

Abstract
Protein tyrosine phosphatase (PTP)-PEST is a critical regulator of cell adhesion and migration. However, the mechanism by which PTP-PEST is regulated in response to oncogenic signaling to dephosphorylate its substrates remains unclear. Here, we demonstrate that activated Ras induces extracellular signal-regulated kinase 1 and 2-dependent phosphorylation of PTP-PEST at S571, which recruits PIN1 to bind to PTP-PEST. Isomerization of the phosphorylated PTP-PEST by PIN1 increases the interaction between PTP-PEST and FAK, which leads to the dephosphorylation of FAK Y397 and the promotion of migration, invasion, and metastasis of v-H-Ras-transformed cells. These findings uncover an important mechanism for the regulation of PTP-PEST in activated Ras-induced tumor progression.
INTRODUCTION
Most cancer deaths are due to tumor invasion and metastasis rather than to primary tumors. Thus, understanding the fundamental mechanisms underlying tumor cell migration, invasion, and metastasis holds the promise of improving therapeutic approaches for cancer (7). Activating mutations of Ras, a small GTP-binding protein, occur in ∼30% of human cancers and promote tumor progression (32). Accordingly, considerable effort has been expended in understanding the functions of Ras proteins in tumor cell migration, invasion, and metastasis (6).
Cell migration is a highly coordinated and dynamic multistep process of leading-edge protrusion, focal adhesion turnover, tractional force generation, and tail retraction and detachment, all of which involve precise regulation of cell-cell adhesion and cell-to-extracellular matrix adhesion. Focal adhesions are specific regions of cells that make close contact with the extracellular matrix (30, 44). Functional regulation of the molecules involved in focal adhesion signaling is a key component of tumor cell motility.
Focal adhesion kinase (FAK) is a ubiquitously expressed nonreceptor protein tyrosine kinase that localizes at focal adhesions, mediates signaling induced by integrins, and plays an important role in many cellular functions (26, 27). Activated FAK, marked by autophosphorylation at Y397, recruits a number of SH2 and SH3 domain-containing proteins, including c-Src. The binding of c-Src to FAK is proposed to disrupt the intramolecular interaction between the c-Src SH2 domain and the negative regulatory carboxy-terminal Y529. Activated Src, in turn, phosphorylates FAK and further enhances FAK activity, thereby forming a positive feedback loop leading to the activation of downstream signaling molecules, such as extracellular signal-regulated kinase 1 and 2 (ERK1/2) and phosphatidylinositol 3-kinase (PI3-K)/AKT (17, 29).
FAK can exert control over the rate of focal adhesion turnover and, therefore, cell motility (29). FAK is a positive regulator of normal cell migration, and its overexpression or activation has been shown to promote cancer cell metastasis of some tumor types (17, 29). However, accumulated evidence shows that in response to certain oncogenic signals, FAK negatively regulates cancer cell migration and invasion (4, 5, 9, 15, 16, 21, 24, 25, 31, 37, 38–40). We previously reported that the activation of epidermal growth factor receptor (EGFR) and Ras promotes cancer cell migration by dephosphorylation of FAK Y397. This dephosphorylation may facilitate focal adhesion turnover at the leading edge of cells, which contributes to the promotion of cell invasion and metastasis (21, 44). FAK Y397 dephosphorylation requires ERK1/2-mediated phosphorylation of FAK S910, which recruits protein interacting with NIMA (never in mitosis A)-1 (PIN1). PIN1, a member of the parvulin subfamily of peptidyl-prolyl cis/trans-isomerases (PPIases), specifically recognizes phosphorylated serine (S) or threonine (T) in pS/TP peptide sequences (20). PIN1 catalyzes the cis/trans isomerization of the peptidyl-prolyl peptide bond at FAK S910/P911, which is required for FAK dephosphorylation at Y397 (45).
Protein tyrosine phosphatase (PTP)-PEST is a ubiquitously expressed cytosolic PTP that is named for its proline-glutamine-serine-threonine-rich motifs (PEST sequences) (19, 35, 41, 42). The tightly regulated activity of PTP-PEST plays instrumental roles in focal adhesion breakdown, cell spreading, and cell motility (1, 14, 19). PTP-PEST dephosphorylates several protein kinases and signaling molecules involved in cell migration and transformation, including FAK, p130CAS (a FAK substrate), c-Src, EGFR, leupaxin, growth factor receptor-bound protein 2 (GRB2), and the p66SHC isoform of SHC-transforming protein 1 (SHC1) (19, 37). In response to Ras activation, PTP-PEST binds to FAK and dephosphorylates it at Y397, thereby reducing the total number of focal contacts of transformed cells and promoting migration of tumor cells (2, 44, 45). However, the mechanisms underlying activated Ras-regulated PTP-PEST dephosphorylation of FAK remain elusive.
In this report, we show that Ras-induced PTP-PEST phosphorylation at S571, which is mediated by ERK1/2, creates an interacting motif for PIN1 binding. Isomerized PTP-PEST then interacts with and dephosphorylates FAK at Y397, leading to increased tumor cell migration, invasion, and metastasis.
MATERIALS AND METHODS
Cells and cell culture conditions.
FAK+/+, FAK−/−, PTP-PEST+/+, and PTP-PEST−/− fibroblasts, U251 glioblastoma cells, and 293T cells were all maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% bovine calf serum (HyClone, Logan, UT).
Materials.
Monoclonal antibodies against phospho-ERK (sc-7383), glutathione S-transferase (GST) (sc-138), and hemagglutinin (HA) (sc-7392) and polyclonal antibodies against mitogen-activated protein kinase kinase 1 (MEK1) (sc-219), ERK2 (sc-154), ERK1 (sc-94), and FAK (sc-558) were from Santa Cruz Biotechnology (Santa Cruz, CA). Monoclonal antibodies against FLAG and tubulin were from Sigma (St. Louis, MO). Monoclonal antibodies against H-Ras and phospho-FAK Y397 and Matrigel were from BD Biosciences (San Diego, CA). The PTP-PEST rabbit polyclonal antibody was from Bethyl Laboratories (Montgomery, TX). Purified active ERK2 protein (P6080) was from New England BioLabs (Ipswich, MA). Purified active GST-ERK5 protein (04-146) was from Carna Biosciences (Kobe, Japan). U0126 was from EMD Biosciences (San Diego, CA). [γ-32P]ATP was from MP Biomedicals (Solon, OH).
DNA constructs and mutagenesis.
A PCR-amplified human PTP-PEST cDNA was cloned into the pcDNA3.1-FLAG vector between the BamHI and NotI restriction enzyme sites. The FLAG-PTP-PEST S571A mutant was generated using the QuikChange site-directed mutagenesis kit (Stratagene, La Jolla, CA).
Transfection.
Cells were plated at a density of 4 × 105 per 60-mm-diameter dish at 18 h prior to transfection. Transfection was performed using HyFect reagents (Denville Scientific, Metuchen, NJ) according to the vendor's instructions. The transfection efficiency for 293T cells was about 90%. Stable cell lines were selected with hygromycin (100 μg/ml) for 10 to 14 days at 37°C. After treatment with hygromycin, antibiotic-resistant colonies (more than 100 colonies) were pooled and expanded for further analysis under selective conditions.
Immunoprecipitation and immunoblotting analysis.
