIzol extraction and the abundance of both strands of gene 10 was evaluated as described in Materials and Methods. Note that the positive strand represents the sum of the mRNA plus the sense strand in the genomic dsRNA, while the negative strand is only present in the genomic segments. The results represent the arithmetic means ± SEM of 3 independent experiments performed in triplicate and are expressed as a percentage of the level of the corresponding (negative- or positive-sense) viral RNA in cells incubated in cell medium free of both MG132 and NEAA. (B) Total RNA was extracted from infected cells incubated with NEAA (C), NEAA plus 10 μM MG132 (M), 20 μM proteasome inhibitor (P), or 7.5 μM lactacystin (L). One microgram of each RNA sample was run in a 1.5% TAE agarose gel, and the RNA was visualized by ethidium bromide staining using a Typhoon Trio scanner. The migration positions of rotavirus dsRNA segments (RV seg) and rRNAs are indicated. One representative experiment of 3 performed is shown.
The intracellular distribution of the viral polymerase VP1 is modified in MG132-treated cells.
As the proteasome inhibitor MG132 reduces the replication of the virus dsRNA genome, which takes place in viroplasms (41, 53), we investigated if the composition and/or formation of these cytoplasmic structures was affected by the inhibitor. We studied the intracellular distribution of viroplasmic proteins in infected cells that had been treated with MG132 in the presence of NEAA, using immunofluorescence microscopy. First we analyzed if viroplasms were formed when proteasome activity was inhibited. The intracellular localizations of NSP2 and NSP5 were similar when control and MG132-treated cells were compared; however, some differences in the sizes and numbers of viroplasms were evident (Fig. 5E and F and a to h). The number of total viroplasms increased 2- to 3-fold in MG132-treated cells, with an overall increase in the number of small viroplasms and a decrease in the large viroplasms (Table 1). The viroplasm size was defined as previously reported (7).
Fig. 5.

Intracellular distribution of viral proteins in the presence of MG132. MA104 cells were infected with RRV at an MOI of 1 and incubated with MEM plus NEAA in the presence (B, D, F, H, b, d, f, and h) or absence (A, C, E, G, a, c, e, and g) of 10 μM MG132. At 8 hpi, the cells were fixed and immunostained as indicated in Materials and Methods with antibodies to VP1 (A and B), VP2 (C and D), VP6 (E and F), or NSP2 (G and H). The cells in panels a to h were all stained with antibodies to NSP5.
Table 1.
Quantification of viroplasms detected with antibodies to NSP5
| Cells | Total no. of cells counted | No. of viroplasms/cella |
||
|---|---|---|---|---|
| Small | Medium | Large | ||
| Control | 75 | 7.0 | 2.3 | 1.4 |
| MG132 | 58 | 23.7 | 2.6 | 0.6 |
The size of viroplasms was defined and determined as previously reported (7).
We also analyzed the intracellular distribution in MG132-treated cells of the viral RNA polymerase VP1 and the capsid protein VP2, both of which are essential for replication of the viral genome, and also of VP6, the intermediate-layer capsid protein required for transcription of the rotavirus dsRNA. All three proteins showed their typical viroplasmic localization in untreated cells. However, in the presence of MG132 and NEAA, the localization in viroplasms of VP1 was drastically reduced, and this protein was found mostly homogeneously distributed throughout the cytoplasm (Fig. 5, compare panels A and B). Under these conditions, the cells conserved the presence of viroplasms as detected with anti-NSP5 antibodies (Fig. 5, compare panels a and b). In the case of VP2 and VP6, a mixed distribution was observed. When the cells were incubated with MG132 and NEAA, the proteins seem to localize mostly in viroplasms, with a punctuated distribution in the cytoplasm more evident that in control cells, and a proportion of the proteins being dispersed throughout the cytoplasm (Fig. 5D and F). To discard that the very limited (VP1) or incomplete (VP2 and VP6) localization of the capsid proteins in viroplasms was due to a failure of these proteins to be synthesized efficiently or because they are being rapidly turned over, we analyzed by Western blotting the levels of VP1, VP2, and VP6 compared to NSP2 and NSP5 in treated and untreated cells. As can be seen in Fig. 3E, no difference was observed in the accumulated proteins. Overall, these data support the role of the UP system in the correct intracellular localization of viroplasmic proteins, particularly of VP1, and offer a mechanistic explanation for the inhibition of replication of the viral dsRNA observed in the presence of MG132.
