

Abstract
Blattella germanica densovirus (BgDNV) is an autonomous parvovirus that infects the German cockroach. BgDNV possesses three mRNAs for NS proteins, two of which are splice variants of the unspliced transcript. The unspliced variant encodes open reading frame 5 (ORF5) (NS3), while NSspl1 encodes ORF3 (NS1) and ORF4 (NS2) and NSspl2 encodes the C-proximal half of NS1. BgDNV possesses three VP transcripts, one of which (VP) is unspliced, while the other two (VPspl1 and VPspl2) are generated by alternative splicing. The unspliced VP transcript contains both ORF1 and ORF2, while in VPspl1, ORF1 and ORF2 are joined in frame. The transcription of NS genes begins at an earlier stage of the virus life cycle than the transcription of VP genes. NS and VP transcripts overlap by 48 nucleotides (nt). BgDNV is characterized by two additional NS transcripts overlapping by more than 1,650 nt with VP-coding transcripts. Four different bands (97, 85, 80, and 57 kDa) corresponding to three BgDNV capsid proteins were detected on SDS-PAGE. Mass spectrometry analysis showed that the amino acid composition of the 85-kDa and 80-kDa proteins is the same. Moreover, both of these proteins are ubiquitinated. The BgDNV PLA2 domain, which is critical for cellular uptake of the virus, is located in ORF2 and is present only in VP1. In contrast to all of the parvoviruses studied in this respect, VP2 has a unique N terminus that is not contained within VP1 and VP3. In situ recognition with NS1- and VP-specific antibodies revealed an uneven pattern of NS1 expression resembling a halo within the nuclear membrane.
INTRODUCTION
The Parvoviridae family comprises animal viruses which are among the smallest and most simply organized and are characterized by linear, single-stranded DNA (ssDNA) genomes encapsidated in 18- to 26-nm nonenveloped icosahedral capsids (9, 18). This family consists of two subfamilies, Parvovirinae and Densovirinae. Densoviruses (densonucleosis viruses or DNVs) are autonomously replicating parvoviruses pathogenic to invertebrates, in particular arthropods (60). Some DNVs are highly species specific, for instance, Galleria mellonella DNV (GmDNV) and Acheta domesticus DNV (AdDNV), which is probably explained by their strict dependence on host cell functions (replication, expression); other DNVs, such as Junonia coenia DNV (JcDNV) and Mythimna loreyi DNV (MlDNV), are polyspecific. Some DNVs, such as GmDNV and JcDNV, infect many tissues (polytropic), whereas others are monotropic (e.g., Bombyx mori DNV [BmDNV]). Almost all DNVs are usually fatal to their hosts (50). About 30 DNVs have been described so far, their hosts belonging to seven orders of the class Insecta and one order of the class Crustacea (45, 50, 51, 55, 61).
Several distinctive features of DNVs, such as high virulence and host specificity, failure to infect vertebrates, and high resistance to extreme environmental conditions, make them potentially effective biological-control agents against populations of agriculturally and medically important pests. Moreover, DNVs could serve as convenient vectors for the genetic manipulation of insects (2, 12, 14, 49). However, to enable their practical use in pest control, it is necessary to understand key features of DNV biology, pathology, species specificity, and especially strategies and regulation of gene expression.
The genome of DNVs, like that of other parvoviruses, is a linear ssDNA molecule 4 to 6 kb in length. It possesses two sets of open reading frames (ORFs); one set codes for 2 to 5 structural, or capsid, proteins, and another set codes for two or three nonstructural regulatory proteins. The DNV genome can be monosense when the two sets of ORFs are located on the same strand or ambisense when NS ORFs occupy the 5′ segment of one strand and VP ORFs occupy the 5′ segment of another strand. The coding part of the genome is flanked by noncoding palindromic sequences which can assume secondary hairpin structures necessary for replication and encapsidation of the viral genome (7, 8, 51).
Based on differences in genome size and structural organization, the structure of terminal sequences, and expression strategies, DNVs are classified into four genera (49), Brevidensovirus (Aedes aegypti DNVs [AeDNVs], Anopheles gambiae DNV, Penaeus stylirostris DNV [PstDNV]) (3, 13, 40, 45), Densovirus (JcDNV, GmDNV, MlDNV) (22, 23, 52), Iteravirus (BmDNV, Casphalia extranea DNV [CeDNV], Dendrolimus punctatus DNV) (24, 32, 56), and Pefudensovirus (Periplaneta fuliginosa DNV [PfDNV]) (60, 61).
Despite the large number of DNVs newly described in the last 2 decades, progress in understanding features of the life cycles of DNVs, particularly strategies and regulation of genome expression and viral proteins and their functions, is rather slow and limited to only certain species: GmDNV (51, 52), MlDNV (23), JcDNV (20, 46), PfDNV (60–62), Culex pipiens DNV (CpDNV) (7), PstDNV (19), and AeDNV (57).
The DNV of the German cockroach, Blattella germanica (BgDNV), was discovered in a cockroach colony that originated from a natural population in a pig farm in North Carolina and maintained in the laboratory for 5 years. The structural organization of this virus was described in our previous work (37).
The purposes of this study were (i) to investigate the BgDNV expression strategy and (ii) to determine the intracellular distribution of structural and nonstructural virus proteins. The expression strategy was found to differ from that of all of the other parvoviruses analyzed thus far. Splicing events are involved in the production of both NS and VP transcripts. Splicing in VP mRNAs results in the joining of two ORFs for capsid proteins into one with an increased coding capacity sufficient to encode the largest VP1 protein. VP2 and VP3, as well as NS2 proteins, are translated according the leaky-scanning mechanism. For the first time, we show monoubiquitination of the DNV capsid proteins and uneven distribution of the NS1 proteins within the nuclei of virus-infected cells.
MATERIALS AND METHODS
Cell culture propagation and virus infection.
For the propagation of BgDNV, a culture of B. germanica BGE-2 cells previously isolated from embryonic tissues was used (30). Cells were grown in 5 ml of antibiotic-free L-15B medium supplemented with 5% fetal bovine serum, 5% tryptose phosphate broth, and 0.1% bovine lipoprotein concentrate, pH 7.0 (39). Cell cultures were incubated at 25°C, and subcultures were made every 10 to 14 days with an initial seeding density of approximately 5 × 105 cells per ml.
To obtain virus-containing extracts, virus-infected cockroaches were lysed in cell culture medium through several consecutive freeze-thaw cycles, homogenized, freeze-thawed once more, and centrifuged at 13,000 ×g for 5 min to remove cellular debris. The resulting supernatant was passed through a 0.22-μm filter and used to infect BGE-2 cells.
Viral RNA purification and Northern blotting.
RNA was extracted from BgDNV-infected BGE-2 cells 3 h, 1 day, 5 days, 7 days, and 20 days postinfection (p.i.) and late in infection (50 to 100 days p.i., passages V1 to V8), when the proportion of virus DNA and genomic DNA in the total DNA extracts remained approximately constant (Fig. 1). Total RNA was extracted using the SV Total RNA Isolation System kit (Promega) according to the manufacturer's instructions. RNA yield and quality were assessed by the absorption value at 260 nm using NanoDrop2000 (Thermo Scientific).
Fig. 1.

Agarose gel electrophoresis of total DNA isolated from virus-infected BGE-2 cells at different times p.i. (3 h, 1 day; and 5 days). V1 to V13 are the numbers of passages through BGE-2 cells, with approximately one passage per 10 to 14 days. The regular arrow marks the total genomic DNA, the single-feathered arrow marks BgDNV genomic DNA ∼5 kb in size, and the double-feathered arrow marks the replicative form of BgDNV DNA. The DNA size markers on the left are in kilobases.
Northern blotting was done as described by Sambrook et al. (43). Promega's RNA Markers were used for size estimation of single-stranded RNA.
All of the probes used were obtained by PCR using respective primer pairs. All of the probes used in our work are listed in Table 1 and depicted in Fig. 2A. The quality of PCR products was verified using electrophoresis on 1% agarose gels in 1× Tris-acetate buffer, and corresponding bands were excised from the gels and purified using the Wizard PCR Preps DNA Purification system kit (Promega) or the MinElute Gel Extraction kit (Qiagen) according to the manufacturer's instructions.
Table 1.
Probes used for Northern blot hybridization
| Probe no. | Description | Name | Primer | BgDNV sequence coordinates (nt) |
|---|---|---|---|---|
| 1 | NS probe ORF3 | pORF3 | ORF3/4 st/ORF3 end | 960–2505 |
| 2 | NS probe ORF5 | pNS5 | ORF5 st/ORF5 end | 346–901 |
| 3 | Whole VP probe | pVPwhole | P1 st/ORF1 end | 2564–5096 |
| 4 | VP probe 5′ half of ORF2 | pVPst | P1 st/ORF2 end | 4642–5096 |
| 5 | VP probe 3′ end of ORF1 | pVPend | lNS_2 F/ORF1 end | 2564–3466 |
Fig. 2.


Schematic illustration of the BgDNV expression strategy. (A) Locations of primers and probes used to map BgDNV VP and NS transcripts and oligopeptides (epitopes) used for antibody production. Primers are indicated by arrows, probes are indicated by dashed lines, and epitopes are indicated by small squares. (B) Summary of BgDNV genome transcription. The BgDNV genome is shown to scale with promoters P1 and P2, polyadenylation [p(A)] signals (vertical double lines), transcription end sites (reverse triangles, empty for VP and shaded for NS), and splice donor (D) and acceptor (A) sites indicated. ITRs are shown as black boxes. mRNAs for VP and NS proteins are transcribed in opposite directions; the direction of transcription is indicated by an arrow in the respective promoter region. RNA species detected in this study are diagrammed with their respective sizes on the left and their names near the end of each transcript. Slanted lines indicate parts of transcripts excised during splicing. BgDNV proteins that are most likely translated from each transcript are indicated on the right. hpVP and hpNStrunc correspond to presumptive proteins encoded by VPspl2 and NSspl2 transcripts, respectively, that were not detected in our study. hpORF6 corresponds to bioinformatically predicted ORF6 located in the VP-coding region but on the opposite strand. For the additional NS transcripts, the endpoint of transcription is presumed to be nt 4139 and in accordance with this, respective mRNA sizes are indicated. Poly(A) tails are indicated by zigzag lines, and in the case of the additional NS transcripts, the existence of poly(A) tails was not confirmed and they are marked by a pair of question marks. (C to H) Schematic representation of NS and VP transcription start and termination sites and splicing events in the corresponding transcripts. For both NS and VP genes, the coding strands and corresponding mRNAs are presented. Normal lowercase letters indicate DNA sequence; lowercase italic letters indicate mRNA sequence. Uppercase letters in both cases indicate sequences corresponding to BgDNV ORFs. For each ORF, the amino acid sequences of the corresponding proteins are in uppercase letters. Numbers throughout the scheme correspond to the transcription start and end sites, donor and acceptor splice sites identified in our study, and ORF start and end sites identified earlier and are indicated in nucleotides. SpDon indicates donor splice sites, and SpAcc indicates acceptor splice sites. Sites of splicing are indicated by vertical lines, and excised mRNA fragments are indicated by slanted dotted lines between them. In panel C, identical sequences at the ends of the first exon and intron are underlined. In panel F, a 48-nt overlap of the NS and VP transcripts is demonstrated. In panel G, in the amino acid sequence for VP1, G with a gray background appears as a result of a junction between the first donor and acceptor sites. For hpVP, V and K amino acid residues with a gray background correspond to a splice junction between the second donor and acceptor sites. (I) Schematic representation of the mechanisms presumably involved in BgDNV VP synthesis. (VP1 and VP3) Leaky-scanning mechanism involved in VP1 and VP3 synthesis from VPspl1 transcript containing ORFspl. A new GGT codon and the corresponding G amino acid residue resulting from a splicing event are underlined. (VP2) Leaky-scanning mechanism presumably involved in VP2 synthesis from the VP unspliced transcript encoding ORF2 and ORF1.
