

Abstract
The initial step in poxvirus morphogenesis, the formation of crescent membranes, occurs within cytoplasmic factories. L2 is one of several vaccinia virus proteins known to be necessary for formation of crescents and the only one synthesized early in infection. Virus replication was unaffected when the L2R open reading frame was replaced by L2R containing an N-terminal epitope tag while retaining the original promoter. L2 colocalized with the endoplasmic reticulum (ER) protein calnexin throughout the cytoplasm of infected and transfected cells. Topological studies indicated that the N terminus of L2 is exposed to the cytoplasm with the hydrophobic C terminus anchored in the ER. Using immunogold labeling and electron microscopy, L2 was detected in tubular membranes outside factories and inside factories near crescents and close to the edge or rim of crescents; a similar labeling pattern was found for the ER luminal protein disulfide isomerase (PDI). The phenotype of L2 conditional lethal mutants and the localization of L2 suggest that it participates in elongation of crescents by the addition of ER membrane to the growing edge. Small amounts of L2 and PDI were detected within immature and mature virions, perhaps trapped during assembly. The repression of L2, as well as A11 and A17, two other proteins that are required for viral crescent formation, profoundly decreased the stability of a subset of viral membrane proteins including those comprising the entry-fusion complex. To avoid degradation, these unstable membrane proteins may need to directly insert into the viral membrane or be rapidly shunted there from the ER.
INTRODUCTION
Poxvirus morphogenesis occurs within the cytoplasm of infected cells culminating in the formation of infectious virions containing a core with a double-stranded DNA genome that is enclosed by lipid-protein membranes (8). The developmental features seen by electron microscopy are similar for all members of the family and have been described in detail for vaccinia virus (VACV). The first recognizable structure is the crescent-shaped membrane comprised of a single lipid bilayer with an external lattice constituted of trimers of the D13 protein (11, 16, 18, 47). The crescents engulf electron-dense material comprising core proteins and a DNA nucleoid to form the spherical immature virion (IV) (28). The next steps involve release of the D13 scaffold, proteolytic processing of major core proteins, and intramolecular disulfide bond formation of membrane proteins to form the infectious brick-shaped mature virion (MV) (5, 29, 41). Some MVs are wrapped by trans-Golgi membranes or endosomal cisternae (17, 40, 51) and transported via microtubules to the periphery of the cell, where they undergo exocytosis and spread to neighboring cells (6, 43).
Neither the origin, mode of formation, or composition of the crescent viral membrane has been determined. The possibility that crescents form de novo was suggested by spatial separation of crescents and intracellular membranes (10). The finding that some viral membrane proteins were associated with the intermediate compartment between the endoplasmic reticulum (ER) and Golgi membrane led to the idea that the crescents were derived from that source (36, 44). Subsequent studies demonstrated that the transport of proteins from the ER to the intermediate compartment and Golgi apparatus is not necessary for IV formation and that there is a transport pathway from the ER to the IV (21, 22, 55). Nevertheless, the mechanisms involved in IV membrane formation remain to be elucidated.
In order to understand the initial steps of morphogenesis, it is necessary to identify the components involved in the process, as well their localization and interactions. Studies with conditional lethal mutants have led to the identification of many VACV proteins (A11 [35], A14 [38, 54], A17 [37, 56], F10 [34, 48, 53], G5 [9], H5 [12], H7 [39], and L2 [27]) that are required for the formation of crescent membranes. Dense masses of viroplasm and in some cases vesicles or tubules accumulate in the absence of these assembly proteins. G5 and H5 have other roles so their effects may be indirect. Repressed expression of the scaffold protein D13 results in the formation of irregular membranes surrounding electron-dense viroplasm similar to effects of the drug rifampin (30, 31, 60).
L2 is the most recent addition to the list of proteins required for crescent formation. In our initial characterization of L2, we determined that it is expressed early in infection and is associated with the detergent soluble fraction of purified virions (27). In addition, a conditional lethal mutant exhibited defects in crescent membrane formation. We were unable to use an L2 antibody for localization of the protein by microscopy because of a cross-reacting viral band that was expressed after viral DNA replication. To circumvent this problem, we constructed a recombinant VACV with a hemagglutinin (HA) epitope-tagged L2 that replicates like wild-type virus. Confocal and electron microscopic images indicated that L2 was associated with the ER throughout the cytoplasm and near the growing edge of membrane crescents and was minimally associated with MVs. In addition, L2 was required for stability of a subset of viral membrane proteins.
MATERIALS AND METHODS
Cells and virus.