The E1 ubiquitin-activating enzyme is necessary for the efficient replication of rotavirus.
To generate further evidence of the involvement of the ubiquitin-proteasome system in rotavirus replication, we used the well-characterized murine fibroblast cell line ts20b, which is temperature sensitive for the activity of the E1 ubiquitin-activating enzyme (9). As shown in Fig. 6, the yield of rotaviral progeny at the restrictive temperature (39°C) in ts20b cells was reduced to about 33% of that obtained in cells infected at 32°C. In contrast, in H38.5 cells, which are ts20b cells stably transfected with the wild-type human E1 ubiquitin-activating enzyme gene (9), the yield of rotavirus progeny was unaffected at the restrictive temperature (Fig. 6). These data suggest that protein ubiquitination plays a role in rotavirus replication.
Fig. 6.
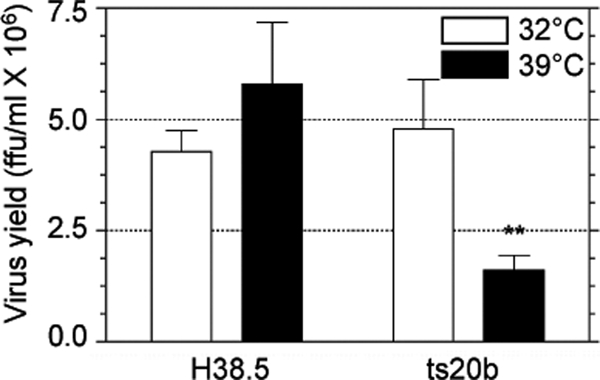
The E1 ubiquitin-activating enzyme has a role in rotavirus replication. Murine fibroblasts ts20b and H38.5 were infected with RRV at an MOI of 1 at the permissive temperature (32°C) for 1 h. The cells were subsequently incubated at either 32°C or 39°C for 8 h, and the yield of infectious viral progeny was determined by an immunoperoxidase assay. The results represent the arithmetic means ± SEM of 3 independent experiments performed in duplicate. **, P < 0.01.
Overexpression of ubiquitin partially reverses the effect of MG132 treatment.
The fact that the activity of E1 ubiquitin-activating enzyme is required for the efficient replication of rotavirus RRV suggests that inhibition of the proteasome activity might cause a depletion or reduction of the pool of free ubiquitin, affecting either (i) the polyubiquitination of cellular antiviral factors that need to be degraded for the efficient replication of the virus or (ii) the ubiquitination of proteins that require to be modified to get activated and effectively participate in the replication of the virus genome. To evaluate if MG132 treatment reduces the pool of free ubiquitin in MA104 control cells, we analyzed by Western blotting the levels of ubiquitin in these cells in the presence or absence of MG132. In control, untreated cells, free ubiquitin was clearly detected, while it was barely detectable in cells that had been treated with MG132 for 8 h (Fig. 7A). In addition, the smear of ubiquitinated proteins became more abundant in cells treated with the inhibitor. These results suggest that inhibition of the proteolytic activity of the proteasome by MG132 leads to a depletion of the pool of free ubiquitin and to an accumulation of ubiquitinated proteins.
Fig. 7.

Overexpression of HA-tagged ubiquitin partially restores virus replication. MA104 cells in 96-well plates were untransfected (A) or transfected with a plasmid coding for HA-tagged ubiquitin (pBJ5-HA-Ub) or the vector alone (pBJ5) (B and C). At 48 h posttransfection, the cells were infected with rotavirus RRV at an MOI of 3 (C) and were treated or not with 10 μM MG132 for 8 h, as indicated. At 8 hpi, the cells were lysed and the cellular proteins analyzed by Western blotting (A and B) using antibodies to ubiquitin (A) or the HA tag (B). The migration positions of both free ubiquitin and HA-tagged ubiquitin are shown. Alternatively, the titer of infectious virus was determined by an immunoperoxidase focus-forming assay (C), as described in Materials and Methods. The results represent the arithmetic means ± SEM of 6 independent experiments performed in duplicate and are expressed as a percentage of the virus titer found in cells incubated in the absence of MG132. *, P < 0.01. In these experiments, NEAA was maintained during the 8 h of incubation with the virus.