All of the probes were 32P labeled using the Prime-a-Gene Labeling System (Promega) and [α-32P]dATP as the radioactive precursor according to the supplier's protocols based on the random hexaribonucleotide technique.
RT-PCR.
The reverse transcription (RT)-PCR method was used to characterize the transcripts detected by Northern blotting. First-strand cDNA was synthesized using the Im-Prom-II Reverse Transcription System kit (Promega) according to the manufacturer's instructions using a poly(A) primer. Then regions of the viral genome corresponding to bioinformatically predicted ORFs were amplified using corresponding pairs of gene-specific primers. All RNA samples were tested with negative controls (no RT) to confirm the absence of contaminating DNA. All of the resulting bands were gel purified using the Wizard PCR Preps DNA purification system kit (Promega), cloned into the pGEM-T Easy vector (Promega), and sequenced according to Sanger et al. (44) with a BigDye Termination kit (Applied Biosystems) on an ABI Prism 310 sequencer. All of the primers used throughout this work are listed in Table 2 and depicted in Fig. 2A.
Table 2.
Primers used in the present work
| Primer | BgDNV sequence coordinates (nt) | Purpose(s) | Primer name |
|---|---|---|---|
| P1 st | 5079–5096 | RT-PCR, Northern blotting probe synthesis | A |
| ORF6 new RACE | 3729–3757 | 5′ RACE | VP_R1 |
| Splice ORF 1_2 | 4408–4438 | 5′ RACE | VP_R2 |
| P1 RACE | 5067–5096 | 3′ RACE | VP_R3 |
| ORF1 end | 2564–2584 | RT-PCR, Northern blotting probe synthesis | B |
| P2 st | 256–275 | RT-PCR | C |
| ORF 3_5 RACE | 2485–2503 | 5′ RACE | NS_R1 |
| P2 RACE | 241–274 | 3′ RACE | NS_R2 |
| ORF6 end | 4046–4063 | RT-PCR | D |
| P3 st | 877–896 | RT-PCR | E |
| ORF3/4st | 2960–2979 | RT-PCR, Northern blotting probe synthesis | P1 |
| ORF3 end | 2486–2505 | RT-PCR, Northern blotting probe synthesis | P2 |
| ORF5 st | 347–367 | RT-PCR, Northern blotting probe synthesis | P3 |
| ORF5 end | 881–900 | RT-PCR, Northern blotting probe synthesis | P4 |
| lNS_2 F | 3444–3466 | Northern blotting probe synthesis | P5 |
| ORF2 end | 4642–4666 | Northern blotting probe synthesis | P6 |
Identification of 5′ and 3′ ends of viral transcripts.
Transcription start and end points were characterized using the SMART RACE cDNA amplification kit (Clontech) according to the manufacturer's recommendations. To identify 5′ ends, first-strand cDNA was synthesized using adaptor primers SMART II A Oligonucleotide and 5′ CDS primer A, and then 5′ ends were amplified using a universal primer and gene-specific primers. Two primers, VP_R1 (nt 3726 to 3757) and VP_R2 (nt 4408 to 4438), for VP transcripts, and one primer, NS_R1 (nt 2485 to 2503), for NS transcripts, were utilized. To identify 3′ ends, first-strand cDNA was synthesized using only 3′ CDS primer A. 3′ ends were amplified using the universal primer and gene-specific primers VP_R3 (nt 5067 to 5096) for VP transcripts and NS_R2 (nt 241 to 274) for NS transcripts. PCR products were separated on 1% agarose gels, all of the visible bands were excised, purified using MinElute Gel Extraction Kit (Qiagen), and cloned into the pGEM-T Easy vector (Promega), and cloned fragments were sequenced.
Production and purification of BgDNV particles.
In order to obtain BgDNV particles, adult B. germanica cockroaches were infected by adding virus-containing lysate to cockroach rearing water. About 100 freshly dead cockroaches were collected, and virus was purified using a CsCl gradient as follows. Cockroaches were homogenized in liquid nitrogen, diluted with 50 ml cold phosphate-buffered saline (PBS) containing 2 mM phenylmethylsulfonyl fluoride, and then centrifuged at 10,000 × g for 30 min to remove cell debris. Polyethylene glycol 8000 and NaCl were added to the supernatant to final concentrations of 8% and 400 mM, respectively. The mixture was stirred for 30 min at 4°C and centrifuged at 15,000 ×g for 20 min. The pellet containing BgDNV particles was resuspended in 1 ml of cold PBS. Following mixing on a shaker at room temperature, the suspension was centrifuged at 9,000 ×g to remove insoluble debris. The supernatant was transferred to a tube containing 4 ml of a 20% sucrose cushion in PBS and centrifuged in a 50.2 Ti rotor (Beckman, Fullerton, CA) at 137,000 × g overnight at 4°C. The resulting pellet was resuspended in 1 ml cold PBS and dissolved overnight at 4°C with intermittent mixing. The resulting suspension was centrifuged once more through a 20% sucrose cushion, the pellet was resuspended in 50 mM Tris-HCl (pH 8.0) and loaded onto a CsCl step gradient, and the gradient was centrifuged at 152,000 ×g for 18 h at 15°C in a Beckman SW41 rotor. The gradient was obtained by successively layering 2 ml of 40% CsCl, 4 ml of 35% CsCl, and 2 ml of 30% CsCl in 50 mM Tris-HCl, pH 8.0, which was overlaid with 1 M sucrose in the same buffer according to a previously described technique (15). Virus bands were visualized by light scattering, collected, and dialyzed against 50 mM Tris-HCl, pH 8.0.
SDS-PAGE.
SDS-PAGE was performed according to the method of Laemmli (31). A suspension of purified virus particles was mixed 1:1 with 2× sample buffer and separated on 7.5% or 10% resolving gels, and protein bands were visualized by staining with Coomassie brilliant blue R250.
Identification of capsid proteins by mass spectrometry.
Bands of interest were excised from the gel and digested with 12 μg/ml trypsin, and samples were mixed with a 2,5-dihydroxybenzoic acid solution on the sample plate. Peptide mass spectra were obtained using a tandem matrix-assisted laser desorption ionization-time of flight-time of flight mass spectrometer Ultraflex II (Bruker) in the positive-ion reflector mode. Virus proteins were identified by peptide mass fingerprinting using the Mascot Search program (Matrix Science Inc., Boston, MA) and the NCBI protein database, taking into consideration possible oxidation of Met residues and Cys modification by acrylamide.
Western blotting.
To obtain total extracts of the virus-infected BGE-2 cells, the cells were harvested and washed three times with cold PBS and the cell pellet was mixed with an appropriate volume of 2× sample buffer, boiled for 5 min at 100°C, and centrifuged at 13,000 ×g to remove residual debris. Samples of virus particles were treated as described above for SDS-PAGE. Proteins were separated on 7.5% or 10% resolving gels and transferred onto PVDF membranes by semidry blotting in Tris-glycine-methanol transfer buffer. Blots containing BgDNV protein samples were either stained with Coomassie brilliant blue R250 or probed with corresponding antibodies.
Rabbit polyclonal antibodies were generated by Genemed Synthesis (San Antonio, TX) against the following oligopeptides, corresponding to virus ORFs: ORF1, CTFDRPYFYGKPQRVLNSVEL; ORF2, HYSEAKSDIDIQRADTEAIG; ORF3, CNDPLEFHSGPEVGDIPARPR; ORF4, CRVLELTDAVKDEIKLLLAE; ORF5, DWLKCDEVVLEEFEELNER. The approximate locations of these oligopeptides are shown in Fig. 2A. To detect ubiquitin, mouse monoclonal anti-ubiquitin antibody P4G7 (3 to 5 mg/ml, 1:850 dilution; Covance) was used. Secondary antibodies were goat anti-rabbit or goat anti-mouse IgGs, respectively, conjugated with alkaline phosphatase (Promega). For immunoblotting, the ProtoBlot II AP System with the Stabilized Substrate kit (Promega) was used and all procedures were performed according to the manufacturer's recommendations.
Immunofluorescence assay.
Virus-infected and control cells were grown on the surface of coverslips placed on the bottom of culture dishes. The cell monolayer was fixed with 4% paraformaldehyde in PBS overnight. Fixed cells were washed three times with PBS, permeabilized with 0.1% Triton X-100 in PBS for 5 min, and washed three times in PBS. The cells were then blocked by incubation in 3% bovine serum albumin (BSA) in PBS at 4°C overnight, incubated with primary rabbit antibodies to ORF2 or ORF3 in 3% BSA in PBS at 4°C for 2 h, and washed three times with PBS. The cells were then incubated with goat anti-rabbit IgG secondary antibodies conjugated with fluorescein isothiocyanate (FITC; 1:200; Jackson ImmunoResearch Laboratories, Baltimore, MD) for 2 h in the dark and washed three times with PBS. Nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI), and fluorescence was detected using an LSM 510 META confocal fluorescence microscope (Carl Zeiss).
Bioinformatic analysis.
The search for homology of BgDNV cDNA nucleotide sequences was made by BLAST (http://blast.ncbi.nlm.nih.gov/Blast.cgi) and prediction of ORFs was made by ORF Finder on the NCBI website (http://www.ncbi.nlm.nih.gov/gorf/gorf.html). Promoter region predictions were made by the Neural Network Promoter Prediction program (http://www.fruitfly.org/seq_tools/promoter.html). Search of NLS signals was performed using the Wolf PSORT program (http://www.psort.org/).
Note that, according to the convention of Armentrout et al. (4), genomes of parvoviruses should be presented as coding, plus strand with NS genes located on the left. As this principle could not be strictly applied to DNVs possessing ambisense genomes, it was decided to define the genome strand encoding NS proteins as plus strand and depict genomes so that NS ORFs are located on the left while VP ORFs are located on the right (52). In our previous work (37), where the complete sequence of the BgDNV genome was presented, we put it in the opposite orientation, so that VP ORFs were on the left. In the present work, the BgDNV genome is given in the conventional orientation.
Nucleotide sequence accession number.
The GenBank sequence accession number for BgDNV is AY189948.
RESULTS
Northern blot detection of BgDNV transcripts.
Total RNA extracted from BgDNV-infected BGE-2 cells late in infection (50 to 100 days) was hybridized with radioactive probes corresponding to different ORFs encoding nonstructural regulatory (NS) or structural (VP) proteins.