BS-C-1 (ATCC CCL-26) and HeLa (ATCC CCL2) cells were grown in minimum essential medium with Earle's salt (E-MEM) and Dulbecco minimum essential medium, respectively, supplemented with 10% fetal bovine serum (FBS), 100 U of penicillin, and 100 μg of streptomycin per ml (Quality Biologicals, Gaithersburg, MD). The VACV Western Reserve (WR) strain, the recombinant VACV vT7lacOI (1), vA9-HA (23), and vL2-HA were propagated as described previously (14). The recombinants vL2Ri (27), vF9Li (7), and vA17Li (56) were grown in the presence of 25 to 100 μM IPTG (isopropyl-β-d-thiogalactopyranoside). MVs were purified from cell lysates by sedimentation twice through 36% sucrose cushions, once in a 25 to 40% sucrose gradient as described previously (15) and once in a CsCl gradient (25).
Antibodies.
Mouse monoclonal antibody (MAb) anti-HA.11 to the influenza virus hemagglutinin (HA) epitope was obtained from Covance (Denver, PA). The sources of rabbit antisera for the following VACV antigens are indicated: L2 (27), A17-N (2), A3 (R. Doms and B. Moss, unpublished data), A4 (13), A30 (46), A21 (52), A28 (32), A26 (19), A11 (35), D13 (45), F13 (17), L1 (26), and F9 (provided by G. Cohen, University of Pennsylvania). AB1.1 anti-D8 mouse MAb (33) and 192C anti-B5 rat MAb (40) were used. Rabbit antibodies to calnexin and ERGIC-53/p58 were from Sigma-Aldrich (St. Louis, MO); anti-β-COP was from Affinity BioReagents (Golden, CO). Mouse protein disulfide isomerase (PDI) MAb was purchased from Stressgen Bioreagents (Ann Arbor, MI). Alexa 594-conjugated anti-mouse immunoglobulin G (IgG), Alexa 488-conjugated anti-rabbit IgG, and Alexa 647-conjugated anti-rabbit IgG were obtained from Invitrogen (Carlsbad, CA).
Plasmid and recombinant VACV construction.
The GFP-L2-HA plasmid was constructed using primers to amplify the L2R ORF tagged with the HA coding sequence at the 5′ end and a separate enhanced green fluorescent protein (GFP) ORF flanked by VACV DNA sequences to allow homologous recombination (see Fig. 1A). The PCR product was cloned using a Zero blunt TOPO PCR cloning kit (Invitrogen). The endogenous L2R open reading frame (ORF) of VACV WR was replaced with an HA epitope-tagged L2R ORF and GFP marker gene by transfecting the GFP-L2-HA plasmid using Lipofectamine 2000 (Invitrogen). The recombinant virus vL2-HA was clonally purified by successive picking of fluorescent plaques.
Fig. 1.

Construction and characterization of a recombinant VACV with HA-epitope tagged L2. (A) Schematic representation of the genome structure of vL2-HA. HA, influenza virus hemagglutinin epitope tag attached to the N terminus of the L2R ORF retaining original L2 promoter; GFP, enhanced green fluorescent protein ORF regulated by the late P11 promoter. Unmodified ORFs encoding L1 and L3 are also shown. Arrows point to the direction of transcription. (B) Plaque phenotype of vL2-HA and wild-type VACV (vWR). BS-C-1 cells were infected with two dilutions of the recombinant virus vL2-HA and with the wild-type virus vWR. After 48 h, the cells were stained with crystal violet. (C) One-step growth curve of vL2-HA. BSC-1 cells were infected at a multiplicity of 3 PFU per cell with wild-type VACV WR or vL2-HA. After 0, 4, 8, 12, 24, 36, and 48 h, the infected cells were harvested, and the virus titers were determined by plaque assay. (D) Synthesis of HA-tagged L2 protein. BSC-1 cells were infected at a multiplicity of 1 PFU per cell with vL2-HA. After 0, 2, 4, 6, 8, 10, and 12 h, the cells were harvested and analyzed by Western blotting with antibody to the HA epitope. M, mock infected. Glyceraldehyde phosphate dehydrogenase (GAPDH) was analyzed as a loading control. Markers in kilodaltons are shown on the left.
Plaque assay.
For plaque assays, BS-C-1 cells monolayers in six-well tissue culture plates were infected with 10-fold dilutions of virus. After a 1-h adsorption, the virus inoculum was removed, the cells were washed with E-MEM–2.5% FBS, and medium containing 0.5% methylcellulose was added. The incubation was continued for 48 h at 37°C. The cells were stained with crystal violet, and the plaques were counted.