If the deleterious effect of MG132 on viral progeny yield is the reduction of the pool of free ubiquitin, overexpression of this protein should reverse the effect of the inhibitor. To determine if this was the case, we evaluated the yield of viral progeny in cells overexpressing HA-tagged ubiquitin. For this, MA104 cells were transfected with plasmid pBJ5-HA-Ub that directs the synthesis of a functional HA-tagged ubiquitin or, as control, with the cloning vector pBJ5. In cells transfected with pBJ5-HA-Ub a pool of free HA-ubiquitin was detected in the absence of MG132, as evaluated with antibodies to the HA tag (Fig. 7B, third lane). Similar to endogenous ubiquitin, the level of free HA-tagged ubiquitin was drastically reduced at 8 h posttreatment with MG132 (Fig. 7B, fourth lane). As shown in Fig. 7C, the inhibitory effect of MG132 on viral progeny production was partially reversed in cells overexpressing HA-ubiquitin. Although the increase in virus yield was discrete, it was highly reproducible and statistically significant. These data suggest that ubiquitination plays a role in the replication cycle of rotavirus, although whether this is required to degrade antiviral cell factors or to activate cellular or viral proteins involved in facilitating the replication of the virus remains to be determined.
The proteasome activity is dispensable for rotavirus entry.
To determine if, in addition to be required for virus genome replication, the proteasome activity was also needed for virus entry, we performed infectivity assays in the presence of MG132. In this case, the cells were pretreated for 1 h with the indicated concentrations of MG132, and the virus was adsorbed to the cell surface for 1 h at 37°C in the presence of the drug. After removing the virus inoculum, the cells were washed twice, and the virus that remained on the cell surface was neutralized with MAb 159. The cells were then incubated with MG132 for 14 h in the presence of NEAA to preclude the effect of the inhibitor on protein synthesis. Under these conditions, MG132 did not affect virus infectivity as determined by a focus-forming immunoperoxidase assay (Fig. 8). As a control, the cells were treated as described previously, but the incubation was performed in the absence of NEAA, conditions under which MG132 produced a clear dose effect reduction of virus infectivity (Fig. 8). These data indicate that the proteasome activity is not required for rotavirus entry into MA104 cells.
Fig. 8.
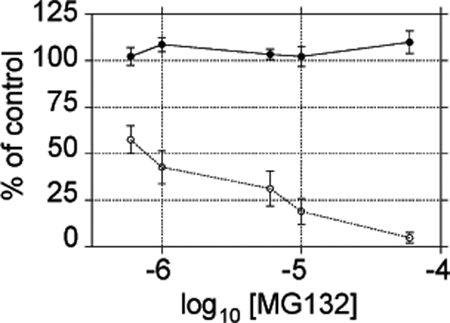
Proteasome activity is not necessary for rotavirus cell entry. MA104 cells were grown in 96-well plates and incubated with the indicated concentration of MG132 1 h before and during virus adsorption. The cells were then maintained at the indicated concentration of MG132 for 14 h in the presence (closed circles) or absence (open circles) of nonessential amino acids. Finally, the infected cells were detected by an immunoperoxidase assay. The results show the arithmetic means ± SEM of 3 independent experiments performed in duplicate and are expressed as the percentage of infected cells incubated in the absence of MG132.
DISCUSSION
The role of the UP system in rotavirus infection has been previously studied in the context of the innate immune response; it is well documented that during rotavirus infection several cellular molecules are degraded by the proteasome, including interferon response elements and the NF-κB regulator IkB (3, 18, 19). These events are relevant to prevent the production of an antiviral response that would limit virus spread (2). However, the role of the UP system during a single virus replication cycle has been poorly characterized. We report in this work that the activity of the UP system is required for efficient replication of rotavirus RRV in MA104 cells.