The NS probe complementary to ORF3 and ORF4 (pORF3, Table 1 and Fig. 2A) revealed two major 2.3- and 1.8-kb transcripts and three additional 4.5-, 4.0-, and 1-kb transcripts (Fig. 3A, lane 1). In Fig. 3A (lane 1), the 1-kb band indicated by a dotted arrow is poorly visible due to a smear in the lower part of the lane. However, it was clearly detected when different exposure times were used (data not shown). The NS probe complementary to ORF5 (pNS5, Table 1 and Fig. 2A) revealed two strong, clearly defined bands corresponding to 4.5- and 2.3-kb transcripts (indicated by arrows in Fig. 3A, lane 2) and a nonspecific smearing in the smaller-molecular-size range.
Fig. 3.

Transcription profiles of BgDNV RNAs corresponding to NS and VP ORFs. (A) Northern blot analysis of NS transcription in BGE-2 cells. The blots were hybridized with 32P-labeled DNA probes corresponding to ORF3 (pORF3) (lane 1) and ORF5 (pNS5) (lane 2). The identified transcripts are indicated by arrows with their respective sizes (kb) on the side. (B) Northern blot analysis of VP transcription. Blots were hybridized with 32P-labeled DNA probes corresponding to the whole VP-coding region (pVPwhole) (lane 1), the 5′ region of ORF2 (nt 4641 to 5096; pVPst) (lane 2), and the end of ORF1 (nt 2564 to 3465; pVPend) (lane 3). Viral RNA bands detected in each case are indicated by arrows with their respective sizes (kb) on the side. The group of additional high-molecular-mass transcripts is indicated by a bracket with one asterisk. Additional lower-molecular-mass transcripts in lanes 1 and 2 are indicated by a bracket with two asterisks, and that in lane 3 is indicated by two asterisks only. (C) Northern blot analysis of the temporal dynamics of transcription. The blots were hybridized with 32P-labeled DNA probes corresponding to NS ORFs (pORF3) (panel 1) and VP ORFs (pVPwhole) (panel 2). Both panels: lane 1, total RNA extracted from cells at 3 h p.i.; lane 2, 1 day p.i.; lane 3, 5 days p.i.; lane 4, 7 days p.i.; lane 5, 25 days p.i. The blot at the bottom of each panel demonstrates the corresponding amounts of rRNA. The identified transcripts are indicated by arrows with their respective sizes (kb) on the side. (D) RACE assay. Total RNA isolated from BgDNV-infected BGE-2 cells was used for first-strand cDNA synthesis with the poly(A) primer and a specific adapter primer as specified in Materials and Methods. The resulting products were used for PCR amplifications with gene-specific and universal primers. Above each lane, the primers used for each case are specified. Panel 1, 5′ RACE to identify NS transcription start sites. Panel 2, 3′ RACE to identify NS and VP transcription termination sites, respectively. Panel 3, 5′ RACE to identify VP transcription start sites. Lane M in each case contains molecular mass markers. Nonspecific amplification products are indicated by dots. The major viral transcripts used to determine the transcription start and end sites are indicated by arrows. cDNAs corresponding to unspliced transcripts in each case are indicated by ordinary arrows, and those for spliced transcripts are indicated by the arrows with vertical lines. cDNA corresponding to both spliced and unspliced VP transcripts in panel 3, lane 2, is indicated by a feathered arrow. The bracket with two asterisks in panel 2, lane 2 (3′ RACE), indicates additional VP transcripts discussed in the text.
The VP probe complementary to the whole region encoding capsid proteins (pVPwhole, Table 1 and Fig. 2A) detected two major 2.6- and 2.4-kb transcripts (indicated by arrows in Fig. 3B, lane 1) and a number of additional minor transcripts (indicated by brackets with asterisks in Fig. 3B, lane 1). Two minor 4.5- and 4.0-kb transcripts (indicated by one asterisk, Fig. 3A, lane 1) were similar in size to transcripts revealed with the NS probes. Among the smaller-sized minor transcripts, 2.0, 1.2, 0.9, and 0.75 kb (indicated by two asterisks), the 2.0-kb transcript was the most abundant. The VP probe complementary to the 5′ segment of ORF2 (pVPst, Table 1 and Fig. 2A) detected the two main 2.6- and 2.4-kb transcripts and all of the minor transcripts except 4.5 and 4.0 kb (Fig. 3B, lane 2), whereas the VP probe complementary to the 3′ end of ORF1 (pVPend, Table 1 and Fig. 2A) hybridized with the two main 2.6- and 2.4-kb transcripts and only one 2.0-kb minor transcript (indicated by two asterisks, Fig. 3B, lane 3). The absence of the 1.2-, 0.9-, and 0.75-kb transcripts upon hybridization with the pVPend probe suggests that they are 3′-truncated variants of the main 2.4- and 2.6-kb VP mRNAs. The presence of these minor transcripts in experiments with different probes indicates that they are not artifacts and may have some functional significance in BgDNV's life cycle.
Note that among both the NS mRNAs and VP mRNAs, the relative amounts of transcripts with larger molecular sizes (2.3 and 2.6 kb, respectively) were considerably lower than the relative amounts of transcripts with smaller molecular sizes (1.8 and 2.4 kb, respectively) (Fig. 3A and B).
Overall, Northern blot experiments demonstrated that BgDNV is characterized by a unique transcription pattern, compared with all of the other DNVs described so far.
Temporal dynamics of transcription of the VP and NS genes.
Total RNA was extracted from BgDNV-infected BGE-2 cells 3 h, 1 day, 5 days, 7 days, and 20 days p.i. and analyzed by Northern blot hybridization. The major bands revealed with NS-specific (pORF3) and VP-specific (pVPwhole) probes are shown in Fig. 3C, panels 1 and 2, respectively. To evaluate the comparative amounts of BgDNV transcripts at different times after infection, the relative amount of rRNA revealed by ethidium bromide staining in each sample was used as a reference (bottom panels of Fig. 3C). As early as 3 h p.i., clearly detectable bands corresponding to the NS transcripts were found, and the amount of mRNA increased appreciably during the following 25 days. At the same time, transcripts of structural genes become visible only 5 days p.i., and the amount of mRNA dramatically increased to 25 days p.i. This result clearly demonstrates that transcription of regulatory genes begins in earlier stages of the virus life cycle than transcription of structural genes. Thus, BgDNV possesses two groups of genes, early (NS) and late (VP).
Mapping of BgDNV transcripts.
3′ and 5′ rapid amplification of cDNA ends (RACE) mapping was used to further analyze the details of BgDNV transcription. Generally, the scheme of the experiments was as follows. Total RNA was isolated from BGE-2 cells late in infection (50 to 100 days). To determine the 3′ ends of all possible NS and VP mRNAs, we used gene-specific primers located nearest to the predicted transcription initiation sites. To determine the 5′ ends of NS transcripts, we used primers located most proximally to the predicted transcription termination site. To determine the 5′ ends of VP mRNAs, a nested set of primers located in the middle of the VP-coding region was used. In all cases, the second universal primer (Clontech) was used (see Materials and Methods). All of the bands resulting from amplification were cloned and sequenced. Some bands appeared to be nonspecific (indicated by dots in Fig. 3D) and were excluded from further analyses. To verify the results of RACE analyses, RT-PCR using primer pairs listed in Table 2 and depicted in Fig. 2A was used.
To ascertain the transcription initiation sites for the NS transcripts, 5′ RACE with the NS_R1 primer (Fig. 2A; Table 2) and the universal primer was performed. As shown in Fig. 3D, panel 1, amplification resulted in one strong band approximately 1.6 kb in size (indicated by an arrow with a vertical line), two weak bands approximately 2.0 and 1.1 kb in size (indicated by a simple arrow and an arrow with a vertical line, respectively), and several nonspecific, lower-molecular-mass bands (indicated by dots). Additional RT-PCR amplifications using the C/P4 primer pair (Fig. 2A; Table 2) confirmed the presence of 2.0-, 1.6-, and 1.1-kb bands (data not shown). These primers were designed so that primer C was located just downstream from the predicted start of NS transcription while primer P4 was located near the end of predicted ORF3.
Subsequent analysis of the clones revealed that the 2.0-kb band contained cDNA corresponding to the unspliced NS transcript, while alternative splicing generated the 1.6- and 1.1-kb NS transcripts (Fig. 2B). In the 1.6-kb transcript, 658 nucleotides (nt) are removed between nt 274 (G) (splice donor site, D NS), which is 7 nt upstream from the AUG start codon of ORF5, and nt 933 (A) (splice acceptor site, Ac NS) just downstream of the ORF5 stop codon (Fig. 2C). This splicing event results in the complete removal of ORF5 and the generation of mRNA encoding almost fully overlapping ORF3 and ORF4 (the ORF3 initiation codon is located only 4 nt upstream of the ORF4 initiation codon, Fig. 2C). In the 1.1-kb NS transcript, the same donor site, but an alternative acceptor site, is engaged. Splicing between nt 274 (G) (D NS) and nt 1705 (G) (splice acceptor site, Ac′ NS) results in the removal of a 1,430-nt intron and the generation of mRNA corresponding to a small, 501-nt ORF identical to the 3′-proximal half of ORF3 (the AUG start codon maps to nt 2025, and the stop codon is identical to the ORF3 stop codon) (Fig. 2D).
The transcription start site for all of the NS transcripts was mapped to position 240, 24 nt downstream of the 5′ inverted terminal repeat (ITR), as predicted earlier, provided that transcription was driven by the P2 promoter (50) (Fig. 2E). The sequence context of the transcription start site, TTAGTTG (transcription start site in bold and underlined), corresponded well to the Inr consensus sequence (5′-Py Py A+1 N T/A Py Py-3′). The 5′ untranslated regions for NS mRNAs appeared to be relatively long at 42 nt for unspliced and 35 nt for spliced transcripts (compare Fig. 2C, D, and E).
Note that RACE analysis did not reveal any transcripts starting with nt 823 (A), as would be anticipated if the bioinformatically predicted P3 promoter (37) were active. Therefore, the P3 promoter is most likely not active and the transcription of all three BgDNV nonstructural genes is controlled by the P2 promoter. For PfDNV, a densovirus infecting the cockroach P. fuliginosa, it was demonstrated that the predicted internal P18 promoter, presumably driving the transcription of ns1/ns2 genes, was nonfunctional, and the transcription of all of the NS genes was regulated by the P3 promoter, an analogue of the BgDNV P2 promoter. Also, as in BgDNV, in PfDNV, the ns3-coding sequence was spliced out of the bigger transcript encoding the overlapping ns1 and ns2 genes (61). In the same manner, only one promoter drives the expression of all of the NS genes and splicing is involved in the generation of mRNAs of nonstructural genes in GmDNV (52) and MlDNV (23).
To identify the transcription termination site for NS transcripts, we performed 3′ RACE with primer NS_R2 and the universal primer (Fig. 2A; Table 2, see Materials and Methods). The result of this experiment is shown in Fig. 3D, panel 2, lane 1. Amplification resulted in one bright band of approximately 1.6 kb (indicated by an arrow with a vertical line), two weak bands with approximate sizes of 1.0 and 2.1 kb (indicated by an arrow with a vertical line and a simple arrow, respectively), and nonspecific minor bands (indicated by dots). Subsequent analysis revealed that the 3′ end mapped to nt 2541, 14 nt downstream from the predicted canonical polyadenylation signal (2521 to 2526, AAUAAA) (37) (Fig. 2F).