Extraction of proteins from cultured cells using a modified lysis buffer was followed by immunoprecipitation and immunoblotting with corresponding antibodies, as described previously (22).
Cell migration assays.
To assess cell migration, we conducted a wound-healing assay. Cells were plated at 70% confluence in DMEM supplemented with 10% fetal bovine serum. At 24 h after seeding, the monolayers were wounded by scoring with a sterile plastic 200-μl micropipette tip, washed, and then incubated in DMEM. After 24 h, cells were photographed using a low-magnification phase-contrast microscope as described previously (21).
GST pulldown assays.
Cell lysates (250 μg/sample) were incubated with 100 ng of GST or GST fusion proteins together with glutathione-agarose beads in a modified buffer consisting of 50 mM Tris-HCl (pH 7.5), 1% Triton X-100, 150 mM NaCl, 1 mM 1,4-dithiothreitol, 0.5 mM EDTA, 0.1 mM phenylmethylsulfonyl fluoride, 12 μg/ml leupeptin, 20 μg/ml aprotinin, 100 μM sodium vanadate, 100 μM sodium pyrophosphate, and 1 mM sodium fluoride at 4°C. The glutathione beads were then washed four times with lysis buffer and the bound proteins eluted with sodium dodecyl sulfate (SDS) sample buffer prior to SDS-polyacrylamide gel electrophoresis (PAGE).
Purification of recombinant proteins.
GST-PIN1 and the GST-PIN1 WW mutant were expressed in BL21(DE3) cells following induction with 1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) for 4 h at 37°C. Protein was purified from bacterial extracts by incubation with glutathione-Sepharose beads followed by extensive washing with 20 mM Tris (pH 7.4), 150 mM NaCl, and 1 mM dithiothreitol. The GST fusion proteins were eluted with reduced glutathione (10 mM) in 50 mM Tris (pH 7.4). The eluted proteins were dialyzed in phosphate-buffered saline (PBS) for 16 h (23).
His-PTP-PEST in the pCold I vector was expressed in BL21(DE3) cells following induction with 1 mM IPTG for 24 h at 16°C. His-PTP-PEST protein was purified from bacterial cell lysate using Ni-nitrilotriacetic acid (NTA) His-binding resin and eluted with 1 M imidazole. The eluted proteins were dialyzed in PBS for 16 h.
In vitro kinase assays.
FLAG-PTP-PEST was transiently expressed in 293T cells and immunoprecipitated with an anti-FLAG antibody. The kinase reactions were done by mixing FLAG-PTP-PEST protein with or without purified ERK2 or ERK5 in kinase assay buffer containing 10 μCi of [γ-32P]ATP, 10 mM Tris-HCl (pH 7.4), 5 mM MnCl2, 1 mM dithiothreitol, and 20 μM ATP for 30 min at 37°C. Reactions were stopped by adding an equal volume of 2× SDS-PAGE sample buffer and boiling for 5 min. Samples were then separated by 8% SDS-PAGE and transferred onto nitrocellulose membranes for exposure to X-ray film (11).
In vitro invasion assays.
Cell invasion was assessed by the invasion of the cells through Matrigel-coated Transwell inserts. Briefly, Transwell inserts with an 8-μm pore size were coated with 100 μl of a final concentration of 1 mg/ml Matrigel in cold serum-free medium. Cells were trypsinized, and cell suspension (500 μl; 1 × 106 cells/ml) was added in triplicate to wells. After a 24-h incubation, cells that invaded the Matrigel and passed through the filter were stained with crystal violet and photographed using a digital camera mounted onto a microscope with a magnification of ×50. The membranes were dissolved in 4% deoxycholic acid and read colorimetrically at 590 nm as previously described (11, 45).
FDP protein phosphatase assays.
The control vector and the vector expressing FLAG-PTP-PEST or the FLAG-PTP-PEST S571A mutant were separately transfected into 293T cells. Cell lysates were prepared in 3-[(3-cholamidopropyl) dimethylammonio]-1-propanesulfonate (CHAPS) buffer consisting of 10 mM HEPES (pH 7.5), 1% CHAPS, 150 mM NaCl, 1 mM dithiothreitol, 0.5 mM EDTA, 0.1 mM phenylmethylsulfonyl fluoride, 12 μg/ml leupeptin, and 20 μg/ml aprotinin. FLAG-PTP-PEST was immunoprecipitated using the FLAG antibody, and its activity toward 3,6-fluorescein diphosphate (FDP) was examined using an Enzolyte FDP protein phosphatase assay kit (AnaSpec Co., Fremont, CA). The fluorescein signal was read using a fluorescence plate reader at excitation and emission wavelengths of 485 and 528 nm, respectively (45).
Pulse-chase analysis.
U251 cells with or without depletion of PIN1 were treated with cycloheximide (CHX) (100 μg/ml). The cells were harvested and analyzed at various time points.
Trypsin digestion of PTP-PEST.
FLAG-PTP-PEST immunoprecipitated from v-H-Ras-expressing PIN1−/− cells was incubated with 10 pM purified GST, wild-type (WT) GST-PIN1, or the GST-PIN1 C113A mutant and was then digested with trypsin (5 ng) for 5 min. The digested fragments were separated by 10% SDS-PAGE, followed by immunoblotting analysis with a PTP-PEST antibody.
Mass spectrometry analysis.
The GelCode Blue (Thermo Fisher Scientific, Waltham, MA)-stained gel band was subjected to in-gel digestion with 200 ng of modified trypsin (sequencing grade) (Promega, Madison, WI) at 37°C for 24 h. The resultant peptides were analyzed by nano-liquid chromatography-coupled ion trap mass spectrometry with on-line desalting on a system consisting of a FAMOS autosampler, an Ultimate Nano liquid chromatography module, and a Switchos precolumn switching device on a 75-μm by 150-mm C18 column (all from Dionex Corp., Sunnyvale, CA). Electrospray ion trap mass spectrometry was performed on an LTQ linear ion trap mass spectrometer (Thermo Fisher Scientific). Proteins were identified by a database search of the fragment spectra against the National Center for Biotechnology Information nonredundant protein database (NCBInr) using Mascot (Matrix Science, London, United Kingdom) and Sequest (Thermo Fisher Scientific). Phosphopeptide matches were manually curated.
Subcutaneous tumor growth and lung metastasis assays.
PTP-PEST+/+ or PTP-PEST−/− v-H-Ras-expressing cells with reconstituted expression of WT PTP-PEST or PTP-PEST S571A were subcutaneously injected into athymic nude mice (5 × 106 cells in 0.3 ml PBS per mouse). Three weeks later, the tumor volumes (V) were estimated by measuring two dimensions (length [a] and width [b]) and calculated using the equation V = ab2/2. For the metastasis assay, these cells were injected into the lateral tail veins of athymic nude mice (1 × 105 cells in 0.1 ml PBS per mouse). After 3 weeks, the mice were sacrificed, and lung tissues were excised and fixed with Bouin's solution. Lung surface tumors were counted under a low-power dissecting microscope. Six mice in each group were used, and the standard errors were calculated as the variation in tumor number. Differences were evaluated for statistical significance at a P value of <0.05 using the two-tailed Student t test. Experiments were done twice.
RESULTS
PTP-PEST and FAK bind to PIN1 in a mutually independent manner.