We found that the most active proteasome inhibitor for reducing the yield of rotaviral progeny was the well-known peptide aldehyde MG132 (46). This drug, as well as other proteasome inhibitors, has been reported to cause cellular stress and to shut down the synthesis of cellular proteins (27, 35, 37). This was the case for MA104 cells, since the drug inhibited the synthesis of both viral and cellular polypeptides (Fig. 2). We also detected an increase in the levels of Hsp70 protein in the presence of the inhibitor, suggesting the induction of a stress response. These data suggested that the reduction of viral protein synthesis was the result of the general effect of this drug on the cell translation machinery. We found that the addition of NEAA restored the efficient translation of both viral and cellular mRNAs. Of interest, addition of only two amino acids, serine and asparagine, was sufficient to restore cell protein synthesis. However, the mechanism through which the addition of amino acids restores protein synthesis remains to be determined. Despite the fact that MG132 no longer affected the synthesis of viral proteins in the presence of NEAA, this drug still reduced the yield of viral progeny by about 20-fold, an amount that correlated with the reduction in the level of the rotavirus genomic negative RNA strand present in cells treated with the inhibitor (Fig. 4), suggesting that the efficient replication of the viral genome requires a functional UP system.
The replication of the rotaviral genome is an event not fully understood, but all available evidence suggests that it takes place in viroplasms (53). Thus, alterations in the assembly of these cytoplamic structures could affect this stage of the virus life cycle. Analysis by immunofluorescence of control and MG132-treated cells showed that infected cells contained viroplasms under both conditions; however, in MG132-treated cells, these viroplasms looked (using either NSP2 or NSP5 as detection antibodies) smaller and were more abundant than those in control, untreated cells, suggesting that the UP system is important for their proper maturation. Not all alterations in the viroplasm structure are reflected in a reduced rotavirus replication. For example, blocking the phosphorylation of NSP5 affects the structure of viroplasms, with a moderate effect on virus replication (6). Thus, we also analyzed the distribution of viroplasmic proteins VP1, VP2, and VP6. The intracellular distribution of VP1 was markedly altered in the presence of the inhibitor, showing a homogeneous distribution throughout the cytoplasm and a poor association with viroplasms, while VP2 and VP6 also had an atypical distribution in MG132-treated cells. These data suggest that an efficient recruitment of viral proteins to viroplasms is dependent on a functional UP system.
The effect of MG132 on the production of viral progeny in the presence of NEAA seems to result, at least in part, from depletion of the free cellular ubiquitin pool (Fig. 7A and B). This conclusion is supported by the observation that the yield of viral progeny in MG132-treated cells transfected with plasmid pBJ5-HA-Ub was about twice that produced in control cells transfected with the plasmid vector pBJ5 (Fig. 7C). The fact that overexpression of ubiquitin did not fully restore the production of infectious virus to the level of untreated controls might be due to several causes. First, MG132 has a wide variety of effects on cells, which include a generalized decrease in protein synthesis. Although in the presence of NEAA, this effect was essentially reversed from a quantitative point of view, qualitative differences must likely occur, as can be appreciated by the preferential synthesis of stress proteins (Fig. 3B). Second, the consumption of ubiquitin in the absence of proteasome activity seems to be very pronounced, since even the overexpressed HA-Ub seems to be depleted in the presence of the inhibitor. Thus, the overexpression of ubiquitin might not be sufficient to suppress all effects caused by MG132. Furthermore, the efficiency of transfection of cells with plasmid pBJ5-HA-Ub was about 20 to 25%, contributing to the low efficiency of viral progeny recovery. Finally, the lack of full recovery of virus yield by ubiquitin overexpression in cells treated with MG132 might also be due to the requirement for a functional proteolytic activity of proteasomes, as has been shown for the late stages of the replication cycle of cytomegalovirus (55) and herpes simplex virus 1 (12, 30), when robust transcription and DNA replication occur and elongating complexes may collide, with proteasomal degradation being required for the resolution of these complexes (30, 55).
The role of ubiquitination in rotavirus replication is furthermore supported by the fact that the yield of rotavirus progeny in ts20b cells, which are temperature sensitive for the expression of an active E1 ubiquitin-activating enzyme, decreased when the cells were infected at the restrictive temperature. In addition, the production of infectious viral progeny at the restrictive temperature was not affected in H38.5 cells stably transfected with a wild-type E1 ubiquitin-activating enzyme. Altogether, these data indicate that ubiquitination is important for the efficient replication of rotavirus.