Radioactive labeling of proteins.
BS-C-1 cells grown in six-well plates were infected with 5 PFU of vL2Ri or vT7LacOI virus per cell for 1 h at 37°C. The inocula were removed, and the cells were incubated with complete E-MEM–2.5% FBS in the presence or absence of 50 μM IPTG. After a period of 5 h and 30 min, the cells were incubated with cysteine- and methionine-free medium for 30 min at 37°C, and pulse-labeled for 5 min with 100 μCi of [35S]methionine-cysteine (Perkin-Elmer, Waltham, MA). After the pulse, the cells were incubated in medium containing excess methionine and cysteine for up to 2 h. The cells were washed with cold phosphate-buffered saline (PBS) containing 20 mM N-ethylmaleimide (NEM). Cells were lysed in 1% NP-40 containing NEM and complete protease inhibitor cocktail (Roche Applied Science, Indianapolis, IN). Proteins in the clarified cell lysate were immunopurified and resolved by SDS-PAGE, dried, and analyzed by autoradiography.
Immunoaffinity purification.
Clarified cell lysates were incubated with primary antibodies for 2 h at 4°C. The complexes were incubated with EZview Red Protein G Affinity gel (Sigma-Aldrich, St. Louis, MO) overnight at 4°C and then washed four times with 1% NP-40–20 mM NEM and protease inhibitor cocktail, lysed in 1× NuPAGE lithium dodecyl sulfate sample loading buffer containing 20 mM NEM, and heated at 70°C for 10 min.
Western blotting.
Proteins were resolved by electrophoresis on 4 to 12% Novex NuPAGE polyacrylamide gels and transferred to nitrocellulose membranes using Mini iBlot gel transfer stacks (Invitrogen). Membranes were blocked for 1 h at room temperature with 5% nonfat dried milk in PBS containing 0.05% Tween 20 and incubated with the primary antibody for 1 h at room temperature. The membranes were washed four times with PBS–0.05% Tween 20 and then incubated with the secondary antibody for 1 h at room temperature. The secondary antibodies used were species-specific, horseradish peroxidase-conjugated antibodies (Pierce, Rockford, IL). The membranes were washed and developed using Dura or Femto chemiluminescent substrate (Pierce).
Confocal microscopy.
HeLa cells grown on coverslips were infected as described above. The cells were fixed at different times postinfection with 4% paraformaldehyde in PBS for 15 min at room temperature and washed with PBS. The cells were permeabilized for 5 min with 0.2% Triton X-100 in PBS at room temperature or with 20 μg of digitonin/ml in PBS at 0°C. Permeabilized cells were blocked with 10% FBS for 30 min and then incubated with the primary antibody in PBS containing 10% FBS for 1 h at room temperature. Cells were washed and incubated with the secondary antibody conjugated to dye (Molecular Probes, Eugene, OR) for 1 h. The coverslips were washed and mounted on a glass slide by using prolong gold (Invitrogen).
For transfection studies, uninfected HeLa cells were grown on coverslips in 24-well plates 1 day before transfection. The cells were transfected with a plasmid that contains the L2-HA gene under an immediate-early cytomegalovirus promoter using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. The cells were fixed at 48 h posttransfection with 4% paraformaldehyde in PBS for 15 min at room temperature, washed with PBS, permeabilized for 15 min with 0.1% Triton X-100 in PBS at room temperature, and blocked with 10% FBS for 30 min. The cells were incubated with the primary and secondary antibodies and prepared for analysis as described above.
Transmission electron microscopy.
For conventional transmission electron microscopy, infected BS-C-1 cells in 60-mm-diameter wells were fixed with 2% glutaraldehyde and embedded in EmBed-182 resin (Electron Microscopy Sciences, Hatfield, PA). Procedures for cryosectioning and immunogold labeling were described previously (42). Cryosections were picked up on grids, thawed, washed free of sucrose, and stained with a mouse MAb to the HA epitope or anti-PDI MAb, followed by rabbit anti-mouse IgG from Cappel-ICN Pharmaceuticals (Aurora, OH) and then protein A conjugated to 10-nm gold spheres (University Medical Center, Utrecht, Netherlands). Specimens were viewed with a FEI Tecnai Spirit transmission electron microscope (FEI, Hillsboro, OR). Double labeling was carried out using 5- and 10-nm gold as described previously (5).
RESULTS
Construction, replication, and expression of a recombinant VACV with HA-epitope tagged L2.