We previously showed that upon Ras activation, PIN1 binds to the complex of FAK and PTP-PEST and is required for the interaction between PTP-PEST and FAK (45). Although FAK is a direct substrate of PIN1 (45), we could not exclude the possibility that PIN1 regulates PTP-PEST simultaneously to promote the binding of PTP-PEST to FAK. To examine whether activated Ras promotes the binding of PIN1 to PTP-PEST independently of FAK, we infected FAK+/+ and FAK−/− cells with a v-H-Ras-expressing adenovirus (Fig. 1A) and performed a GST-PIN1 pulldown assay. As shown in Fig. 1B, the level of PTP-PEST associated with PIN1 was moderate in the absence of v-H-Ras but was significantly enhanced by v-H-Ras expression in both FAK+/+ and FAK−/− cells; the levels of enhanced expression were comparable. The GST-PIN1 WW domain mutant, which contains W11A and W34A mutations in the binding domain, was incapable of binding to PTP-PEST (Fig. 1B). Immunoblotting analysis of the immunoprecipitated PIN1 from FAK−/− cells with an anti-PTP-PEST antibody showed that expression of v-H-Ras resulted in an increased association of endogenous PIN1 with endogenous PTP-PEST (Fig. 1C). These results indicate that Ras activation promotes the interaction between the PIN1 WW domain and PTP-PEST in a FAK-independent manner. Similarly, expression of v-H-Ras (Fig. 1D) induced the interaction between GST-PIN1 and FAK in PTP-PEST−/− cells to a level comparable to that in PTP-PEST+/+ cells (Fig. 1E). These results indicate that both PTP-PEST and FAK can bind to PIN1 independently.
Fig. 1.

PTP-PEST and FAK bind to PIN1 in a mutually independent manner. Western blotting (WB) and immunoprecipitation (IP) analyses were performed with the indicated antibodies. Tubulin was used as a loading control. (A) FAK+/+ and FAK−/− cells were infected with v-H-Ras-expressing adenovirus for 12 h. Cell lysates were prepared from the indicated cells. (B) GST pulldown assays were performed by incubating GST-, GST-PIN1-, or GST-PIN1 WW mutant-bound agarose beads with lysates of FAK+/+ and FAK−/− cells infected with or without v-H-Ras-expressing adenovirus for 12 h. (C) FAK−/− cells were infected with or without v-H-Ras-expressing adenovirus for 12 h. PIN1 was immunoprecipitated with a specific antibody. (D) PTP-PEST+/+ and PTP-PEST−/− cells were infected with or without v-H-Ras-expressing adenovirus for 12 h. Cell lysates were prepared from the indicated cells. (E) GST pulldown assays were performed by incubating GST-, GST-PIN1-, or GST-PIN1 WW mutant-bound agarose beads with lysates of PTP-PEST+/+ or PTP-PEST−/− cells infected with or without v-H-Ras-expressing adenovirus for 12 h.
cis-trans isomerization of PTP-PEST by PIN1 is required for the interaction between PTP-PEST and FAK and subsequent FAK dephosphorylation.
Depletion of PIN1 from U251 glioblastoma cells (Fig. 2A) largely inhibited v-H-Ras-induced FAK dephosphorylation (Fig. 2B), which was in line with our previous findings that PIN1 is required for v-H-Ras-regulated FAK phosphorylation (45). A pulse-chase experiment showed that the presence or absence of v-H-Ras or PIN1 did not significantly change the expression or half-life of PTP-PEST (Fig. 2C), suggesting that PTP-PEST-dependent FAK dephosphorylation is not due to regulation of the PTP-PEST expression level. Incubation of FAK immunoprecipitated from v-H-Ras-expressing PTP-PEST−/− cells with FLAG-PTP-PEST immunoprecipitated from v-H-Ras-expressing PIN1−/− cells did not result in increased association of FAK with PTP-PEST or FAK dephosphorylation (Fig. 2D). However, preincubation of purified WT PIN1, but not of an inactive PIN1 C113A mutant, with immunoprecipitated FLAG-PTP-PEST enabled PTP-PEST to bind to and dephosphorylate FAK (Fig. 2D). An in vitro phosphatase assay showed that PTP-PEST phosphatase activity toward 3,6-fluorescein diphosphate (FDP) was not affected by incubation with purified WT PIN1 or the PIN1 C113A mutant (Fig. 2E), which suggests that PIN1 promotes the binding of PTP-PEST to FAK without affecting its PTP-PEST activity. To test whether PTP-PEST is a direct substrate of PIN1 for cis-trans isomerization, we incubated FLAG-PTP-PEST immunoprecipitated from v-H-Ras-expressing PIN1−/− cells with purified GST, WT GST-PIN1, or the GST-PIN1 C113A mutant and then digested the samples using trypsin. Immunoblotting with an anti-PTP-PEST antibody showed that PTP-PEST incubated with WT GST-PIN1 had less full-length protein after digestion than those incubated with GST or GST-PIN1 C113A and lacked an ∼75-kDa digested product (Fig. 2F). These results strongly suggest that PIN1 isomerase activity alters the conformation of PTP-PEST and leads to a change of protease susceptibility.
Fig. 2.

cis-trans isomerization of PTP-PEST by PIN1 is required for the interaction between PTP-PEST and FAK and subsequent FAK dephosphorylation. Western blotting and immunoprecipitation analyses were performed with the indicated antibodies. (A and B) U251 cells expressing a control or PIN1 short hairpin RNA (shRNA) (A) were infected with or without v-H-Ras-expressing adenovirus for 12 h (B). (C) U251 cells with or without depletion of PIN1 or with or without infection with v-H-Ras-expressing adenovirus were treated with CHX (100 μg/ml). The cells were harvested at the indicated time points. (D) FLAG-PTP-PEST immunoprecipitated from v-H-Ras-expressing PIN1−/− cells was incubated with or without soluble GST, WT GST-PIN1, or GST-PIN1 C113A, which was followed by PBS washings. FLAG-PTP-PEST was then eluted from protein G agarose beads with FLAG peptide and incubated with FAK immunoprecipitated from v-H-Ras-expressing PTP-PEST−/− cells. (E) FLAG-PTP-PEST immunoprecipitated from v-H-Ras-expressing PIN1−/− cells was incubated with or without soluble GST, WT GST-PIN1, or GST-PIN1 C113A, which was followed by PBS washings. PTP-PEST activity toward FDP was measured. Data represent the means ± standard deviations from three independent experiments. (F) FLAG-PTP-PEST immunoprecipitated from v-H-Ras-expressing PIN1−/− cells was incubated with purified GST, WT GST-PIN1, or GST-PIN1 C113A mutant and then digested using trypsin (5 ng) for 5 min.
ERK1/2 phosphorylate PTP-PEST and are required for PIN1 to bind to PTP-PEST.
PIN1 binds to phosphorylated serine or threonine residues in pS/TP-peptide sequences in its substrates (20). To test whether PTP-PEST is phosphorylated upon Ras activation, we transfected FLAG-PTP-PEST alone or in combination with v-H-Ras into 293T cells, immunoprecipitated PTP-PEST, and performed an in vitro kinase assay. Figure 3A shows that PTP-PEST was significantly phosphorylated by PTP-PEST-associated protein kinases only in the presence of v-H-Ras expression. These results suggest that Ras expression activates a downstream protein kinase that associates with and phosphorylates PTP-PEST.
Fig. 3.