In contrast to our findings that assign a positive role to the UP system in the replication of rotavirus, it has been reported that the proteins of bovine rotavirus strain RF in Caco-2 cells are degraded by proteasome in an Hsp70-dependent pathway (5); in that work, silencing the expression of Hsp70 by RNA interference (RNAi) enhanced the production of viral progeny, and addition of proteasome inhibitors increased the accumulation of viral proteins. In all of the experiments carried out in our work, MG132 was added immediately after the adsorption period and was maintained in the cell medium during the duration of the experiment. In contrast, Broquet et al. added the drug for 2 h starting at 10 hpi. Thus, the differences between their work and ours most probably result from the different times at which the drugs were added. However, differences due to the cell lines or rotavirus strains used cannot be ruled out.
Recently, Contin et al. reported that proteasome inhibitors MG132 and bortezomib abolish the accumulation of viral proteins and viral RNA and decrease the yield of infectious virus in MA104 cells and concluded that the crucial step impaired by proteasome inhibitors was the assembly of new viroplasms (10). Their results, however, are difficult to interpret, since their experiments were carried out under conditions where we found a marked inhibition of cellular protein synthesis by MG132 (Fig. 2B), suggesting that the inhibition in the synthesis of viral protein they observed might be the consequence of a general inhibition of cellular translation, rather than a specific inhibitory effect on the synthesis of rotaviral polypeptides.
The relevance of the UP system has been shown for several other viruses at different stages of their life cycles (26, 45). For instance, it has been shown to be required for the early steps of infection of influenza virus (28, 59), coronavirus (44), and the parvovirus minute virus of mice (47, 48), as well as during the cell egress of Sendai virus (58), vesicular stomatitis virus (23, 58), rabies virus (23), human immunodeficiency virus (52), and Rous sarcoma virus (40). In many cases, the expression of viral proteins has been shown to be affected by proteasome-inhibiting drugs, as observed in the case of West Nile virus (15, 16), coxsackievirus (33), human respiratory syncytial virus (34), reovirus (8), vesicular stomatitis virus (37), and vaccinia virus (50). However, the effect of proteasome inhibitors on the synthesis of cellular proteins has been evaluated in only a few cases (34, 37, 50), and in all those cases, the inhibition of the proteasome activity for several hours also inhibited the cellular translation machinery. Thus, it is possible that the effect of proteasome inhibitors on viral protein translation observed in previous reports (8, 15, 16, 33, 34, 37, 50) could be due to a generalized, nonspecific inhibition of both cellular and viral protein syntheses, mediated by different mechanisms, including phosphorylation of the α subunit of eukaryotic initiation translation factor (eIF2-α) (27, 35, 37).
In summary, the data presented in this work suggest that the UP system has a very complex interaction with the rotavirus life cycle, being involved in several stages of viral replication. The activity of the UP system is not involved in virus entry, but is required for efficient cellular and viral protein synthesis, and is involved, in an unknown manner, in the recruitment of VP1 and probably VP2 and VP6 to viroplasms. The latter observation could explain the requirement of the rotavirus genome replication for a functional UP system; however, other more direct effects should not be discarded. In this regard, the identification of the E3 ubiquitin-ligases and the protein targets of ubiquitination involved in virus replication will be relevant to understand the role of the UP system during the various stages of the rotavirus replication life cycle.
ACKNOWLEDGMENTS
This work was partially supported by grants 55005515 from the Howard Hughes Medical Institute, SCAI3377 from CONACyT, and IN-2192078 and IN-212211-3 from DGAPA-UNAM. D.S.-A. is recipient of a scholarship from CONACyT.
We are grateful to Suman Das for plasmid pBJ5 and pBJ5-HA-Ub and Harvey Ozer for ts20b and H38.5 cells. We thank Pedro Romero and Rafaela Espinosa for technical assistance.
Footnotes
Published ahead of print on 7 September 2011.