We constructed a recombinant VACV in which the endogenous L2R ORF was replaced with L2R containing an HA epitope sequence at the N terminus, while retaining the natural promoter (Fig. 1A). To facilitate recombinant virus isolation, the GFP ORF regulated by the P11 late promoter was inserted adjacent to L2R from the same plasmid. The integrity of the recombinant gene was confirmed by sequencing (data not shown). vL2-HA and wild-type virus formed plaques that were indistinguishable in size (Fig. 1B) and exhibited similar replication kinetics (Fig. 1C). Expression of L2-HA protein was demonstrated by Western blotting. A faint band of the expected size (25) was detected at 2 h and progressively increased in amount (Fig. 1D).
L2 colocalized with calnexin, an ER-resident protein.
For initial confocal microscopy analysis, cells infected with vL2-HA were fixed at 9 h after infection and stained with a mouse MAb to the HA epitope tag on L2 and rabbit polyclonal antibodies to the late proteins A11, L1, and A17 for comparison. In addition, DNA in the nucleus and viral factories was stained with DAPI. L2 was distributed throughout the cytoplasm in a reticular manner and was less abundant in the viral factory areas (Fig. 2). In contrast, A11, L1, and A17 localized predominantly in factories (Fig. 2). One reason for this difference could be that L2 has an early promoter and therefore is expressed from the infecting virus particles, whereas A11, L1, and A17 have late promoters (57, 58) and are synthesized in the factory.
Fig. 2.

Confocal microscopy of HeLa cells infected with vL2-HA. After 9 h, the infected cells were fixed, permeabilized, and stained with a polyclonal primary antibody for A11, L1, or A17 and the mouse anti-HA MAb to L2-HA, followed by goat anti-rabbit IgG and goat anti-mouse IgG coupled to Alexa Fluor 647 and Alexa Fluor 594, respectively, and DAPI. The viral protein (A11, L1, or A17) detected in the third (viral protein) column is shown on the left.
The reticular staining pattern of L2 suggested that it was associated with the ER. To confirm this localization, HeLa cells were infected with vL2-HA and fixed at 2, 4, 6, and 8 h postinfection. Fixed cells were costained with the anti-HA MAb to detect L2 protein, polyclonal anti-calnexin antibody to visualize the ER and DAPI to detect DNA in nuclei and viral factories. The factories appeared as small spherical bodies at 2 h and increased in size and became less regular in shape and more dispersed with time (Fig. 3A). L2 colocalized with calnexin throughout the cytosol, as well as the nuclear envelope at each time (Fig. 3A). Both L2 and calnexin appeared to more intensely stain the periphery of the DAPI-staining factories than the interior particularly up to 6 h, which is consistent with the enclosure of factories by ER membrane during this time (50). L2 did not specifically colocalize with β-COP, a marker for the Golgi apparatus, or ERGIC-53, a marker for the intermediate compartment, although there was some overlap in staining (Fig. 3B).
Fig. 3.

Colocalization of L2 with calnexin. (A) HeLa cells were infected with vL2-HA. After 2, 4, 6, and 8 h, the infected cells were fixed, permeabilized, and stained with the rabbit anti-calnexin polyclonal antibody and mouse anti-HA MAb to detect L2, followed by goat anti-rabbit IgG and goat anti-mouse IgG coupled to Alexa Fluor 488 and Alexa Fluor 594, respectively, and DAPI. (B) HeLa cells were infected with vL2-HA as in panel A. After 9 h, the infected cells were fixed, permeabilized, and stained with the rabbit polyclonal primary antibody for calnexin, β-COP, or ERGIC-53 (detected in the third cellular protein column as indicated on the left) and the mouse anti-HA MAb to detect L2, followed by goat anti-mouse IgG and goat anti-rabbit IgG coupled to Alexa Fluor 594 and Alexa Fluor 647, respectively, and DAPI. The cells were visualized by confocal microscopy.
Topology of L2.
Hydrophobicity analysis suggested that L2 has two closely spaced transmembrane domains near the C terminus and a hydrophilic N terminus (27). The N-terminal HA epitope tag was used to investigate the orientation of L2 in cytoplasmic membranes. Cells infected with vL2-HA were fixed and treated with digitonin, which selectively permeabilizes the plasma membrane or with Triton X-100, which permeabilizes all membranes, or left unpermeabilized. The cells were stained with a mouse MAb to HA followed by a secondary antibody and DAPI. L2 was detected in cells permeabilized with digitonin or Triton X-100, but there was only a faint background in nonpermeabilized cells (Fig. 4), indicating that the N terminus was oriented to the cytoplasm. As a control to ensure that digitonin only permeabilized the plasma membrane, the cells were also stained with a rat MAb to the luminal/extracellular domain of B5, which is present in the ER, Golgi apparatus, vesicles, wrapped virions, and plasma membrane. The plasma membrane of nonpermeabilized or digitonin-treated cells stained with the MAb to B5 (Fig. 4). However, staining of B5 on intracellular membranes required Triton X-100 (Fig. 4).