ERK1/2 phosphorylate PTP-PEST and are required for PIN1 to bind to PTP-PEST. Western blotting analyses were performed with the indicated antibodies. (A) A vector expressing FLAG-PTP-PEST was cotransfected with or without a vector expressing v-H-Ras in 293T cells. FLAG-PTP-PEST was immunoprecipitated and analyzed by in vitro kinase assay. (B) Immunoprecipitated FLAG-PTP-PEST, which was boiled to inactivate any associated protein kinases, was incubated with purified ERK2 or ERK5 in an in vitro kinase assay. (C) A vector expressing FLAG-PTP-PEST was cotransfected with or without a vector expressing v-H-Ras in 293T cells. Immunoprecipitation of ERK2 was followed by immunoblotting with an anti-FLAG antibody. (D) A vector expressing HA-ERK2 was cotransfected with or without a vector expressing constitutively active MEK1 (MEK1 Q56P) in 293T cells expressing FLAG-PTP-PEST. Immunoprecipitation of FLAG-PTP-PEST by an anti-FLAG antibody was followed by immunoblotting with an anti-HA antibody. (E) GST pulldown assays were performed by incubating GST-PIN1-bound agarose beads with lysates of FAK−/− cells, which were treated with or without U0126 (20 μM) and infected with or without v-H-Ras-expressing adenovirus for 12 h.
Given that ERK1/2 are downstream from Ras signaling and can phosphorylate FAK, we examined whether ERK2 is able to also phosphorylate PTP-PEST. Incubation of purified active ERK2 or ERK5 with immunoprecipitated, heat-inactivated FLAG-PTP-PEST showed that ERK2, but not ERK5, phosphorylated PTP-PEST (Fig. 3B). In addition, immunoblotting of immunoprecipitated ERK2 with an anti-FLAG antibody showed that v-H-Ras expression significantly increased the interaction between ERK2 and FLAG-PTP-PEST (Fig. 3C). Furthermore, ERK activation by coexpression of a constitutively active MEK1 mutant (MEK1 Q56P) resulted in enhanced association of PTP-PEST with HA-ERK2, which was demonstrated by immunoblotting of the immunoprecipitated FLAG-PTP-PEST with an anti-HA antibody (Fig. 3D). These results indicate that Ras-induced ERK2 activation increases the binding of ERK2 to PTP-PEST.
To test the effect of ERK1/2 activation on activated Ras-induced association between PIN1 and PTP-PEST, we treated FAK−/− cells expressing v-H-Ras with the MEK inhibitor U0126. U0126, which inhibited v-H-Ras-induced ERK1/2 phosphorylation and subsequent activation, blocked Ras-induced GST-PIN1 binding to PTP-PEST (Fig. 3E). These results suggest that ERK1/2-mediated PTP-PEST phosphorylation is required for PIN1 to bind to PTP-PEST.
ERK1/2 phosphorylate PTP-PEST at S571, which is required to recruit PIN1.
Analysis of the PTP-PEST amino acid sequence revealed that PTP-PEST has 17 putative ERK1/2 phosphorylation sites. To identify the ERK1/2-mediated phosphorylation sites of PTP-PEST, we analyzed a tryptic digest of purified PTP-PEST phosphorylated by ERK2 by mass spectrometry. One candidate phosphopeptide spanning amino acids 565 to 592 was detected by nano-liquid chromatography online to mass spectrometry. S571 was identified as the likely phosphorylation site in this peptide (Fig. 4A). The S571A mutation reduced ERK2-dependent phosphorylation of PTP-PEST in vitro (Fig. 4B). These results indicate that ERK2 phosphorylates PTP-PEST primarily at S571. Consistent with the results showing that ERK activation is required for Ras-induced interaction between PIN1 and PTP-PEST, the PTP-PEST S571A mutation reduced PTP-PEST's binding to GST-PIN1 (Fig. 4C). These results indicate that ERK2 phosphorylates PTP-PEST at S571 and that this phosphorylation is required for the interaction between PTP-PEST and PIN1.
Fig. 4.

ERK1/2 phosphorylate PTP-PEST at S571, which is required for recruiting PIN1. (A) Purified His-PTP-PEST was phosphorylated by ERK2 in vitro and was analyzed by mass spectrometry. Mass spectrometric analysis of a tryptic fragment at m/z 1050.2 matched the triply charged peptide 565-TVSLTPpSPTTQVETPDLVDHDNTSPLFR-592, suggesting that S571 was phosphorylated. The Sequest scores for this match were Xcorr = 2.29 and ppep = 1.0. (B) FLAG-PTP-PEST or FLAG-PTP-PEST S571A was immunoprecipitated from 293T cells expressing v-H-Ras with an anti-FLAG antibody. The immunoprecipitated proteins were boiled to inactivate any associated protein kinases and then incubated with purified ERK2 for an in vitro kinase assay. (C) GST pulldown assays were performed by incubating purified GST-PIN1 with lysates of 293T cells expressing v-H-Ras and WT FLAG-PTP-PEST or FLAG-PTP-PEST S571A. Western blotting analyses were performed with the indicated antibodies.
PTP-PEST S571 phosphorylation promotes the interaction between PTP-PEST and FAK and subsequent dephosphorylation of FAK Y397.
Ras activation induces the binding and dephosphorylation of FAK by PTP-PEST (45). To test the effect of ERK1/2-mediated PTP-PEST phosphorylation on FAK Y397 dephosphorylation, we stably transfected WT FLAG-PTP-PEST or the FLAG-PTP-PEST S571A mutant into PTP-PEST−/− cells (Fig. 5A). Consistent with our previous observations (45), expression of v-H-Ras resulted in FAK dephosphorylation at Y397 in WT PTP-PEST-expressing cells, and this dephosphorylation was blocked by U0126 treatment (Fig. 5B). In contrast, v-H-Ras-induced FAK Y397 dephosphorylation was largely inhibited in the cells expressing the PTP-PEST S571A mutant. These results indicate that Ras-induced PTP-PEST S571 phosphorylation is required for PTP-PEST to dephosphorylate FAK Y397.
Fig. 5.

PTP-PEST S571 phosphorylation promotes the interaction between PTP-PEST and FAK and subsequent dephosphorylation of FAK Y397. Western blotting analyses were performed with the indicated antibodies. (A) Cell lysates were prepared from PEST-PEST−/− cells stably expressing an empty vector, WT FLAG-PTP-PEST, or the FLAG-PTP-PEST S571A mutant. (B and D) FAK was immunoprecipitated from PTP-PEST−/− cells with reconstituted expression of WT FLAG-PTP-PEST or the FLAG-PTP-PEST S571A mutant. Cells were treated with or without U0126 (20 μM) for 30 min before being infected with v-H-Ras-expressing adenovirus for 12 h. (C) Vectors expressing WT FLAG-PTP-PEST or FLAG-PTP-PEST S571A were cotransfected with a vector expressing v-H-Ras in 293T cells. WT FLAG-PTP-PEST and the FLAG-PTP-PEST S571A mutant were immunoprecipitated with an anti-FLAG antibody, and their activity toward FDP was measured. Data represent the means ± standard deviations from three independent experiments.