Data obtained for NS transcripts and their exact sizes are summarized in Fig. 2B. The sizes, 2,302, 1,644, and 872 nt, are in reasonable agreement with the respective sizes of ∼2,300, ∼1,800, and ∼1,000 nt obtained by Northern blotting (Fig. 3A, lane 1). The fact that hybridization with the ORF5 probe (pNS5) did not reveal ∼1,800- and ∼1,000-nt transcripts (Fig. 3A, lane 2) confirms that ORF5 is contained only within the largest unspliced mRNA and excised from the others during splicing events.
5′ RACE with the VP_R1 primer, located approximately in the middle of the predicted ORF1 sequence (nt 3729 to 3757) (Fig. 2A; Table 2), and the universal primer, performed in order to identify the transcription start sites for VP transcripts, provided two major bands with approximate sizes of 1.4 and 1.2 kb (Fig. 3D, panel 3, lane 1, indicated by a simple arrow and an arrow with a vertical line, respectively) and a number of lower-molecular-mass nonspecific bands (indicated by dots in the same lane). This result was confirmed by RT-PCR amplifications with primers A and B (Fig. 2A; Table 2) that resulted in two bands with respective sizes of approximately 2.3 and 2.5 kb (data not shown).
Analysis of the 1.2-kb cDNAs revealed that two different splicing events were responsible for the generation of VP transcripts (Fig. 2B). In one case, 229 nt were excised, resulting in the junction of ORF2 and ORF1 in frame. This new ORF (ORFspl) is 778 amino acids (aa) in size and has a capacity to encode an 85.3-kDa protein. The donor site (D1 VP) is the G at nt 4421, and the acceptor site (Ac VP) is the G at nt 4191. D1 VP is located 7 nt upstream of the AUG start codon for ORF1 and 16 nt upstream of the TAA stop codon for ORF2, overlapping the beginning of ORF1 by 11 nt (Fig. 2G). In another case, an alternative donor site (D2 VP), the G at nt 4431, 10 nt upstream of D1 VP (Fig. 2E), was exploited. This splicing did not generate any new ORF, but it slightly extended ORF2 (40 aa). Because the cDNAs resulting from these alternative splicing events differed by only 10 nt, they appeared together as one 1.2-kb band.
The larger 1.4-kb cDNA corresponded to an unspliced VP transcript containing both ORF2 and ORF1.
5′ RACE designed to facilitate VP mRNA 5′-end identification with the universal primer and the VP_R2 primer (Fig. 2A; Table 2) nested relative to VP_R1 and located in ORF2, yielded one band of approximately 750 nt (Fig. 3D, panel 3, lane 2, indicated by a feathered arrow) and two nonspecific bands (indicated by dots). Because the VP_R2 primer is located just upstream of the VP D2 splice donor site, the resulting single 750-nt band was common to the spliced and unspliced transcript 5′ ends, which was further confirmed by sequencing of the corresponding cDNA.
The transcription start for all of the VP transcripts mapped to nt 5096, 23 nt downstream from the 3′ ITR (Fig. 2H), in accordance with the bioinformatically predicted start point, provided that transcription is driven by the P1 promoter (37). Therefore, in contrast to NS transcripts, the 5′ untranslated region for VP mRNAs is very short and comprises only 3 nt, exactly the same as for CpDNV (7). A very short 5′ leader sequence was also described for GmDNV and MlDNV (23, 52). The sequence context of the VP transcription start site, TTAGTATG (transcription start site in bold and underlined), corresponds quite well to the consensus Inr sequence.
To identify transcription stop sites for VP mRNAs, we performed 3′ RACE using the universal primer and the VP_R3 primer, located close to the VP transcription start site (Fig. 2A; Table 2). In addition to the expected two bands corresponding to the unspliced and spliced transcripts (indicated by a simple arrow and an arrow with a vertical line, respectively), amplification yielded a number of bright bands differing in size (indicated by a bracket with two asterisks), as shown in Fig. 3D, panel 2, lane 2. We cloned and sequenced all of the bands; subsequent analysis revealed that the largest two (approximately 2.2 and 2.4 kb, indicated by an arrow with a vertical line and a simple arrow, respectively) indeed corresponded to the full spliced and unspliced transcripts, respectively, with a transcription stop point at nt 2494, 19 nt downstream from the predicted polyadenylation signal (AAUAAA, nt 2513 to 2518) (37) (Fig. 2F). The exact sizes of VP mRNAs, taking into account two different splicing events, are 2,603, 2,374, and 2,364 nt (Fig. 2B) and are in good agreement with the 2,600- and 2,400-nt bands obtained by Northern blotting (Fig. 3B, lane 1). Since the size difference between the two spliced mRNAs is just 10 nt, they appear on the blots as one band. All of the other 3′ RACE bands proved to contain products of transcription termination at the internal sites: 4,031, 3,989, 3,824, and 3,387 nt. The relative sizes of the resulting mRNAs, 840, 882, 1,043, and 1,481 nt, matching respective cDNAs (indicated by a bracket with two asterisks in Fig. 3D, panel 2, lane 2), corresponded to the sizes of the minor transcripts group (2,000, 1,200, 900, and 750 nt) detected by Northern blotting (Fig. 3B, lane 1, indicated by a bracket with two asterisks), suggesting that they are authentic products of viral origin rather than products of artifactual PCR amplification. Additional support would come from the results of Northern blot hybridization with probe pVPend (Fig. 2A; Table 1), which overlapped the 3′ end of the longest 1,481-nt additional transcript; it should not detect shorter transcripts. As described above, this was indeed the only transcript detected by this probe (Fig. 3B, lane 3, band with asterisk). At the same time, probe pVPst, targeted to the 5′ part of the transcripts, was able to detect all of them, confirming the presence of a shared common transcription start point (Fig. 3B, lane 2). Nevertheless, the functional role of these additional transcripts is unclear, as they do not contain any new ORF, so the only difference among them is the size of the N-terminal parts of VP ORFs. They might result from some errors in transcription termination or the polyadenylation machinery recognizing A-rich sequences or A stretches, which are highly abundant in the BgDNV genome. However, analysis of sequences surrounding termination points for these transcripts revealed that they contain a set of conservative polyadenylation-driving signals [poly(A) signal, A-rich sequences, GT-rich sequences]. Moreover, their relative number compared to that of the main transcripts is considerable, and although such transcripts are not detected in NS genes, the NS-coding region is also rich in A stretches. All of these observations suggest that these additional transcripts are not artifactual and that they may play some role that has yet to be determined. No such transcripts were described so far for any other DNVs. A summary of the VP transcripts is given in Fig. 2B.
Note that in our previous work (37), an additional polyadenylation signal for VP genes at positions 4394 to 4399 was predicted that, if functional, could generate mRNA containing only ORF2. But in our present work, we did not detect any transcripts with a transcription stop point corresponding to this polyadenylation site. The relative amount of unspliced mRNAs for VP, as well as for NS transcripts, is poorly comparable to the amount of the spliced ones, as estimated by Northern blotting and RT-PCR. Finally, identification of the 3′ ends of transcripts showed that the NS and VP transcripts overlapped by 48 nt (Fig. 2B). This is characteristic of DNVs possessing ambisense genomes, as demonstrated for GmDNV, MlDNV, CpDNV, and others (7, 23, 52).
Identification of long NS transcripts.
The sizes of the two largest additional NS transcripts (about 4,000 and 4,500 nt) detected by Northern blotting (Fig. 3A, lane 1) suggest that they may result from transcription that does not terminate at the identified NS transcription termination point but rather proceeds farther, so the resulting transcripts may comprise almost the whole BgDNV genome. The 4,000- and 4,500-nt bands do not result from the hybridization of contaminant BgDNV genome DNA, since hybridization of the same probes with mRNA samples containing BgDNV genomic DNA demonstrated that the band corresponding to the whole genome is larger and is accompanied by two distinct bands of approximately 4,000 and 4500 nt (data not presented). The natural origin of the additional transcripts was supported by the fact that the same bands were detected with probes to the whole VP-coding region, pVPwhole (Fig. 3B, lane 1, indicated by a bracket with one asterisk), but not with the probe pVPst, corresponding to the very beginning of ORF2 (the probe closest to the 5′ end of the genome) (Fig. 3B, lane 2). Bioinformatic analysis of the BgDNV genome revealed the presence of one more ORF (ORF6, nt 3684 to 4079) located in the VP-coding part of genome but on the NS-coding strand, with a predicted size of 396 nt and the coding capacity for a 131-aa (13.6-kDa) protein (Fig. 2A). Comparison with the NCBI database showed no significant homology with any known proteins.
We proposed that ORF6 may be encoded by large additional NS transcripts, but its functional role is unclear. RT-PCR amplifications with primer pairs C/D and E/D (Fig. 2A; Table 2) confirmed that transcripts containing ORF6 and extending to the VP-coding region of the BgDNV genome did exist (data not shown). We also showed that a portion of these transcripts is spliced and that splicing occurred between the D NS donor sites and A NS acceptor sites described earlier. This agreed well with the fact that only the 4,500-nt transcript was revealed by hybridization with probe pNS5 (Fig. 3A, lane 2).
The bioinformatic analysis of the BgDNV genome revealed the presence of three additional polyadenylation sites for NS transcripts at positions 4209 to 4214, 4533 to 4538, and 4620 to 4625 (data not shown). We used 3′ RACE to determine the 3′ ends of the transcripts, but unfortunately, we encountered some difficulties due to a great amount of nonspecific products resulting from amplifications with different gene-specific primers that made isolation of the sought-for specific amplification products difficult. We found only one polyadenylated transcript that terminated at nt 4139, which is before the first hypothesized polyadenylation site at nt 4209 to 4214. We hypothesize that a larger amount of transcripts may terminate at some point(s) closer to the 5′ end of the BgDNV genome, after the predicted additional polyadenylation signals. Moreover, it is also possible that these long transcripts are, in fact, not polyadenylated, which hindered the identification of the stop point by the RACE method. Features of long NS transcripts are summarized in Fig. 2B.
Therefore, BgDNV possesses two groups of nonstructural transcripts with the same promoter and transcription start sites but with different lengths and alternative transcription stop points.
To summarize the data obtained, BgDNV possesses three mRNAs for NS proteins, two of which (NSspl1 and NSspl2) are spliced variants of the unspliced transcript (NS) having the same donor (D NS) and different acceptor (Ac NS and Ac′ NS, respectively) splice sites. The unspliced variant most likely encodes ORF5 (NS3), while NSspl1 encodes ORF3 (NS1) and ORF4 (NS2) and NSspl2 most likely encodes the C-proximal half of NS1, designated hpNStrunc. BgDNV is characterized by two additional NS transcripts (lNS and lNSspl1) with the same transcription start point and splicing donor (D NS) and acceptor (Ac NS) sites as the main NS transcripts but overlapping VP-coding transcripts by more than 1,650 nt.