Fig. 4.

Topology of L2. HeLa cells were infected with vL2-HA and fixed after 9 h. The cells were permeabilized for 5 min at room temperature with 0.2% Triton X-100 (upper row), not permeabilized (N.P., middle row), or permeabilized for 5 min at 0°C with 20 μg of digitonin/ml (lower row). The cells were stained with anti-HA mouse MAb and with anti-B5R rat MAb, followed by goat anti-mouse IgG and goat anti-rat IgG coupled to Alexa Fluor 594 and Alexa Fluor 647, respectively, and DAPI.
The intracellular distribution of L2-HA in transfected cells was also determined. HeLa cells were transfected with a plasmid containing the L2-HA gene regulated by an RNA polymerase II-specific promoter. After 48 h, the cells were fixed and costained with anti-HA mouse MAb and polyclonal anti-calnexin rabbit antibody. The location of L2 overlapped with calnexin, indicating that L2 does not need other viral components for ER localization (Fig. 5A). Subtle differences in the intensity of L2 and calnexin staining of some areas of the cytoplasm in the merged images (Fig. 5A) could suggest a preference of L2 for certain ER domains or variation due to the expression of L2 by transfection. The transfected cells were also costained with antibody to HA and ERGIC-53. In the majority of cells there appeared to be no specific colocalization with ERGIC, although there was some overlap of staining (Fig. 5B).
Fig. 5.

Localization of L2 in transfected cells. HeLa cells were transfected with a plasmid expressing L2-HA regulated by the cytomegalovirus early promoter (A and C) or with the transfection reagent (B and D), fixed and permeabilized with Triton X-100, and stained with mouse anti-HA MAb to detect L2 and rabbit anti-calnexin (A and B) or ERGIC-53 (C and D) antibodies, followed by goat anti-mouse IgG coupled to Alexa Fluor 488 and goat anti-rabbit IgG coupled to Alexa Fluor 594 and DAPI.
L2 was present in tubular membranes adjacent to viral crescents.
Immunogold labeling and transmission electron microscopy was used to detect L2 at higher magnifications. BS-C-1 cells infected with vL2-HA for 20 h were fixed and stained with MAb to HA, followed by rabbit anti-mouse IgG and then protein A conjugated to 10-nm gold spheres. Gold grains were present in tubular membranes throughout the cytoplasm including the perinuclear envelope (Fig. 6A and B), consistent with ER staining determined by confocal microscopy. In addition, L2-labeled tubular membranes were found near viral crescent membranes and close to the free edges of crescents (Fig. 6C to F). A small number of gold grains were also found within IVs (Fig. 6G) and virions (Fig. 6H). Similar immunogold staining of tubules near crescents and close to the edges of crescents occurred with a mouse MAb to the ER luminal protein PDI (Fig. 6I and J). Like L2, some PDI was also found within IVs and virions (data not shown). Localization of L2 and PDI on the same membranes was confirmed by double labeling with 10- and 5-nm gold grains, respectively (Fig. 6K and L).
Fig. 6.

Localization of L2 by immunogold transmission electron microscopy. BSC-1 cells were infected with vL2-HA. After 20 h, the cells were fixed, cryosectioned, and stained with anti-HA MAb (A to H) or anti-PDI MAb (I and J), followed by rabbit anti-mouse IgG and protein A conjugated to 10-nm gold spheres. (K and L) In addition, 10- and 5-nm gold was used to label L2 and PDI in the same sections. Arrows point to gold particles labeling L2; arrowheads point to PDI. Magnifications are indicated by scale bars at the bottom of each panel.
The electron micrographs (Fig. 6) suggested that there was only minimal association of L2 with virions compared to other VACV membrane proteins such as A9, which was also detected with antibody to an HA epitope tag (59). In order to more directly compare the relative amounts of L2 and A9, sucrose and cesium chloride gradient-purified vL2-HA and vA9-HA viruses were analyzed by Western blotting with the same anti-HA antibody. With the same amounts of the two viruses, L2-HA was a faint band, whereas A9-HA was intense (Fig. 7). Assuming that the antibody to HA reacted equally with the same tag on the two proteins in Western blots, the data suggested that there is much less L2 than A9 associated with purified virions. Equal-intensity L2 bands were detected in purified vL2-HA and vA9-HA with polyclonal antibody, confirming that similar amounts of the two virions had been used (Fig. 7).