PIN1 promotes the binding of PTP-PEST to FAK without affecting PTP-PEST activity (Fig. 2D and E), suggesting that ERK-dependent phosphorylation of PTP-PEST affects PTP-PEST's ability to bind FAK but not PTP-PEST's phosphatase activity. To further validate this implication, we immunoprecipitated FLAG-PTP-PEST and FLAG-PTP-PEST S571A from 293T cells expressing v-H-Ras and measured PTP-PEST phosphatase activity toward FDP. As shown in Fig. 5C, the PTP-PEST S571A mutant had phosphatase activity comparable to that of WT PTP-PEST, which indicates that ERK-dependent phosphorylation of PTP-PEST does not significantly alter its phosphatase activity. To determine whether the PTP-PEST S571A mutation affects the binding of PTP-PEST to FAK, we immunoprecipitated FAK from v-H-Ras-expressing PTP-PEST−/− cells that reconstituted the expression of WT PTP-PEST or the S571A mutant. Immunoblotting with an anti-FLAG antibody showed that expression of v-H-Ras dramatically enhanced the association between FAK and WT PTP-PEST and that this association was blocked by U0126 treatment. In contrast, PTP-PEST S571A failed to bind to FAK in response to v-H-Ras expression (Fig. 5D). These results indicate that ERK1/2-mediated PTP-PEST phosphorylation promotes the interaction between PTP-PEST and FAK and thus facilitates the dephosphorylation of FAK Y397 by PTP-PEST.
PTP-PEST S571 phosphorylation mediates v-H-Ras-regulated cell focal adhesion, migration, and invasion.
The reconstituted expression levels of WT PTP-PEST and the PTP-PEST S571A mutant in PTP-PEST−/− cells were comparable to the expression level of endogenous PTP-PEST in PTP-PEST+/+ cells (Fig. 6A). To examine the effect of ERK1/2-mediated PTP-PEST S571 phosphorylation on v-H-Ras-induced regulation of focal adhesion, we immunostained vinculin, a marker of focal adhesions, and showed that expressing v-H-Ras in PTP-PEST+/+ cells changed the cell morphology to spindle shaped and reduced the number of focal adhesions (Fig. 6B). In contrast, PTP-PEST−/− cells, which had more focal adhesions than PTP-PEST+/+ cells, were largely resistant to v-H-Ras-induced morphological changes and reductions in the number of total focal adhesions. However, this resistance was abrogated by reconstituted expression of WT PTP-PEST, but not of the PTP-PEST S571A mutant, in PTP-PEST−/− cells (Fig. 6B). These results strongly suggest that v-H-Ras-induced PTP-PEST phosphorylation is instrumental in the v-H-Ras-induced changes in focal adhesions and cell morphology. To examine the role of PTP-PEST in v-H-Ras-induced cell migration, we next conducted a monolayer wound-healing assay. Compared with PTP-PEST+/+ cells, PTP-PEST−/− cells showed a reduced migration rate in response to infection with adenovirus expressing v-H-Ras (Fig. 6C). This migration inhibition was rescued by reconstituted expression of WT PTP-PEST but not of the PTP-PEST S571A mutant. Similarly, a Matrigel invasion Transwell assay showed that PTP-PEST deficiency blocked v-H-Ras-induced cell invasion, and this effect was abrogated by reconstituted expression of WT PTP-PEST but not by expression of the PTP-PEST S571A mutant (Fig. 6D). These results indicate that ERK1/2-mediated PTP-PEST S571 phosphorylation promotes v-H-Ras-induced cell migration and invasion.
Fig. 6.

PTP-PEST S571 phosphorylation mediates v-H-Ras-regulated cell focal adhesion, migration, and invasion. (A) PEST-PEST+/+ and PEST-PEST−/− cells with or without stable expression of WT FLAG-PTP-PEST or the FLAG-PTP-PEST S571A mutant. Western blotting analyses were performed with the indicated antibodies. (B) The indicated cells were infected with or without v-H-Ras-expressing adenovirus for 12 h. Immunofluorescence analysis was performed with an antivinculin antibody. (C) The indicated cells were infected with v-H-Ras-expressing adenovirus for 12 h, and the cell lawns were wounded by scraping with a micropipette tip. Pictures were taken with a digital camera mounted on a microscope with a magnification of ×50. (D) The indicated cells with or without the expression of v-H-Ras were plated on the top surface of a Matrigel insert. Twelve hours after plating, cells that migrated to the opposite side of the insert were stained with crystal violet. Representative photomicrographs were taken with a digital camera mounted on a microscope with a magnification of ×50 (left panels). The Matrigel membranes that contained invading cells were dissolved in 4% deoxycholic acid and read colorimetrically at 590 nm for quantification of invasion (right panel). Data represent the means ± standard deviations from three independent experiments.
PTP-PEST S571 phosphorylation promotes v-H-Ras-regulated lung metastasis.
We did not find any obvious changes in cell proliferation when growing v-H-Ras-expressing PTP-PEST+/+, PTP-PEST−/−, or PTP-PEST−/− cells with reconstituted expression of WT PTP-PEST or the PTP-PEST S571A mutant in 1% or 10% serum (Fig. 7A). All these cell types induced similar tumor growth rates when subcutaneously injected into athymic nude mice (Fig. 7B). These results indicate that ERK1/2-mediated PTP-PEST S571 phosphorylation does not affect v-H-Ras-promoted cell proliferation or tumorigenesis.
Fig. 7.

PTP-PEST S571 phosphorylation promotes v-H-Ras-regulated metastasis. (A) v-H-Ras was stably expressed in PEST-PEST+/+ and PEST-PEST−/− cells with or without stable expression of FLAG-PTP-PEST or the FLAG-PTP-PEST S571A mutant. Western blotting analyses were performed with the indicated antibodies (left panel). The indicated cells were cultured in either 1% (middle panel) or 10% (right panel) bovine calf serum and counted at the indicated time points. (B) The indicated cells expressing v-H-Ras were subcutaneously injected into six athymic nude mice per group. Three weeks later, the tumor volumes were measured. Data represent the means ± standard deviations from two independent experiments. (C) The indicated cells expressing v-H-Ras were injected into the lateral tail veins of six athymic nude mice per group. After 3 weeks, the mice were sacrificed, and lung metastasis nodules were counted under a low-power dissecting microscope. Data represent the means ± standard deviations from two independent experiments (P < 0.05, indicating significant differences in nodule numbers in post hoc comparisons to the group of mice injected with PTP-PEST+/+ cells expressing v-H-Ras). The arrow points to a metastasis nodule. (D) p130CAS was immunoprecipitated from the lysates of lung metastasis nodules derived from the indicated cell types. Immunoblotting analyses were performed with the indicated antibodies.
To examine the effect of PTP-PEST S571 phosphorylation on v-H-Ras-induced cell metastasis, we injected the same cell types into the tail veins of athymic nude mice. Dissection of the mice 3 weeks later revealed that PTP-PEST−/− cells expressing v-H-Ras had generated fewer metastatic nodules in the lungs of the mice than PTP-PEST+/+ cells that expressed v-H-Ras (Fig. 7C). Compared to the reconstituted expression of WT PTP-PEST, expression of the PTP-PEST S571A mutant largely inhibited Ras-induced metastasis. In addition, immunoblotting analysis of tumor tissues showed that the phosphorylation levels of FAK Y397 and p130CAS were increased in tumors induced by injecting mice with v-H-Ras-expressing PTP-PEST−/− cells with or without expression of the PTP-PEST S571A mutant. Conversely, tumors induced by injecting v-H-Ras-expressing PEST+/+ or PTP-PEST−/− cells with reconstitution of WT PTP-PEST did not show increased FAK Y397 phosphorylation (Fig. 7D). These results indicate that ERK1/2-mediated PTP-PEST S571 phosphorylation promotes v-H-Ras-induced metastasis.