BgDNV also possesses three VP transcripts. One of them (VP) is unspliced, while the other two are generated by alternative splicing using different donor sites (D2 VP, VPspl2; D1 VP, VPspl1) and the same acceptor site (Ac VP). The VP unspliced transcript contains both ORF1 and ORF2, while in VPspl1, ORF1 and ORF2 are joined in frame to produce new ORFspl, and ORF2 in VPspl2 is slightly longer. A summary of BgDNV transcripts is given in Fig. 2B.
Purification of BgDNV particles and identification of capsid proteins.
Centrifugation of BgDNV particles in a CsCl buoyant density gradient (see Materials and Methods) yielded three visible bands. By collecting these bands into separate fractions and further purification of the viral DNA, we demonstrated that the upper band contained empty particles while the two lower bands contained full BgDNV particles differing in buoyant density. Both kinds of full particles appeared to contain plus and minus viral DNA strands in the same proportions. These findings were the same as described previously for GmDNV, viral particle preparations of which also contained two types of full particles, DNVI and DNVII. Thorough investigation of this phenomenon supported the idea that the difference between the two types of particles may be related to protein content and quaternary structure (53).
The two fractions containing full viral particles were combined, and the polypeptide composition of the purified BgDNV capsids was analyzed by SDS-PAGE. Coomassie staining of the gel revealed several discrete protein bands (Fig. 4A). The largest band corresponded to an ∼100-kDa protein (arrow in Fig. 4A), two groups of bands (single and double asterisks, respectively, Fig. 4A) corresponded to ∼85- and ∼80-kDa and ∼56- and ∼55-kDa proteins, and the last group of bands (marked by three asterisks in Fig. 4A) was represented by several low-molecular-mass proteins (∼41, ∼36, and ∼27 kDa). The ∼56-kDa protein was the most abundant among BgDNV capsid proteins, while the others were present in considerably lower quantities. After SDS-PAGE separation, BgDNV capsid proteins were transferred onto PVDF membrane and stained with Coomassie to confirm complete transfer (Fig. 4B, lane 1, bands are marked as in Fig. 4A). Then a series of Western blot assays was carried out. Assay with antibodies against the supposed common C terminus of the capsid proteins (C terminus of ORF1, Fig. 2A) revealed a pattern similar to that shown by SDS-PAGE (Fig. 4B, lane 2), except that only one low-molecular-mass band (∼36 kDa, feathered arrow in Fig. 4B, lane 2) was detected. Probing with an antibody against the N terminus of the protein encoded by the VPspl1 mRNA (Fig. 2B) revealed only one clear band corresponding to the ∼100-kDa protein (Fig. 4B, lane 3), suggesting that it was the only protein containing the respective antibody determinants. No other protein with a molecular mass corresponding to that of the ORF2-encoded protein (24.8 kDa) or its extended variant (30 kDa) encoded by the VPspl2 mRNA (Fig. 2B) was recognized by this antibody (Fig. 4B, lane 3). Therefore, the Western blotting results showed that BgDNV capsid proteins possessed identical C ends, while the largest protein was characterized by a unique N terminus, which agrees well with the supposition that BgDNV capsid proteins could be translated by a leaky-scanning mechanism from the same transcript.
Fig. 4.

Characterization of BgDNV structural proteins. (A) SDS-PAGE of BgDNV structural proteins obtained by infection of B. germanica adults and purification in a CsCl gradient. Protein molecular mass markers are indicated on the left in kilodaltons. The protein bands observed with Coomassie brilliant blue R250 are indicated as follows: ∼100-kDa band, arrow; ∼85-kDa and ∼80-kDa bands, small bracket and one asterisk; ∼57-kDa and ∼56-kDa bands, small bracket with two asterisks; ∼41-kDa, 36-kDa, and 27-kDa bands, large bracket with three asterisks. (B) Western blot analysis of BgDNV capsid proteins. In lane 1, BgDNV capsid proteins were resolved by SDS-PAGE, blotted onto PVDF membrane, and then stained with Coomassie brilliant blue R250. All of the detected bands are indicated as in panel A. In lane 2, the same blot was probed with antibodies to the common C terminus of the capsid proteins. The ∼100-kDa band is indicated by an arrow and designated VP1, the ∼85-kDa and ∼80-kDa bands are indicated by a bracket and designated VP2, the ∼57-kDa band is indicated by an arrow and designated VP3, and the ∼36-kDa band and several bands near VP3 are indicated by feathered arrows. In lane 3, the same blot as in lane 1 was probed with antibody to the unique VP1 N terminus. In lane 4, detection of ubiquitin conjugation with VP2 capsid protein. The same blot as in lane 1 was probed with monoclonal antibody to ubiquitin (P4G7 clone; Covance). Two bands corresponding to VP2 are indicated by a bracket. Protein molecular mass markers are indicated in kilodaltons on the left. (C) SDS-PAGE of BgDNV capsid proteins in BGE-2 cells. Protein extracts were obtained from BGE-2 cells infected with BgDNV, subjected to 10% SDS-PAGE, and blotted onto PVDF membranes. Lane 1, immunoreactivity with antibodies to the common C terminus. Lane 2, immunoreactivity with antibodies to the unique VP1 N terminus. BgDNV capsid proteins are indicated as in panel B, lane 2. Unique BgDNV capsid proteins (presumably resulting from intracellular protease digestion of the native capsid proteins) are indicated by a large bracket and four asterisks. Protein molecular mass markers are indicated in kilodaltons. (D) Examples of mass spectroscopy-detected peptides for virus proteins VP1, VP2, and VP3 matching the corresponding BgDNV ORFs. Matched peptides are shown in bold. The methionine residue (M) driving the synthesis of VP1, VP2, and VP3 is shown in larger bold font. The amino acid sequence where two ORFs are joined during a splicing event to produce ORFspl is underlined. The G shown in italics in ORFspl originates as a consequence of the junction between splice donor and acceptor sites.
Mass spectrometry with subsequent peptide mass fingerprinting using Mascot Search software showed that the largest ∼100-kDa protein, named VP1 (Fig. 4B, lane 2, arrow), started with the first AUG codon of the VPspl1 transcript (Fig. 2B, I) and was fully encoded by the corresponding ORF, ORFspl1 (Fig. 4D; see Fig. S1 in the supplemental material).
Surprisingly, two proteins of ∼80 and ∼85 kDa, named VP2 (Fig. 4B, lane 2, bracket), proved to be identical and entirely corresponded to an ORF1 translation product (Fig. 4D; see Fig. S2 in the supplemental material).
Mass spectrometric analysis demonstrated that the coding sequence for the ∼56-kDa protein named VP3 (Fig. 4B, lane 2, marked by an arrow) began with the second in-frame AUG codon of ORFspl1 (nt 4060 of the BgDNV genome) (Fig. 4D, the corresponding Met is represented by a bold capital letter M in the ORF1-encoded protein sequence; see Fig. S3 in the supplemental material) and that this protein had the same C terminus as VP1.
We hypothesized that the additional low-molecular-mass proteins (∼41, ∼36, and ∼27 kDa), as well as proteins forming bands surrounding the VP3 band (Fig. 4A and B, lane 2), were products of protease cleavage of BgDNV capsid proteins. Probably a C-terminal part of the ∼41- and ∼27-kDa proteins was cleaved, also explaining why they were not visualized with antibodies against the common C terminus, while the C termini of the other cleavage products remained intact (feathered arrow in Fig. 4B, lane 2). Additional support in favor of this hypothesis was obtained by mass spectroscopic analysis of the ∼41-, ∼36-, and ∼27-kDa proteins (see Fig. S4 in the supplemental material). The results show that the proteins analyzed represent different parts of the capsid BgDNV proteins, although they were isolated from native purified capsids. Moreover, BgDNV capsid proteins found in virus-infected BGE-2 cell lysate are characterized by a stable, unique proteolytic degradation pattern, which can be detected by Western blotting with C terminus-specific antibodies (Fig. 4C).
Taken together, these data show that the BgDNV capsid is composed of three different proteins, namely, VP1, VP2, and VP3, and the latter is the basic structural element since it is the most abundant.
Based on mass spectrometry data (Fig. 4D; see Fig. S1 and S4 in the supplemental material), we infer that VP1 and VP3 BgDNV capsid proteins could both be translated from the VPspl1 transcript which is formed as a result of the splicing of the long transcript contained in both ORF1 and ORF2 (Fig. 2A and B). VP1 is translated from the first AUG codon, while VP3 is translated from the second AUG codon by a leaky-scanning mechanism (Fig. 2I). The same translation strategy of the capsid proteins was described for several other DNVs, such as CpDNV (7) and GmDNV (52). Amino acid sequence analysis of BgDNV VP1 revealed a PLA2 domain in the unique N-terminal part (aa 134 to 192) common to almost all parvoviruses. The PLA2 domain of BgDNV is located in ORF2 and is therefore present only in VP1.
Mass spectrometry data (Fig. 4D; see Fig. S2 and S3 in the supplemental material) showed that the amino acid sequence of the VP2 capsid protein completely corresponded to ORF1. The bioinformatic analysis of the nucleotide sequence of the VP-coding part of the BgDNV genome revealed two additional promoter regions with transcription start points at nt 4854 and 4462, respectively, that could produce transcripts encoding ORF1 only. However, a detailed 5′ RACE-assisted search failed to detect any such transcripts in sufficient amounts. Besides, the absence of a difference between Northern blot hybridizations with probes pVPst and pVPwhole (Fig. 3B, lanes 1 and 2) further supports the assumption that no separate ORF1-encoding transcripts exist. Therefore, the VP2 protein is most likely translated from the unspliced VP transcript (Fig. 2B) by the so-called leaky-scanning mechanism. It is clear that the first AUG codon of ORF2 is functional, as it serves as the start codon for VP1 protein in ORFspl1. But the 5′ untranslated region in VP transcripts is just 3 nt long, and the context of the first AUG codon is not very favorable (AGTATGT, where the start codon is in bold and the −3 and +4 positions are in bold and underlined) (30), which causes the ribosome to pass it through and to start translating the VP2 protein from the first AUG codon of ORF1. In other words, the start AUG for ORF2 may, in some cases, be “unseen” by the ribosome due to its proximity to the 5′ end, so the ribosome can “jump” to the second AUG codon of this transcript, corresponding to the first ATG codon of ORF1. A hypothetical scheme for the VP2 translation mechanism is presented in Fig. 2I. In contrast to all of the parvoviruses studied in this respect, VP2 has a unique N terminus that is not contained within VP1 and VP3.
VP2 protein ubiquitination.
According to the BgDNV translation strategy described above, the predicted molecular masses of the VP1 to VP3 capsid proteins should be 85.3, 69.7, and 56.3 kDa, respectively. Surprisingly, the molecular masses of VP1 and VP2 (but not VP3) estimated by SDS-PAGE did not correspond to the predicted values and were ∼100 and ∼80 to 85 kDa, respectively. Moreover, the VP2 protein was represented by two bands. These two deviances are most likely due to posttranslational modifications of these proteins, such as glycosylation, phosphorylation, and/or ubiquitination. The last hypothesis was tested experimentally by Western blotting using BgDNV particles and an anti-ubiquitin antibody possessing extensive cross-reactivity from yeasts to human. As shown in Fig. 4B, lane 4, the antiubiquitin antibody recognized two protein bands corresponding to VP2 proteins, suggesting that they indeed are ubiquitinated, while VP1 and VP3 are not.