REFERENCES
- 1. Ayala-Bretón C., et al. 2009. Analysis of the kinetics of transcription and replication of the rotavirus genome by RNA interference. J. Virol. 83: 8819–8831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Barro M., Patton J. T. 2005. Rotavirus nonstructural protein 1 subverts innate immune response by inducing degradation of IFN regulatory factor 3. Proc. Natl. Acad. Sci. U. S. A. 102: 4114–4119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Barro M., Patton J. T. 2007. Rotavirus NSP1 inhibits expression of type I interferon by antagonizing the function of interferon regulatory factors IRF3, IRF5, and IRF7. J. Virol. 81: 4473–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ben-Saadon R., et al. 2004. The tumor suppressor protein p16(INK4a) and the human papillomavirus oncoprotein-58 E7 are naturally occurring lysine-less proteins that are degraded by the ubiquitin system. Direct evidence for ubiquitination at the N-terminal residue. J. Biol. Chem. 279: 41414–41421 [DOI] [PubMed] [Google Scholar]
- 5. Broquet A. H., et al. 2007. Hsp70 negatively controls rotavirus protein bioavailability in Caco-2 cells infected by the rotavirus RF strain. J. Virol. 81: 1297–1304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Campagna M., et al. 2007. Impaired hyperphosphorylation of rotavirus NSP5 in cells depleted of casein kinase 1alpha is associated with the formation of viroplasms with altered morphology and a moderate decrease in virus replication. J. Gen. Virol. 88: 2800–2810 [DOI] [PubMed] [Google Scholar]
- 7. Carreño-Torres J. J., Gutiérrez M., Arias C. F., López S., Isa P. 2010. Characterization of viroplasm formation during the early stages of rotavirus infection. Virol. J. 7: 350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Chen Y. T., Lin C. H., Ji W. T., Li S. K., Liu H. J. 2008. Proteasome inhibition reduces avian reovirus replication and apoptosis induction in cultured cells. J. Virol. Methods 151: 95–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Chowdary D. R., Dermody J. J., Jha K. K., Ozer H. L. 1994. Accumulation of p53 in a mutant cell line defective in the ubiquitin pathway. Mol. Cell. Biol. 14: 1997–2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Contin R., Arnoldi F., Mano M., Burrone O. R. 2011. Rotavirus replication requires a functional proteasome for effective assembly of viroplasms. J. Virol. 85: 2781–2792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Craiu A., et al. 1997. Lactacystin and clasto-lactacystin beta-lactone modify multiple proteasome beta-subunits and inhibit intracellular protein degradation and major histocompatibility complex class I antigen presentation. J. Biol. Chem. 272: 13437–13445 [DOI] [PubMed] [Google Scholar]
- 12. Dai-Ju J. Q., Li L., Johnson L. A., Sandri-Goldin R. M. 2006. ICP27 interacts with the C-terminal domain of RNA polymerase II and facilitates its recruitment to herpes simplex virus 1 transcription sites, where it undergoes proteasomal degradation during infection. J. Virol. 80: 3567–3581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Delboy M. G., Roller D. G., Nicola A. V. 2008. Cellular proteasome activity facilitates herpes simplex virus entry at a postpenetration step. J. Virol. 82: 3381–3390 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Estes M. K., Kapikian A. Z. 2007. Rotaviruses, p. 1917–1974 In Knipe D. M., et al. (ed.), Fields virology, 5th ed., vol. 2 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 15. Fernández-García M. D., et al. 2011. Appraising the roles of CBLL1 and the ubiquitin/proteasome system for flavivirus entry and replication. J. Virol. 85: 2980–2989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gilfoy F., Fayzulin R., Mason P. W. 2009. West Nile virus genome amplification requires the functional activities of the proteasome. Virology 385: 74–84 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. González R. A., Espinosa R., Romero P., López S., Arias C. F. 2000. Relative localization of viroplasmic and endoplasmic reticulum-resident rotavirus proteins in infected cells. Arch. Virol. 145: 1963–1973 [DOI] [PubMed] [Google Scholar]