DISCUSSION
Ras is either mutated or activated in many types of human cancer (6, 8). However, the mechanism by which Ras regulates focal contacts and consequently promotes tumor cell motility remains to be further elucidated. We have demonstrated here that activated Ras regulates the binding of PTP-PEST to FAK via ERK1/2-mediated phosphorylation and PIN1-dependent isomerization of PTP-PEST. The increased interaction between PTP-PEST and FAK leads to FAK dephosphorylation at Y397 in v-Ras-transformed cells and promotes cell migration, invasion, and metastasis.
PTP-PEST has been implicated in regulation of focal adhesion disassembly and cell migration (1, 14). PTP-PEST−/− mouse fibroblasts have more focal adhesions, spread faster on fibronectin substrate, and have slower migration than PTP-PEST+/+ cells (1), all of which suggest that PTP-PEST promotes focal adhesion turnover and cell migration. Mutations of PTP-PEST that alter its catalytic activity have been found in both breast and squamous carcinoma cells (34), which implies a role for misregulation of PTP-PEST in tumor cell motility. PTP-PEST can be posttranslationally modified, which affects its catalytic activity or its access to substrates. It has been shown that treatment of HeLa cells with 12-O-tetradecanoylphorbol-13-acetate (TPA), forskolin, or 3-isobutyl-1-methylxanthine results in phosphorylation of the S39 and S435 residues of PTP-PEST, which is regulated by cyclic AMP-dependent protein kinase A (PKA) and protein kinase C (PKC) (13). Phosphorylation of S39 in vitro reduces PTP-PEST's affinity for its substrate. In addition, PTP-PEST immunoprecipitated from TPA-treated cells displays reduced phosphatase activity (13). In line with the fact that PTP-PEST can be regulated by phosphorylation, we have shown here that activation of ERK1/2 by Ras results in phosphorylation of PTP-PEST at S571. Phosphorylation of S571, which by itself does not alter the catalytic activity of PTP-PEST toward phosphopeptide substrates, recruits PIN1 for the cis-trans isomerization of PTP-PEST. The conformational changes of PTP-PEST induced by PIN1, which may alter the structure of the protein-interacting domains adjacent to S571 (10, 13), increased PTP-PEST's association with FAK and led to FAK dephosphorylation. The fact that mutation of S571 in PTP-PEST inhibits v-H-Ras-induced FAK Y397 dephosphorylation and cell migration, invasion, and metastasis highlights the significance of ERK1/2-mediated PTP-PEST phosphorylation in v-H-Ras-enhanced tumor cell motility.
PIN1 isomerizes the phosphorylated pS/TP bonds in its substrate proteins and catalytically induces their conformational change, which can be involved in regulating protein-protein interactions (18, 46). We previously showed that activation of Ras and ERK1/2 results in the colocalization and interaction of PIN1, PTP-PEST, and FAK at the lamellipodia, the front leading edge of cells (45). ERK1/2 phosphorylate FAK S910 and recruit PIN1 for FAK cis-trans isomerization, which leads to interaction between FAK and PTP-PEST (45). However, Ras can still induce the interaction between PIN1 and ERK1/2-phosphorylated PTP-PEST in FAK−/− cells. In addition, Ras promotes the association between PIN1 and ERK1/2-phosphorylated FAK in PTP-PEST−/− cells. These results indicate that ERK1/2- and PIN1-mediated phosphorylation and the isomerization of both FAK and PTP-PEST occur simultaneously in a mutually independent manner. Nevertheless, both events are coordinated in the regulation of the interaction between FAK and PTP-PEST and subsequent FAK Y397 phosphorylation.
Increased FAK expression and activity, both of which promote tumor growth and metastasis, have been detected in several types of human cancer (17, 26, 29, 33). In contrast, paradoxical evidence shows that FAK expression was reduced in metastatic tumors compared to their corresponding primary tumors from patients with colorectal adenocarcinoma, cervical cancer, and intrahepatic cholangiocarcinoma (3, 12, 28). Previously, we and other groups showed that activation of the ErbB family members EGFR, ErbB2, ErbB3, and ErbB4 and of the insulin-like growth factor 1 (IGF-1) receptor induced Tyr dephosphorylation of FAK, inactivation of FAK, and increased tumor cell motility, invasion, and metastasis (4, 5, 9, 15, 16, 21, 24, 25, 36, 38). In response to IGF-1 signaling, SHP2 is instrumental in tumor cell invasion (25), and SHP2 activity is required for FAK Tyr dephosphorylation (25, 43). Notably, activation of EGFR and Ras results in a reduction of total focal adhesions, with limited focal contacts remaining at the leading-edge lamellipodia and tail regions (21, 44, 45). Moreover, FAK phosphorylation can be dynamically regulated because EGFR- and Ras-induced FAK dephosphorylation is overridden by activated integrin signaling during cell attachment. However, integrin-induced FAK phosphorylation occurs only during the process of adhesion and is downregulated by EGFR and Ras signaling thereafter (21, 44). On the basis of all these findings, we proposed that overexpression of FAK in human tumor cells might contribute to malignancy by promoting survival through the activation of a positive FAK-c-Src feedback loop and subsequently of downstream tumor-promoting molecules, such as ERK and AKT (17, 29, 45). However, some other tumors that have aberrant Ras activity, which does not require Src for its transforming ability (data not shown), can activate cell proliferation molecules such as ERK and AKT in a FAK-c-Src-independent manner. Activated Ras signaling inhibits FAK activity, which leads to reduced cell adhesion and increased cell migration. Importantly, FAK activity can be dynamically regulated by integrated Ras and integrin signaling, which may play an instrumental role in the regulation of lamellipodial dynamics and turnover of focal adhesions at the leading edge of the cells during migration (44, 45). Thus, FAK might have different roles in different tumors and at different stages of tumor progression.
In summary, we have uncovered an important mechanism of PTP-PEST-regulated FAK dephosphorylation in response to oncogenic Ras signaling (Fig. 8). Activation of Ras results in phosphorylation of FAK S910 and PTP-PEST S571 by ERK1/2, which leads to the recruitment of PIN1 and the cis-trans isomerization of both FAK and PTP-PEST. The conformational alterations of FAK and PTP-PEST enable them to bind each other to facilitate subsequent FAK Y397 dephosphorylation and enhanced tumor cell migration, invasion, and metastasis.
Fig. 8.