To date, monoubiquitination has not been shown for densovirus capsid proteins. The exact function of ubiquitin conjugation of BgDNV VP2 proteins is unclear. At the same time, based on the proposed roles of capsid ubiquitination in various other viruses, it is possible that the observed modification of the BgDNV capsid proteins provides a means for (i) cell defense against BgDNV infection, (ii) viral entry into cockroach cells, and (iii) viral capsid assembly during late phases of infection and viral release from the cell (see Discussion).
Western blot assay identification of BgDNV regulatory proteins.
To identify BgDNV regulatory proteins, we performed Western blot assays of cellular extracts of virus-infected BGE-2 cells with antibodies raised against the NS1, NS2, and NS3 proteins encoded by ORF3, ORF4, and ORF5, respectively.
Antibodies to NS1 and NS2 revealed clear bands with approximate sizes of 60 and 29 kDa, corresponding well to the expected molecular masses (60.2 and 30.3 kDa, respectively) (Fig. 5, lanes 1 and 2).
Fig. 5.
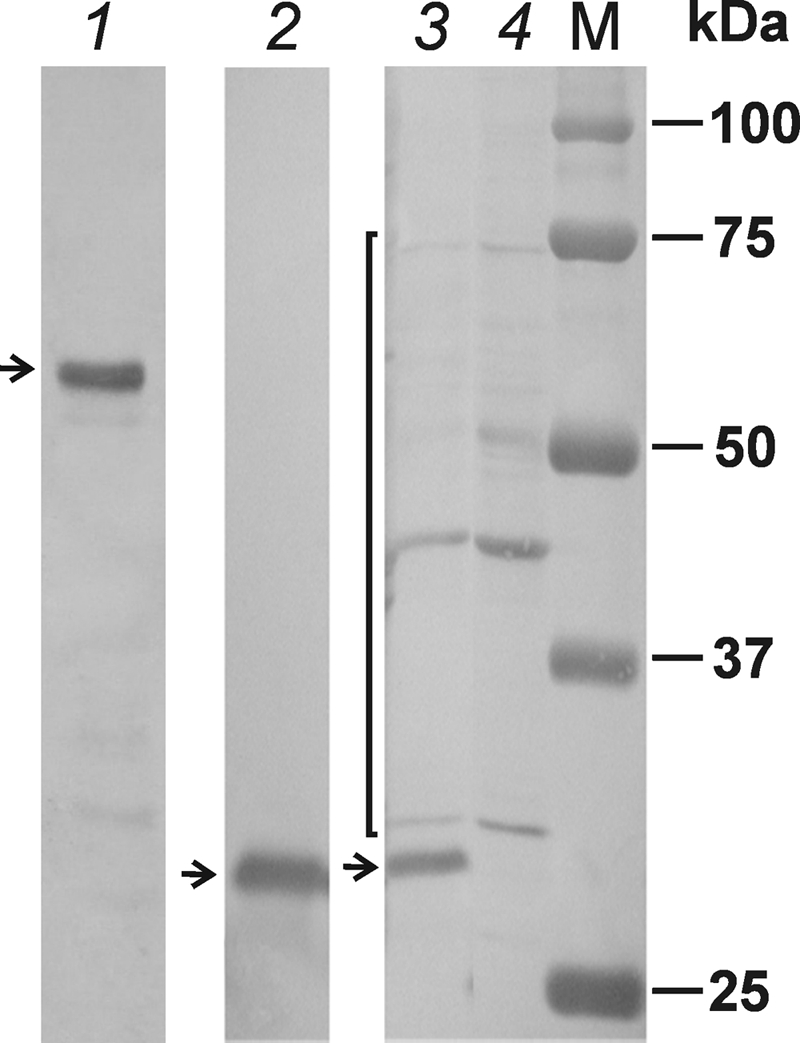
Western blot analysis of BgDNV nonstructural proteins. BGE-2 cells were infected with BgDNV and harvested, and protein extracts were obtained by boiling with SDS-PAGE loading buffer. Samples were separated by 7.5% (for NS1) or 10% (for NS2 and NS3) SDS-PAGE, electroblotted onto PVDF membranes, and probed with the respective antibodies to nonstructural proteins. Lane 1, antibodies to NS1. Lane 2, antibodies to NS2. The ∼60 kDa (NS1) and ∼29 kDa (NS2) bands are indicated by arrows. Lane 3, protein extracts from infected BGE-2 cells probed with antibodies to NS3. Lane 4, protein extracts from control, uninfected cells probed with the same antibodies. The band corresponding to NS3 (∼30 kDa) is indicated by an arrow. Nonspecific band are indicated by a bracket. M, protein molecular mass markers (in kilodaltons).
Antibody to NS3 protein revealed one major band with an approximate size of 31 kDa and several other bands of different sizes (Fig. 5, lane 3). Control experiments with an uninfected cell extract revealed the same “additional” bands but not the 31-kDa protein (Fig. 5, lane 4). We suggest that these bands are the result of nonspecific binding due to the high reactivity of the NS3 antibody. The size of the major protein detected in infected cells exceeds the size of the predicted protein (25.9 kDa), which could be explained by some posttranslational modifications.
A summary of the BgDNV proteins and their relationships to viral transcripts is given in Fig. 2B.
Finally, we did not check the predicted protein (19.3 kDa) potentially encoded by the NSspl2 RNA (hpNStrunc in Fig. 2B).
Intracellular localization of capsid and regulatory proteins.
In order to determine the precise intracellular localization of BgDNV-encoded proteins during the infection of BGE-2 cells, we performed in situ immunofluorescence assays using virus-specific antibodies and FITC-conjugated secondary antibodies to facilitate visualization.
Earlier, we demonstrated that a huge number of BgDNV particles were located in both the nuclei and in cytoplasm of virus-infected BGE-2 cells (37). VP-specific antibodies used in this work revealed clear nuclear localization of the corresponding virus proteins (Fig. 6A). We assume that these antibodies are able to recognize only unbound BgDNV proteins because after capsid formation, specific epitopes could be hidden inside. The fact that viruses assemble in the host cell nucleus explains the accumulation of capsid proteins within this organelle.
Fig. 6.

Confocal microscopy of BgDNV VP1 (A) and NS1 (B) distribution in infected BGE-2 cells. Proteins were visualized using antibodies specified to epitopes predicted for ORF2 (A) and ORF3 (B); the secondary antibodies were conjugated with FITC. Part 1, BGE-2 cells infected with BgDNV; part 2, BGE-2 cell nuclei stained with DAPI; part 3, VP1 (A) and NS1 (B) proteins visualized using the corresponding antibodies; part 4, DAPI and protein-specific staining superimposed in infected BGE-2 cells. The scale is indicated by the bar at the bottom of each image.
Immunofluorescence assays with NS1-specific antibody demonstrated that NS1 is also localized in the nuclei of infected cells (Fig. 6B). This result is in agreement with the functions of NS1 protein during the viral life cycle, which includes regulation of transcription, replication, and viral DNA packaging (5, 9, 21, 29). Our data demonstrate for the first time that the pattern of NS1 expression resembles a halo within the nuclear membrane, similar to those of parvovirus minute virus of mice (MVM) NS2 (10) and densovirus AeDNV NS2 (6). However, the NS1 proteins of all of the other parvoviruses and DNVs described so far are uniformly distributed throughout the nucleus (6, 64). Presumably, association of the BgDNV NS1 protein with the nuclear membrane could be explained by particularities of its unknown functional activity.
Sequence analysis of BgDNV capsid and NS1 proteins using Wolf PSORT (http://www.psort.org/) software revealed two possible nuclear localization signals (NLSs) in the VP1, VP2, and VP3 proteins (PTKKNKP and KKWK) and one NLS in the NS1 protein (RRRKRR, aa 243 to 248). These NLSs can play an important role in the passing of the corresponding proteins through the nuclear membrane.
The absence of positive signals from the newly synthesized VP and NS proteins in the cytoplasm of infected cells could be explained by the low concentration of the proteins in the cytoplasm in comparison with their concentration inside nuclei.
Finally, we were not able to obtain any definite results for intracellular NS2 and NS3 distribution, probably because the corresponding antibodies did not recognize the nondenatured proteins.
DISCUSSION
The main purposes of our present study were to identify the strategy and temporal sequence of BgDNV genome expression, characterize proteins expressed from BgDNV transcripts, and determine their intracellular localization. Our results suggest that some characteristics of the German cockroach densovirus BgDNV resemble those of other viruses in the subfamily Densovirinae, while others are unique and, to our knowledge, have not yet been described.
Organization and expression strategy of the BgDNV genome.
The densovirus of the German cockroach B. germanica belongs to the subfamily Densovirinae and possesses a 5,335-nt-long ambisense genome that contains five basic ORFs. Two of them (ORF1 and ORF2) on one strand code for structural proteins, and three (ORF3 to ORF5) on another strand code for nonstructural proteins (37). Moreover, one additional ORF, ORF6, was found to be located on the same strand as ORF3 and ORF5 but within the VP-coding region. The BgDNV coding sequence is bracketed by ITRs 216 nt (left) and 217 nt (right) long forming simple I-like structures. The overall organization of the BgDNV genome is similar to that of the cricket densovirus AdDNV genome, while the genome sequence is very close to that of another cockroach densovirus, PfDNV (8, 60).
Like other DNVs in the genus Densovirus (GmDNV, MlDNV) (23, 52) and like PfDNV (61), with a genome sequence most similar to that of BgDNV, BgDNV utilizes splicing to express alternative ORFs for nonstructural proteins. The NS3 protein (ORF5) can thus be translated from unspliced mRNA species, while two other nonstructural proteins, NS1 (ORF3) and NS2 (ORF4), are both expressed from spliced transcripts; furthermore, NS2 (ORF4) is translated by an alternative translation initiation mechanism. The latter conjecture is supported by the fact that after splicing, the AUG start codon for ORF3, the first AUG in frame in the transcript, is in a less favorable context (ACGUAGAUGA) than the AUG codon for ORF4 (UGAAUUAUGG) (the −3 and +4 positions are in bold, and the start codon is in bold and underlined). Therefore, the 40S ribosomal subunit can bypass the first AUG codon while scanning and initiate translation from the ORF4 start codon to synthesize the NS2 protein. BgDNV is characterized by a group of additional NS mRNAs transcribed from the promoter for nonstructural proteins but extending beyond the common stop point for NS transcripts into the VP protein-encoding region. To our knowledge, such transcripts have not been previously described for any densovirus. Although additional high-molecular-mass mRNAs were detected by Northern blotting in PstDNV (19), their nature and origin have not been investigated. These additional transcripts may encode a small ORF, ORF6, bioinformatically predicted in the BgDNV genome, which is located in the VP-coding region, but the functional role of ORF6 in BgDNV's life cycle is unclear, and no similarities to any other known proteins have been found. In a number of DNVs, namely, AeDNV (3), PstDNV (45), and PfDNV (25), small ORFs like BgDNV ORF6 located opposite to the genome coding strand were predicted. They may have some functional roles such as, for example, in the case of the Erythrovirus 11-kDa and 7.5-kDa proteins (34, 47, 63), but these roles are still to be elucidated.