- 18. Graff J. W., Ewen J., Ettayebi K., Hardy M. E. 2007. Zinc-binding domain of rotavirus NSP1 is required for proteasome-dependent degradation of IRF3 and autoregulatory NSP1 stability. J. Gen. Virol. 88: 613–620 [DOI] [PubMed] [Google Scholar]
- 19. Graff J. W., Ettayebi K., Hardy M. E. 2009. Rotavirus NSP1 inhibits NFkappaB activation by inducing proteasome-dependent degradation of beta-TrCP: a novel mechanism of IFN antagonism. PLoS Pathog. 5: e1000280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Guerrero C. A., Zárate S., Corkidi G., López S., Arias C. F. 2000. Biochemical characterization of rotavirus receptors in MA104 cells. J. Virol. 74: 9362–9371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gutiérrez M., et al. 2010. Different rotavirus strains enter MA104 cells through different endocytic pathways: the role of clathrin-mediated endocytosis. J. Virol. 84: 9161–9169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Haglund K., Dikic I. 2005. Ubiquitylation and cell signaling. EMBO J. 24: 3353–3359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Harty R. N., et al. 2001. Rhabdoviruses and the cellular ubiquitin-proteasome system: a budding interaction. J. Virol. 75: 10623–10629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Hershko A., Ciechanover A. 1998. The ubiquitin system. Annu. Rev. Biochem. 67: 425–479 [DOI] [PubMed] [Google Scholar]
- 25. Hoeller D., et al. 2006. Regulation of ubiquitin-binding proteins by monoubiquitination. Nat. Cell Biol. 8: 163–169 [DOI] [PubMed] [Google Scholar]
- 26. Isaacson M. K., Ploegh H. L. 2009. Ubiquitination, ubiquitin-like modifiers, and deubiquitination in viral infection. Cell Host Microbe 5: 559–570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jiang H. Y., Wek R. C. 2005. Phosphorylation of the alpha-subunit of the eukaryotic initiation factor-2 (eIF2alpha) reduces protein synthesis and enhances apoptosis in response to proteasome inhibition. J. Biol. Chem. 280: 14189–14202 [DOI] [PubMed] [Google Scholar]
- 28. Khor R., McElroy L. J., Whittaker G. R. 2003. The ubiquitin-vacuolar protein sorting system is selectively required during entry of influenza virus into host cells. Traffic 4: 857–868 [DOI] [PubMed] [Google Scholar]
- 29. Lawton J. A., Estes M. K., Prasad B. V. 1997. Three-dimensional visualization of mRNA release from actively transcribing rotavirus particles. Nat. Struct. Biol. 4: 118–121 [DOI] [PubMed] [Google Scholar]
- 30. Li L., Johnson L. A., Dai-Ju J. Q., Sandri-Goldin R. M. 2008. Hsc70 focus formation at the periphery of HSV-1 transcription sites requires ICP27. PLoS One 3: e1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Livak K. J., Schmittgen T. D. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- 32. López T., Rojas M., Ayala-Bretón C., López S., Arias C. F. 2005. Reduced expression of the rotavirus NSP5 gene has a pleiotropic effect on virus replication. J. Gen. Virol. 86: 1609–1617 [DOI] [PubMed] [Google Scholar]
- 33. Luo H., et al. 2003. Proteasome inhibition reduces coxsackievirus B3 replication in murine cardiomyocytes. Am. J. Pathol. 163: 381–385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Lupfer C., Pastey M. K. 2010. Decreased replication of human respiratory syncytial virus treated with the proteasome inhibitor MG-132. Virus Res. 149: 36–41 [DOI] [PubMed] [Google Scholar]
- 35. Mazroui R., Di Marco S., Kaufman R. J., Gallouzi I. E. 2007. Inhibition of the ubiquitin-proteasome system induces stress granule formation. Mol. Biol. Cell 18: 2603–2618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mosesson Y., et al. 2003. Endocytosis of receptor tyrosine kinases is driven by monoubiquitylation, not polyubiquitylation. J. Biol. Chem. 278: 21323–21326 [DOI] [PubMed] [Google Scholar]
- 37. Neznanov N., et al. 2008. Different effect of proteasome inhibition on vesicular stomatitis virus and poliovirus replication. PLoS One 3: e1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Okumoto K., et al. 2011. Cysteine ubiquitination of PTS1 receptor Pex5p regulates Pex5p recycling. Traffic 12: 1067–1083 [DOI] [PubMed] [Google Scholar]