Fig. 7.

Minimal amounts of L2 are associated with virions. HeLa cells were infected with vL2-HA or vA9-HA, and the virions produced were purified through a sucrose gradient (S) and a cesium gradient (C). For each virus, the same amounts of cell extract and purified particles were resolved by SDS-PAGE and analyzed by Western blotting with MAb to the HA epitope tag (α-HA) and polyclonal antibody to L2 (α-L2). Proteins were detected by chemiluminescence.
Effect of repression of L2 on the stability of a subset of viral membrane proteins.
In our previous studies with the conditional lethal inducible L2 mutant vL2Ri, we noted that processing of the A17 membrane protein and the abundant core proteins were reduced, consistent with the early block in virion assembly demonstrated by electron microscopy (25). However, the overall [35S]methionine labeling pattern was similar in the presence or absence of IPTG, indicating no gross affect on viral protein synthesis. We were curious as to the fate of viral membrane proteins when viral membrane formation was prevented. To address this question, BS-C-1 cells were infected with vL2Ri in the presence or absence of IPTG. After 10 h, the cells were lysed, and the proteins were resolved by SDS-PAGE and analyzed by Western blotting. Cells were infected with T7LacOI, the parent of vL2Ri, in the presence or absence of rifampin as a control. Under the latter conditions, viral membranes form without the D13 lattice and morphogenesis is arrested. In addition to L2, we analyzed 12 viral late proteins by Western blotting. These included several MV transmembrane proteins (A17, A21, A28, D8, F9, and L1), a protein indirectly associated with the MV membrane (A26), the IV scaffold protein (D13), core proteins (A3 and A4), a nonmembrane protein required for crescent membrane formation (A11), and a protein associated with the EV membrane (F13). Analysis of proteins made in cells infected with vT7LacOI showed that rifampin had little effect except for reducing the processing of A3 (Fig. 8). In contrast, there were remarkable differences when the proteins from cells infected with vL2i in the presence or absence of IPTG were compared. As expected, L2 was not detected, and the processing of A3 and A17 was reduced in the absence of IPTG (Fig. 8). More interestingly, some proteins (A11, A21, A26, A28, F9, and L1) were greatly reduced in amount, A17 was slightly reduced, and others (A3, A4, D8, D13, and F13) seemed unaffected. The proteins that were greatly reduced in amount included proteins associated with the entry-fusion complex (A21, A28, F9, and L1) and the formation of viral membranes (A11).
Fig. 8.
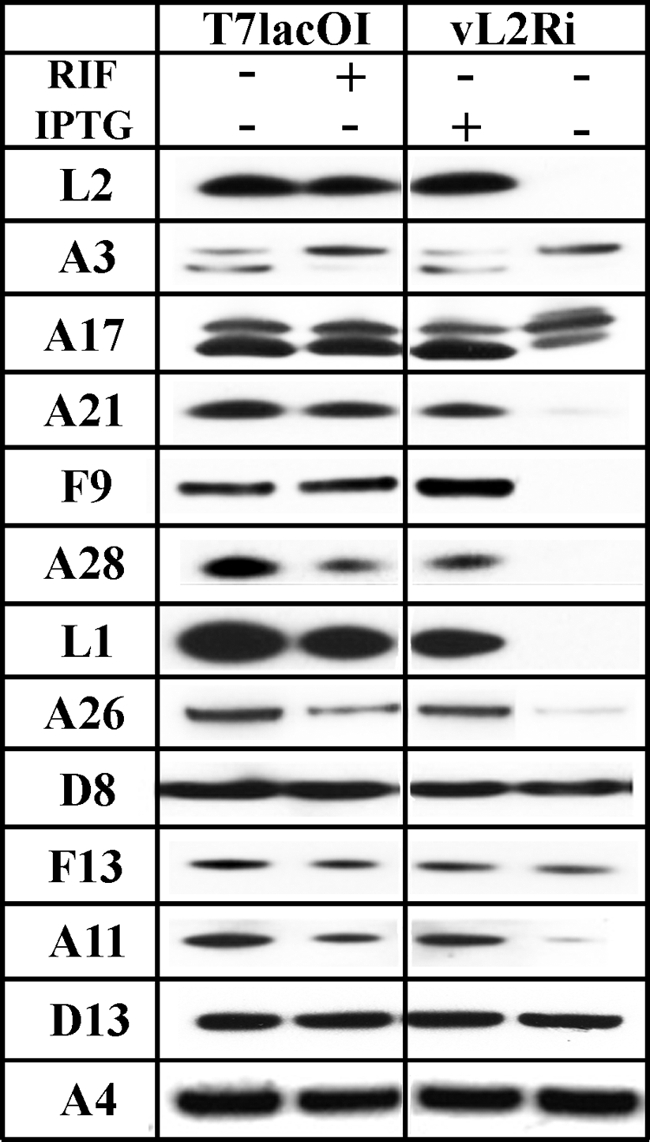
Instability of a subset of proteins when synthesis of L2 is repressed. (A) BS-C-1 cells were infected at a multiplicity of 3 PFU per cell with vT7lacOI in presence (+) or absence (−) of 100 μg of rifampin/ml (RIF) or with the recombinant vL2Ri in presence (+) or absence (−) of 50 μM IPTG. After 12 h, the cells were lysed, and the proteins were resolved by SDS-PAGE and analyzed by Western blotting with antibodies for L2, A3, A17, A21, F9, A28, L1, A26, D8, F13, A11, D13, and A4. Proteins were detected by chemiluminescence.