It is worth noting that long NS transcripts have no less than 1,600 nt of overlap with mRNAs for VP proteins. This overlap may lead to the formation of a relatively long region of double-stranded RNA (dsRNA) during the virus life cycle, which in turn can induce a cellular antiviral response by means of RNA interference. In this case, RNA interference can impair the transcription of VP genes and therefore the expression of capsid proteins and subsequent capsid formation, which would prevent a productive infection. Involvement of RNA interference in the antiviral response is common and has been demonstrated for plant and animal viruses belonging to different families (49, 54, 58).
Moreover, VP and NS transcripts overlap by 48 nt at their 3′ ends, which may also result in the formation of viral dsRNAs in the host cells and induce a defense response of the infected cell to the viral infection by the RNA interference mechanism.
It is important to note, however, that overlap of RNA sequences is required, but not sufficient, for the induction of RNA interference. The role of RNA interference during BgDNV infection remains unknown.
Another distinctive feature of BgDNV's expression strategy is the presence of alternative splicing in nonstructural mRNAs that leads to the appearance of transcripts encoding only the SF3 helicase domain of the NS1 protein, which is analogous to the adeno-associated virus type 2 rep40 protein; unfortunately, in the present work, we did not have the reagents to detect this protein by Western blotting.
BgDNV possesses two ORFs encoding capsid proteins and utilizes splicing to combine them into one large (85.3 kDa) reading frame that is analogous to the only VP ORF encoded in the genomes of other DNVs. Besides BgDNV, a VP-coding region split into two or three ORFs has been described for a number of other DNVs, such as PfDNV (60), Myzus persicae DNV (MpDNV) (55), Planococcus citri DNV (50), and AdDNV (8). However, the involvement of splicing in the generation of mRNAs for capsid proteins has been described only for PfDNV and MpDNV (55). As in BgDNV, a splicing event leads two PfDNV VP ORFs (ORF1 and ORF2) to connect in frame, generating a new ORF with a similar coding capacity (86 kDa). In the case of PfDNV, as well as BgDNV, an unspliced mRNA is also present, but in contrast to BgDNV, a number of additional splicing donor and acceptor sites that generate mRNAs varying in ORF composition have been described (60).
As demonstrated in our study, the BgDNV capsid is composed of three unique proteins (VP1 to VP3), while the VP2 protein is present in two forms presumably differing in some posttranslational modifications. As a result, four different bands (97, 85, 80, and 57 kDa) corresponding to VP proteins are detected on SDS-PAGE. It makes BgDNV similar to other representatives of the genera Densovirus (52) and Iteravirus (24), which also possess four capsid proteins but with a different molecular mass distribution of VP proteins (i.e., 98, 69, 59, and 49 kDa for Densovirus GmDNV and two doublets with 82 and 74 kDa and 54 and 49 kDa for Iteravirus CeDNV and BmDNV-1). Only for PfDNV (28) have capsid proteins with molecular masses similar to those of BgDNV been found (105, 82, 79, 56, and 52 kDa), and like BgDNV, this virus also possesses two proteins with very similar molecular masses (82 and 79 kDa). It is difficult to further compare the properties of BgDNV capsid proteins with those of PfDNV because their features and functions have not been completely determined.
In the present work, some unique features of BgDNV structural protein production were discovered. Mass spectrometric analysis demonstrated that VP1 and VP3 are most probably translated from the first two AUG codons of a large ORF resulting from the joining of ORF1 and ORF2, as in GmDNV and MlDNV. On the other hand, VP2 is fully encoded by ORF1 and presumably translated from unspliced VP mRNA that so far has not been described for DNVs.
Like the majority of DNVs, BgDNV possesses only one promoter and one termination site for mRNAs for both capsid and nonstructural proteins. BgDNV possesses two groups of genes, early (NS) and late (VP). This situation is apparently characteristic of parvoviruses and was described for mammalian parvoviruses, for example, MVM (17) and porcine parvovirus (PPV) (35), as well as for GmDNV (48). This temporal order of transcription is in agreement with the functions of the corresponding proteins in the parvoviral life cycle (8, 9). Regulatory proteins, particularly NS1 and NS3 (1), are necessary at early stages of infection. NS1 acts as the initiator protein of viral DNA replication that leads to the accumulation of replicative forms serving as templates for further transcription and synthesis of viral genomic ssDNA. NS1 is also capable of regulating and transactivating the VP promoter (21, 33, 41). VP proteins make up the viral capsid, and apparently VP transcription and capsid formation begin along with the accumulation of replicative forms of the viral genome. Our data on DNA temporal dynamics (Fig. 1) demonstrate that a considerable amount of an additional 5-kb DNA corresponding to the product of annealing of plus and minus strands of the BgDNV genome appears clearly only 20 days p.i., which is later than the beginning of VP transcription. This observation is consistent with the fact that viral ssDNA formation and encapsulation occurs only in the presence of partially or fully formed capsids (18).
Ubiquitination.
Ubiquitination of the VP2 protein is another prominent feature of BgDNV previously undescribed for any DNVs. Ubiquitination could serve to facilitate virus entry into the cell by assisting endocytosis in some way opposite to retrovirus egress or serve as some sort of viral ligand for cellular recognition factors. Or, as suggested by Hingamp et al. (27), it could influence virus entry as a primer for the formation of a polyubiquitin chain that could target now multiubiquitinated proteins for degradation, thus participating in virus uncoating. Or ubiquitination may play some roles in viral capsid assembly during late phases of infection and virus exit from the cell. The latter presumed mechanism is partly supported by results of coimmunoprecipitation experiments showing that PPV capsid proteins were ubiquitinated from early-stage infection up to 12 h p.i. It was also demonstrated that proteosome activity was important for a virus-producing infection (11). Furthermore, proteosome function after endosomal escape was required for MVM intranuclear penetration, as proteosome inhibition led to viral perinuclear accumulation and arrest of replication and capsid disassembly. The same phenomenon was shown for canine parvovirus, but no ubiquitination of viral capsid proteins was detected in these cases (42).
It was recently shown that protein VI, an internal capsid protein of the adenoviruses, is rapidly exposed after cell surface attachment and internalization and remains partially associated with the capsid during intracellular transport. The PPxY motif within protein VI recruits Nedd4 E3 ubiquitin ligases to bind and ubiquitylate protein VI. This PPxY motif is involved in rapid, microtubule-dependent intracellular movement of protein VI. Adenoviruses with a mutated PPxY motif can efficiently escape endosomes but are defective in microtubule-dependent trafficking toward the nucleus. Likewise, depletion of Nedd4 ligases attenuates the nuclear accumulation of incoming virus particles and infection (59). This was the first evidence that virus-encoded PPxY motifs are required during virus entry, which may be of significance for several other pathogens. Interestingly, BgDNV VP2 protein contains a PPxY motif (PPPY, aa 8 to 11) at its N terminus, which may also function during entry and interact with novel cellular pathways for efficient viral transduction.
Ubiquitination may also play a role in the host cell defense against BgDNV infection (16). However, monoubiquitination does not usually lead to proteosomal degradation, as does polyubiquitination, but rather plays a role in regulation of protein activity, structure, or location (e.g., directing endocytosis and sorting of transmembrane proteins), regulation of the functions of proteins involved in endosome pathway, and histone modification (26, 36, 38). Such monoubiquitination could play a primer role for the host antiviral defense system. Upon BgDNV entry into cells, additional ubiquitin molecules could be attached to produce polyubiquitin chains and direct the respective proteins to a degradation pathway.
All said, the functional role of ubiquitin conjugation of DNV proteins, as described for the first time in this paper, remains unknown.
In conclusion, the expression strategy of the BgDNV genome combines features characteristic of representatives of the genera Densovirus and Pefudensovirus. As shown by comparison of sequences of corresponding ORFs, nonstructural proteins show maximal sequence similarity to those of PfDNV, while capsid proteins demonstrate maximum similarity to structural proteins of other members of the genus Densovirus.
Supplementary Material
ACKNOWLEDGMENTS
We thank Timothy J. Kurtti, who kindly provided the B. germanica cell culture. We thank Peter Tijssen for critical review of an earlier draft of the manuscript and three anonymous reviewers for their constructive comments.
This project was supported in part by the Presidium of Russian Academy of Sciences grant Biodiversity (subprogram Gene Pools and Biodiversity) and Foundation for Basic Research grant 11-04-01776-a to D.V.M., the National Institute of Food and Agriculture, U.S. Department of Agriculture (award 2009-35302-05303 to C.S.), and the Blanton J. Whitmire endowment at North Carolina State University.
Footnotes
Supplemental material for this article may be found at http://jvi.asm.org/.
Published ahead of print on 7 September 2011.