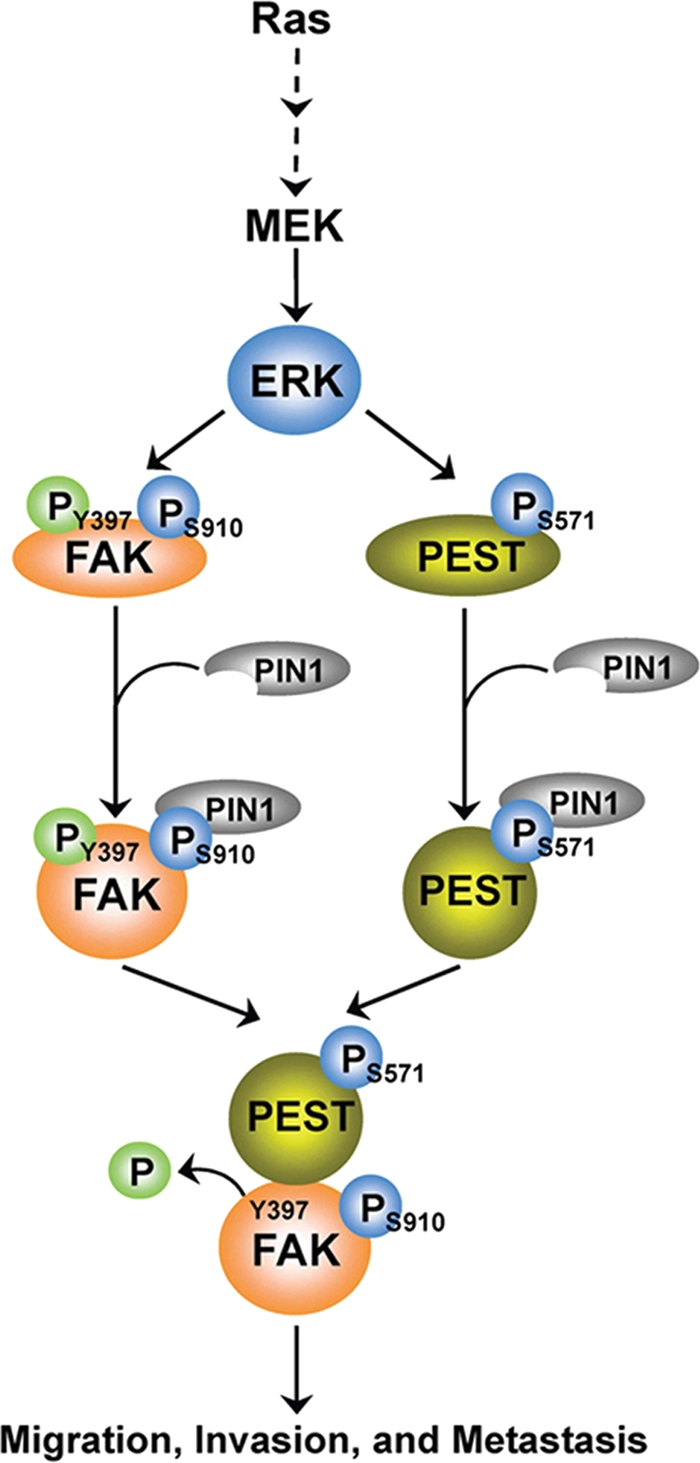
A proposed mechanism for activated Ras-induced and ERK1/2- and PIN1-dependent FAK Y397 dephosphorylation by PTP-PEST. Activated Ras results in ERK1/2-dependent phosphorylation of PTP-PEST S571 and FAK S910 and subsequent recruitment of PIN1 for cis-trans isomerization of both PTP-PEST and FAK. This leads to increased interaction between PTP-PEST and FAK, dephosphorylation of FAK Y397, and enhanced tumor cell migration, invasion, and metastasis.
ACKNOWLEDGMENTS
We thank Tony Hunter (The Salk Institute for Biological Studies) for FAK knockout cells, Anthony Means (Duke University) for PIN1 knockout cells and constructs, Michel L. Tremblay (McGill University) for PTP-PEST knockout cells, Steven Reed (The Scripps Research Institute) for a vector expressing pGEX-4T GST-PIN1 WT and the WW mutant, Kevin Pumiglia (Albany Medical Center) for the v-H-Ras adenovirus, and Zach Bohannan for critical reading of the manuscript.
This work was supported by National Cancer Institute grants 5R01CA109035 (Z.L.), 5 P50 CA127001-03, and CA016672 (Cancer Center Support Grant), research grant RP110252 (Z.L.) from the Cancer Prevention and Research Institute of Texas, American Cancer Society Research Scholar Awards RSG-09-277-01-CSM (Z.L.) and RSG-08-288-01-GMC (D.X.L.), an institutional research grant from The University of Texas MD Anderson Cancer Center (Z.L.), and Department of Defense grant W81XWH-09-1-0349 (D.X.L.).
Footnotes
Published ahead of print on 29 August 2011.
REFERENCES
- 1. Angers-Loustau A., et al. 1999. Protein tyrosine phosphatase-PEST regulates focal adhesion disassembly, migration, and cytokinesis in fibroblasts. J. Cell Biol. 144:1019–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Antonyak M. A., Cerione R. A. 2009. Ras and the FAK paradox. Mol. Cell 35:141–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ayaki M., et al. 2001. Reduced expression of focal adhesion kinase in liver metastases compared with matched primary human colorectal adenocarcinomas. Clin. Cancer Res. 7:3106–3112 [PubMed] [Google Scholar]
- 4. Bose R., et al. 2006. Phosphoproteomic analysis of Her2/neu signaling and inhibition. Proc. Natl. Acad. Sci. U. S. A. 103:9773–9778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Caceres M., Guerrero J., Martinez J. 2005. Overexpression of RhoA-GTP induces activation of the epidermal growth factor receptor, dephosphorylation of focal adhesion kinase and increased motility in breast cancer cells. Exp. Cell Res. 309:229–238 [DOI] [PubMed] [Google Scholar]
- 6. Campbell P. M., Der C. J. 2004. Oncogenic Ras and its role in tumor cell invasion and metastasis. Semin. Cancer Biol. 14:105–114 [DOI] [PubMed] [Google Scholar]
- 7. Chambers A. F., Groom A. C., MacDonald I. C. 2002. Dissemination and growth of cancer cells in metastatic sites. Nat. Rev. Cancer 2:563–572 [DOI] [PubMed] [Google Scholar]
- 8. Chin L., et al. 1999. Essential role for oncogenic Ras in tumour maintenance. Nature 400:468–472 [DOI] [PubMed] [Google Scholar]
- 9. Cui Y., Liao Y. C., Lo S. H. 2004. Epidermal growth factor modulates tyrosine phosphorylation of a novel tensin family member, tensin3. Mol. Cancer Res. 2:225–232 [PubMed] [Google Scholar]
- 10. Davidson D., Veillette A. 2001. PTP-PEST, a scaffold protein tyrosine phosphatase, negatively regulates lymphocyte activation by targeting a unique set of substrates. EMBO J. 20:3414–3426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fang D., et al. 2007. Phosphorylation of beta-catenin by AKT promotes beta-catenin transcriptional activity. J. Biol. Chem. 282:11221–11229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gabriel B., et al. 2006. Weak expression of focal adhesion kinase (pp125FAK) in patients with cervical cancer is associated with poor disease outcome. Clin. Cancer Res. 12:2476–2483 [DOI] [PubMed] [Google Scholar]
- 13. Garton A. J., Tonks N. K. 1994. Ptp-Pest—a protein-tyrosine-phosphatase regulated by serine phosphorylation. EMBO J. 13:3763–3771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Garton A. J., Tonks N. K. 1999. Regulation of fibroblast motility by the protein tyrosine phosphatase PTP-PEST. J. Biol. Chem. 274:3811–3818 [DOI] [PubMed] [Google Scholar]
- 15. Guo H. B., Randolph M., Pierce M. 2007. Inhibition of a specific N-glycosylation activity results in attenuation of breast carcinoma cell invasiveness-related phenotypes: inhibition of epidermal growth factor-induced dephosphorylation of focal adhesion kinase. J. Biol. Chem. 282:22150–22162 [DOI] [PubMed] [Google Scholar]
- 16. Guvakova M. A., Surmacz E. 1999. The activated insulin-like growth factor I receptor induces depolarization in breast epithelial cells characterized by actin filament disassembly and tyrosine dephosphorylation of FAK, Cas, and paxillin. Exp. Cell Res. 251:244–255 [DOI] [PubMed] [Google Scholar]
- 17. Hanks S. K., Ryzhova L., Shin N. Y., Brabek J. 2003. Focal adhesion kinase signaling activities and their implications in the control of cell survival and motility. Front. Biosci. 8:d982–d996 [DOI] [PubMed] [Google Scholar]
- 18. Hunter T. 1998. Prolyl isomerases and nuclear function. Cell 92:141–143 [DOI] [PubMed] [Google Scholar]
- 19. Julien S. G., Dube N., Hardy S., Tremblay M. L. 2011. Inside the human cancer tyrosine phosphatome. Nat. Rev. Cancer 11:35–49 [DOI] [PubMed] [Google Scholar]