Pulse-chase experiments were carried out to determine whether the reduced amounts of certain proteins were due to decreased biosynthesis or stability. L1 and F9 were chosen as examples of MV membrane proteins that were unstable and D8 as stable in the absence of L2. Both L1 and F9 are required for virus entry into cells. L1 is myristoylated and has three intramolecular disulfide bonds that are formed shortly after synthesis (3, 4). F9 is related to L1 (20% sequence identity) and also has three intramolecular disulfide bonds but is not myristoylated (7, 41). D8 is a transmembrane protein that enhances virus attachment by binding to chondroitin sulfate on the cell surface (20). BS-C-1 cells were infected with vL2Ri in the presence or absence of IPTG and 6 h later were pulsed for 5 min with 100 μCi of [35S]methionine-cysteine and then chased with excess unlabeled methionine and cysteine. The cells were lysed at intervals during the chase and L1, F9, and D8 were immunoaffinity purified and resolved by SDS-PAGE and autoradiography. By reacting free SH groups with NEM just prior to lysis to prevent oxidation, we were able resolve the slower migrating non-disulfide-bonded L1 and F9 precursors from the more rapidly migrating intramolecular disulfide-bonded forms (Fig. 9) as previously shown for L1 (3). In the presence of IPTG, both forms of L1 were detected immediately after the chase, and the disulfide-bonded form accumulated during the 2-h chase. In the absence of IPTG, the two forms were also detected at zero time. While the precursor decreased with time, there was little increase in the disulfide-bonded form, and the latter disappeared between 60 and 120 min. The result with F9 was similar to L1 except that there was even less of the more rapidly migrating intramolecular disulfide-bonded form in the absence of IPTG (Fig. 9). In contrast, D8 appeared stable in the presence or absence of IPTG (Fig. 9). Thus, L1 and F9 are synthesized but unstable in the absence of L2.
Fig. 9.

Pulse-chase analysis. BS-C-1 cells were mock infected (M) or infected at a multiplicity of 5 PFU per cell with vL2Ri in presence (+) or absence (−) of 50 μM IPTG. After 6 h, the infected cells were labeled with 100 μCi of [35S]methionine-cysteine for 5 min. The cells were washed twice with PBS and chased with an excess unlabeled methionine and cysteine for the indicated times. The cells were lysed, and L1, F9, and D8 proteins were affinity purified, resolved by SDS-PAGE, and detected by autoradiography.
Effect of repression of other proteins on stability.
We investigated whether the instability of certain membrane proteins was specifically due to the absence of L2 or a general phenomenon resulting from the inhibition of viral membrane biogenesis. We took advantage of inducible VACV mutants previously constructed in our laboratory. We tested the effects of repressing two proteins needed for crescent formation: A17 (56) and A11 (35). In addition, we determined the effect of repressing the F9 protein associated with the entry-fusion complex embedded in the MV membrane. BS-C-1 cells were infected with the parental control virus vT7LacOI, vF9Li, vA11Ri, or vA17Li in presence or absence of IPTG. After 12 h, the cells were lysed, and the proteins were resolved by SDS-PAGE and analyzed by Western blotting with the entire set of antibodies used in Fig. 8 plus antibody to the A30 protein that is required for the association of viroplasm with crescent membranes (49). The results obtained by repressing A11 and A17 were virtually identical to that obtained by repressing L2 (Fig. 10). Thus, F9, A21, A28, L1, and A26 were greatly reduced in cells infected with vA17Li and vA11Ri in the absence of IPTG but not in cells infected with vT7LacOI or vF9Li. Other proteins including A3, A4, D8, F13, D13, and A30 were not affected or reduced by only a small amount. Interestingly, both L2 and A11 were stable when A17 was repressed. These results indicated that specific late MV membrane proteins were unstable when the viral membranes were not formed due to repression of L2, A11, or A17.