REFERENCES
- 1. Abd-Alla A., et al. 2004. NS-3 protein of the Junonia coenia densovirus is essential for viral DNA replication in an Ld 652 cell line and Spodoptera littoralis larvae. J. Virol. 78: 790–797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Afanasiev B., Carlson J. 2000. Densovirinae as gene transfer vehicles, p. 33–58 In Faisst S., Rommelaere J. (ed.), Parvoviruses: from molecular biology to pathology and therapeutic uses. Karger, Basel, Switzerland: [DOI] [PubMed] [Google Scholar]
- 3. Afanasiev B. N., Galyov E. E., Buchatsky L. P., Kozlov Y. V. 1991. Nucleotide sequence and genomic organization of Aedes densonucleosis virus. Virology 185: 323–336 [DOI] [PubMed] [Google Scholar]
- 4. Armentrout R., et al. 1978. A standardized nomenclature for restriction endonuclease fragments, p. 523–526 In Ward D., Tattersall P. (ed.), Replication of mammalian parvoviruses. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY [Google Scholar]
- 5. Astell C. R., et al. 1996. Minute virus of mice cis-acting sequences required for genome replication and the role of the trans-acting viral protein, NS-1. Prog. Nucleic Acid Res. Mol. Biol. 55: 245–285 [DOI] [PubMed] [Google Scholar]
- 6. Azarkh E., et al. 2008. Mosquito densonucleosis virus non-structural protein NS2 is necessary for a productive infection. Virology 374: 128–137 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Baquerizo-Audiot E., et al. 2009. Structure and expression strategy of the genome of Culex pipiens densovirus, a mosquito densovirus with an ambisense organization. J. Virol. 83: 6863–6873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bergoin M., Tijssen P. 2000. Molecular biology of Densovirinae, p. 1–11 In Faisst S., Rommelaere J.(ed.), Parvoviruses: from molecular biology to pathology and therapeutic uses. Karger, Basel, Switzerland [Google Scholar]
- 9. Berns K. I. 1990. Parvovirus replication. Microbiol. Rev. 54: 316–329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bodendorf U., Cziepluch C., Jauniaux J. C., Rommelaere J., Salome N. 1999. Nuclear export factor CRM1 interacts with nonstructural proteins NS2 from parvovirus minute virus of mice. J. Virol. 73: 7769–7779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Boisvert M., Fernandes S., Tijssen P. 2010. Multiple pathways involved in porcine parvovirus cellular entry and trafficking toward the nucleus. J. Virol. 84: 7782–7792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bossin H., et al. 2003. Junonia coenia densovirus-based vectors for stable transgene expression in Sf9 cells: influence of the densovirus sequences on genomic integration. J. Virol. 77: 11060–11071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boublik Y., Jousset F. X., Bergoin M. 1994. Complete nucleotide sequence and genomic organization of the Aedes albopictus parvovirus (AaPV) pathogenic for Aedes aegypti larvae. Virology 200: 752–763 [DOI] [PubMed] [Google Scholar]
- 14. Carlson J., Suchman E., Buchatsky L. 2006. Densoviruses for control and genetic manipulation of mosquitoes. Adv. Virus Res. 68: 361–392 [DOI] [PubMed] [Google Scholar]
- 15. Chen K. C., et al. 1986. Complete nucleotide sequence and genome organization of bovine parvovirus. J. Virol. 60: 1085–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Ciechanover A. 1998. The ubiquitin-proteasome pathway: on protein death and cell life. EMBO J. 17: 7151–7160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Clemens K. E., Pintel D. 1987. Minute virus of mice (MVM) messenger-RNAs predominately polyadenylate at a single site. Virology 160: 511–514 [DOI] [PubMed] [Google Scholar]
- 18. Cotmore S. F., Tattersall P. 1996. Parvovirus DNA replication, p. 799–813 In DePamphilis M.(ed.), DNA replication in eukaryotic cells. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 19. Dhar A. K., Lakshman D. K., Natarajan S., Allnutt F. C. T., van Beek N. A. M. 2007. Functional characterization of putative promoter elements from infectious hypodermal and hematopoietic necrosis virus (IHHNV) in shrimp and in insect and fish cell lines. Virus Res. 127: 1–8 [DOI] [PubMed] [Google Scholar]
- 20. Ding C., Urabe M., Bergoin M., Kotin R. M. 2002. Biochemical characterization of Junonia coenia densovirus nonstructural protein NS-1. J. Virol. 76: 338–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Doerig C., Hirt B., Beard P., Antonietti J. P. 1988. Minute virus of mice non-structural protein NS-1 is necessary and sufficient for trans-activation of the viral p39-promoter. J. Gen. Virol. 69: 2563–2573 [DOI] [PubMed] [Google Scholar]
- 22. Dumas B., Jourdan M., Pascaud A. M., Bergoin M. 1992. Complete nucleotide sequence of the cloned infectious genome of Junonia coenia densovirus reveals on organization unique among parvoviruses. Virology 191: 202–222 [DOI] [PubMed] [Google Scholar]
- 23. Fédière G., El-Far M., Li Y., Bergoin M., Tijssen P. 2004. Expression strategy of densonucleosis virus from Mythimna loreyi. Virology 320: 181–189 [DOI] [PubMed] [Google Scholar]
- 24. Fédière G., Li Y., Zadori Z., Szelei J., Tijssen P. 2002. Genome organization of Casphalia extranea densovirus, a new iteravirus. Virology 292: 299–308 [DOI] [PubMed] [Google Scholar]
- 25. Guo H., Zhang J., Hu Y. 2000. Complete sequence and organization of Periplaneta fuliginosa densovirus genome. Acta Virol. 44: 315–322 [PubMed] [Google Scholar]
- 26. Hicke L. 2001. Protein regulation by monoubiquitin. Nat. Rev. Mol. Cell Biol. 2: 195–201 [DOI] [PubMed] [Google Scholar]
- 27. Hingamp P. M., et al. 1995. Characterization of a ubiquitinated protein which is externally located in African swine fever virions. J. Virol. 69: 1785–1793 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hu Y., Zheng J., Iizuka T., Bando H. 1994. A densovirus newly isolated from the smoky-brown cockroach Periplaneta fuliginosa. Arch. Virol. 138: 365–372 [DOI] [PubMed] [Google Scholar]
- 29. Ihalainen T. O., et al. 2007. Dynamics and interactions of parvoviral NS1 protein in the nucleus. Cell. Microbiol. 9: 1946–1959 [DOI] [PubMed] [Google Scholar]
- 30. Kozak M. 1995. Adherence to the first-AUG rule when a second AUG codon follows closely upon the first. Proc. Natl. Acad. Sci. U. S. A. 92: 2662–2666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Laemmli U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227: 680–685 [DOI] [PubMed] [Google Scholar]
- 32. Li Y., et al. 2001. Genome organization of the densovirus from Bombyx mori (BmDNV-1) and enzyme activity of its capsid. J. Gen. Virol. 82: 2821–2825 [DOI] [PubMed] [Google Scholar]
- 33. Lorson C., Burger L. R., Mouw M., Pintel D. J. 1996. Efficient transactivation of the minute virus of mice P38 promoter requires upstream binding of NS1. J. Virol. 70: 834–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Luo W. X., Astell C. R. 1993. A novel protein encoded by small RNAs of parvovirus B19. Virology 195: 448–455 [DOI] [PubMed] [Google Scholar]
- 35. Molitor T. W., Joo H. S., Collett M. S. 1985. Identification and characterization of a porcine parvovirus nonstructural polypeptide. J. Virol. 55: 554–559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Mosesson Y., Yarden Y. 2006. Monoubiquitylation: a recurrent theme in membrane protein transport. Isr. Med. Assoc. J. 8: 233–237 [PubMed] [Google Scholar]
- 37. Mukha D. V., Chumachenko A. G., Dykstra M. J., Kurtti T. J., Schal C. 2006. Characterization of a new densovirus infecting the German cockroach, Blattella germanica. J. Gen. Virol. 87: 1567–1575 [DOI] [PubMed] [Google Scholar]
- 38. Mukhopadhyay D., Riezman H. 2007. Proteasome-independent functions of ubiquitin in endocytosis and signaling. Science 315: 201–205 [DOI] [PubMed] [Google Scholar]
- 39. Munderloh U. G., Kurtti T. J. 1989. Formulation of medium for tick cell culture. Exp. Appl. Acarol. 7: 219–229 [DOI] [PubMed] [Google Scholar]
- 40. Ren X. X., Hoiczyk E., Rasgon J. L. 2008. Viral paratransgenesis in the malaria vector Anopheles gambiae. PLoS Pathog. 4(8): e1000135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Rhode S. L. 1985. trans-activation of parvovirus p38 promoter by 76K noncapsid protein. J. Virol. 55: 886–889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Ros C., Kempf C. 2004. The ubiquitin-proteasome machinery is essential for nuclear translocation of incoming minute virus of mice. Virology 324: 350–360 [DOI] [PubMed] [Google Scholar]
- 43. Sambrook J., Russell D. W. 2001. Molecular cloning: a laboratory manual, 3rd ed Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY [Google Scholar]
- 44. Sanger F., Nicklen S., Coulson A. R. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74: 5463–5467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shike H., et al. 2000. Infectious hypodermal and hematopoietic necrosis virus of shrimp is related to mosquito brevidensoviruses. Virology 277: 167–177 [DOI] [PubMed] [Google Scholar]
- 46. Shirk P. D., Bossin H., Furlong R. B., Gillett J. L. 2007. Regulation of Junonia coenia densovirus P9 promoter expression. Insect Mol. Biol. 16: 623–633 [DOI] [PubMed] [Google Scholar]
- 47. St. Amand J., Astell C. R. 1993. Identification and characterization of a family of 11-kDa proteins encoded by the human parvovirus B19. Virology 192: 121–131 [DOI] [PubMed] [Google Scholar]
- 48. Tal J., Attathom T. 1993. Insecticidal potential of the insect parvovirus GmDNV. Arch. Insect Biochem. Physiol. 22: 345–356 [Google Scholar]
- 49. Tattersall P., et al. 2006. Family Parvoviridae, p. 353–369 In Faquet C. M., Mayo M. A., Maniloff J., Desselberger U., Ball L. A.(ed.), Virus taxonomy: classification and nomenclature of viruses. Eighth report of the International Committee on the Taxonomy of Viruses Elsevier Academic Press, London, United Kingdom [Google Scholar]
- 50. Thao M. L., Wineriter S., Buckingham G., Baumann P. 2001. Genetic characterization of a putative densovirus from the mealybug Planococcus citri. Curr. Microbiol. 43: 457–458 [DOI] [PubMed] [Google Scholar]
- 51. Tijssen P., Bergoin M. 1995. Densonucleosis viruses constitute an increasingly diversified subfamily among the parvoviruses. Semin. Virol. 6: 347–355 [Google Scholar]
- 52. Tijssen P., et al. 2003. Organization and expression strategy of the ambisense genome of densonucleosis virus of Galleria mellonella. J. Virol. 77: 10357–10365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Tijssen P., Tijssenvanderslikke T., Kurstak E. 1977. Biochemical, biophysical, and biological properties of densonucleosis virus (Parvovirus). II. Two types of infectious virions. J. Virol. 21: 225–231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vanitharani R., Chellappan P., Fauquet C. M. 2005. Geminiviruses and RNA silencing. Trends Plant Sci. 10: 144–151 [DOI] [PubMed] [Google Scholar]
- 55. van Munster M., et al. 2003. A new virus infecting Myzus persicae has a genome organization similar to the species of the genus Densovirus. J. Gen. Virol. 84: 165–172 [DOI] [PubMed] [Google Scholar]
- 56. Wang J. P., et al. 2005. Nucleotide sequence and genomic organization of a newly isolated densovirus infecting Dendrolimus punctatus. J. Gen. Virol. 86: 2169–2173 [DOI] [PubMed] [Google Scholar]
- 57. Ward T. W., Kimmick M. W., Afanasiev B. N., Carlson J. O. 2001. Characterization of the structural gene promoter of Aedes aegypti densovirus. J. Virol. 75: 1325–1331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wilkins C., et al. 2005. RNA interference is an antiviral defence mechanism in Caenorhabditis elegans. Nature 436: 1044–1047 [DOI] [PubMed] [Google Scholar]
- 59. Wodrich H., et al. 2010. A capsid-encoded PPxY-motif facilitates adenovirus entry. PLoS Pathog. 6(3): e1000808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Yamagishi J., Hu Y., Zheng J., Bando H. 1999. Genome organization and mRNA structure of Periplaneta fuliginosa densovirus imply alternative splicing involvement in viral gene expression. Arch. Virol. 144: 2111–2124 [DOI] [PubMed] [Google Scholar]
- 61. Yang B., et al. 2008. Characterization of the promoter elements and transcription profile of Periplaneta fuliginosa densovirus nonstructural genes. Virus Res. 133: 149–156 [DOI] [PubMed] [Google Scholar]
- 62. Yang B., et al. 2006. Biochemical characterization of Periplaneta fuliginosa densovirus non-structural protein NS1. Biochem. Biophys. Res. Commun. 342: 1188–1196 [DOI] [PubMed] [Google Scholar]
- 63. Zhi N., et al. 2006. Molecular and functional analyses of a human parvovirus B19 infectious clone demonstrates essential roles for NS1, VP1, and the 11-kilodalton protein in virus replication and infectivity. J. Virol. 80: 5941–5950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zhou W. D., Zhang J. M., Yang B., Zhou L., Hu Y. Y. 2009. The nuclear localization signal of the NS1 protein is essential for Periplaneta fuliginosa densovirus infection. Virus Res. 145: 134–140 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.