- 20. Lu K. P., Zhou X. Z. 2007. The prolyl isomerase PIN1: a pivotal new twist in phosphorylation signalling and disease. Nat. Rev. Mol. Cell. Biol. 8:904–916 [DOI] [PubMed] [Google Scholar]
- 21. Lu Z., Jiang G., Blume-Jensen P., Hunter T. 2001. Epidermal growth factor-induced tumor cell invasion and metastasis initiated by dephosphorylation and downregulation of focal adhesion kinase. Mol. Cell. Biol. 21:4016–4031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lu Z., et al. 1998. Activation of protein kinase C triggers its ubiquitination and degradation. Mol. Cell. Biol. 18:839–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lu Z., Xu S., Joazeiro C., Cobb M. H., Hunter T. 2002. The PHD domain of MEKK1 acts as an E3 ubiquitin ligase and mediates ubiquitination and degradation of ERK1/2. Mol. Cell 9:945–956 [DOI] [PubMed] [Google Scholar]
- 24. Lynch L., Vodyanik P. I., Boettiger D., Guvakova M. A. 2005. Insulin-like growth factor I controls adhesion strength mediated by alpha5beta1 integrins in motile carcinoma cells. Mol. Biol. Cell 16:51–63 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Manes S., et al. 1999. Concerted activity of tyrosine phosphatase SHP-2 and focal adhesion kinase in regulation of cell motility. Mol. Cell. Biol. 19:3125–3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McLean G. W., et al. 2005. The role of focal-adhesion kinase in cancer. A new therapeutic opportunity. Nat. Rev. Cancer 5:505–515 [DOI] [PubMed] [Google Scholar]
- 27. Mitra S. K., Hanson D. A., Schlaepfer D. D. 2005. Focal adhesion kinase: in command and control of cell motility. Nat. Rev. Mol. Cell Biol. 6:56–68 [DOI] [PubMed] [Google Scholar]
- 28. Ohta R., et al. 2006. Reduced expression of focal adhesion kinase in intrahepatic cholangiocarcinoma is associated with poor tumor differentiation. Oncology 71:417–422 [DOI] [PubMed] [Google Scholar]
- 29. Parsons J. T. 2003. Focal adhesion kinase: the first ten years. J. Cell Sci. 116:1409–1416 [DOI] [PubMed] [Google Scholar]
- 30. Ridley A. J., et al. 2003. Cell migration: integrating signals from front to back. Science 302:1704–1709 [DOI] [PubMed] [Google Scholar]
- 31. Schaller M. D. 2004. FAK and paxillin: regulators of N-cadherin adhesion and inhibitors of cell migration? J. Cell Biol. 166:157–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Schubbert S., Shannon K., Bollag G. 2007. Hyperactive Ras in developmental disorders and cancer. Nat. Rev. Cancer 7:295–308 [DOI] [PubMed] [Google Scholar]
- 33. Sood A. K., et al. 2004. Biological significance of focal adhesion kinase in ovarian cancer: role in migration and invasion. Am. J. Pathol. 165:1087–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Streit S., et al. 2006. PTP-PEST phosphatase variations in human cancer. Cancer Genet. Cytogenet. 170:48–53 [DOI] [PubMed] [Google Scholar]
- 35. Takekawa M., et al. 1992. Cloning and characterization of a human cDNA encoding a novel putative cytoplasmic protein-tyrosine-phosphatase. Biochem. Biophys. Res. Commun. 189:1223–1230 [DOI] [PubMed] [Google Scholar]
- 36. Thelemann A., et al. 2005. Phosphotyrosine signaling networks in epidermal growth factor receptor overexpressing squamous carcinoma cells. Mol. Cell Proteomics 4:356–376 [DOI] [PubMed] [Google Scholar]
- 37. Tonks N. K. 2006. Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 7:833–846 [DOI] [PubMed] [Google Scholar]
- 38. Vadlamudi R. K., Adam L., Nguyen D., Santos M., Kumar R. 2002. Differential regulation of components of the focal adhesion complex by heregulin: role of phosphatase SHP-2. J. Cell Physiol. 190:189–199 [DOI] [PubMed] [Google Scholar]
- 39. Wang F. M., et al. 2005. SHP-2 promoting migration and metastasis of MCF-7 with loss of E-cadherin, dephosphorylation of FAK and secretion of MMP-9 induced by IL-1 beta in vivo and in vitro. Breast Cancer Res. Treat. 89:5–14 [DOI] [PubMed] [Google Scholar]
- 40. Yamanaka I., et al. 2003. Epidermal growth factor increased the expression of alpha 2 beta 1-integrin and modulated integrin-mediated signaling in human cervical adenocarcinoma cells. Exp. Cell Res. 286:165–174 [DOI] [PubMed] [Google Scholar]
- 41. Yang Q., Co D., Sommercorn J., Tonks N. K. 1993. Cloning and expression of Ptp-Pest—a novel, human, nontransmembrane protein tyrosine phosphatase. J. Biol. Chem. 268:6622–6628 [PubMed] [Google Scholar]
- 42. Yi T. L., Cleveland J. L., Ihle J. N. 1991. Identification of novel protein tyrosine phosphatases of hematopoietic-cells by polymerase chain-reaction amplification. Blood 78:2222–2228 [PubMed] [Google Scholar]
- 43. Yu D. H., Qu C. K., Henegariu O., Lu X., Feng G. S. 1998. Protein-tyrosine phosphatase Shp-2 regulates cell spreading, migration, and focal adhesion. J. Biol. Chem. 273:21125–21131 [DOI] [PubMed] [Google Scholar]
- 44. Zheng Y., Lu Z. 2009. Paradoxical roles of FAK in tumor cell migration and metastasis. Cell Cycle 8:3474–3479 [DOI] [PubMed] [Google Scholar]
- 45. Zheng Y. H., et al. 2009. FAK phosphorylation by ERK primes Ras-induced tyrosine dephosphorylation of FAK mediated by PIN1 and PTP-PEST. Mol. Cell 35:11–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Zhou X. Z., Lu P. J., Wulf G., Lu K. P. 1999. Phosphorylation-dependent prolyl isomerization: a novel signaling regulatory mechanism. Cell. Mol. Life Sci. 56:788–806 [DOI] [PMC free article] [PubMed] [Google Scholar]