- 39. Pando V., Isa P., Arias C. F., López S. 2002. Influence of calcium on the early steps of rotavirus infection. Virology 295: 190–200 [DOI] [PubMed] [Google Scholar]
- 40. Patnaik A., Chau V., Wills J. W. 2000. Ubiquitin is part of the retrovirus budding machinery. Proc. Natl. Acad. Sci. U. S. A. 97: 13069–13074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Patton J. T., Silvestri L. S., Tortorici M. A., Vasquez-Del Carpio R., Taraporewala Z. F. 2006. Rotavirus genome replication and morphogenesis: role of the viroplasm. Curr. Top. Microbiol. Immunol. 309: 169–187 [DOI] [PubMed] [Google Scholar]
- 42. Patton J. T., Spencer E. 2000. Genome replication and packaging of segmented double-stranded RNA viruses. Virology 277: 217–225 [DOI] [PubMed] [Google Scholar]
- 43. Poruchynsky M. S., Atkinson P. H. 1991. Rotavirus protein rearrangements in purified membrane-enveloped intermediate particles. J. Virol. 65: 4720–4727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Raaben M., et al. 2010. The ubiquitin-proteasome system plays an important role during various stages of the coronavirus infection cycle. J. Virol. 84: 7869–7879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Randow F., Lehner P. J. 2009. Viral avoidance and exploitation of the ubiquitin system. Nat. Cell Biol. 11: 527–534 [DOI] [PubMed] [Google Scholar]
- 46. Rock K. L., et al. 1994. Inhibitors of the proteasome block the degradation of most cell proteins and the generation of peptides presented on MHC class I molecules. Cell 78: 761–771 [DOI] [PubMed] [Google Scholar]
- 47. Ros C., Burckhardt C. J., Kempf C. 2002. Cytoplasmic trafficking of minute virus of mice: low-pH requirement, routing to late endosomes, and proteasome interaction. J. Virol. 76: 12634–12645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ros C., Kempf C. 2004. The ubiquitin-proteasome machinery is essential for nuclear translocation of incoming minute virus of mice. Virology 324: 350–360 [DOI] [PubMed] [Google Scholar]
- 49. Sánchez-San Martín C., López T., Arias C. F., López S. 2004. Characterization of rotavirus cell entry. J. Virol. 78: 2310–2318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Satheshkumar P. S., Anton L. C., Sanz P., Moss B. I. 2009. Inhibition of the ubiquitin-proteasome system prevents vaccinia virus DNA replication and expression of intermediate and late genes. J. Virol. 83: 2469–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Schägger H. 2006. Tricine—SDS-PAGE. Nat. Protoc. 1: 16–22 [DOI] [PubMed] [Google Scholar]
- 52. Schubert U., et al. 2000. Proteasome inhibition interferes with gag polyprotein processing, release, and maturation of HIV-1 and HIV-2. Proc. Natl. Acad. Sci. U. S. A. 97: 13057–13062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Silvestri L. S., Taraporewala Z. F., Patton J. T. 2004. Rotavirus replication: plus-sense templates for double-stranded RNA synthesis are made in viroplasms. J. Virol. 78: 7763–7774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Thrower J. S., Hoffman L., Rechsteiner M., Pickart C. M. 2000. Recognition of the polyubiquitin proteolytic signal. EMBO J. 19: 94–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Tran K., Mahr J. A., Spector D. H. 2010. Proteasome subunits relocalize during human cytomegalovirus infection, and proteasome activity is necessary for efficient viral gene transcription. J. Virol. 84: 3079–3093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vabulas R. M., Harti F. U. 2005. Protein synthesis upon acute nutrient restriction relies on proteasome function. Science 310: 1960–1963 [DOI] [PubMed] [Google Scholar]
- 57. Wang X., et al. 2007. Ubiquitination of serine, threonine, or lysine residues on the cytoplasmic tail can induce ERAD of MHC-I by viral E3 ligase mK3. J. Cell Biol. 177: 613–624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Watanabe H., et al. 2005. Cell-specific inhibition of paramyxovirus maturation by proteasome inhibitors. Microbiol. Immunol. 49: 835–844 [DOI] [PubMed] [Google Scholar]
- 59. Widjaja I., et al. 2010. Inhibition of the ubiquitin-proteasome system affects influenza A virus infection at a postfusion step. J. Virol. 84: 9625–9631 [DOI] [PMC free article] [PubMed] [Google Scholar]