Fig. 10.
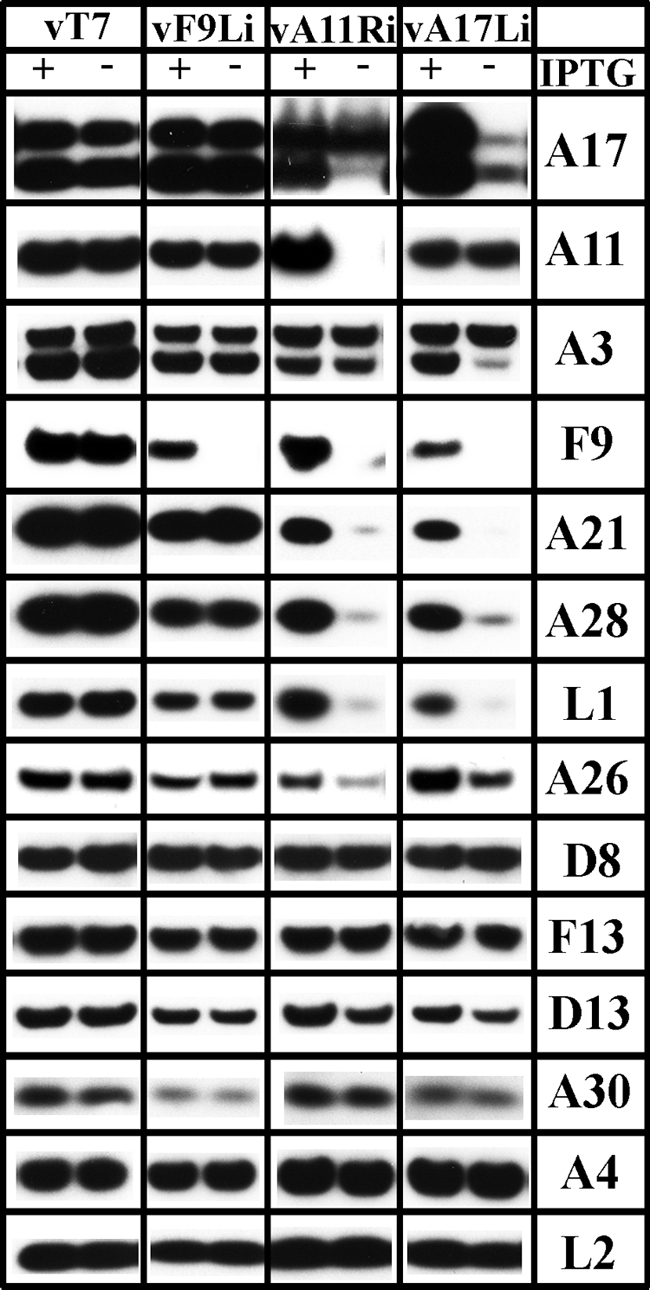
Effects of repression of F9, A11, and A17 on protein stabilities. BS-C-1 cells were infected at a multiplicity of 3 PFU per cell with vT7lacOI (vT7) as a control, vF9Li, vA11Ri, and vA17Li in the presence (+) or absence (−) of IPTG. After 12 h, the cells were lysed, and the proteins were resolved by SDS-PAGE and analyzed by Western blotting with antibodies for A17, A11, A3, F9, A21, A28, L1, A26, D8, F13, D13, A30, A4, and L2. Proteins were detected by chemiluminescence.
DISCUSSION
The construction of a recombinant VACV with a HA-tagged L2 (vL2-HA) under its natural promoter allowed the use of a specific MAb to study the localization of the protein during infection. Since L2 is essential for replication, the ability to construct such a recombinant virus indicated that the tag did not significantly perturb the function of L2. Moreover, there was no difference in plaque size or virus yield between the wild type and vL2-HA. Upon examination of cells infected with the vL2-HA by confocal microscopy, we determined that L2 localized with the ER-resident protein calnexin throughout the cytoplasm and the perinuclear envelope, as well as the perimeter of viral factories, with only light staining in the interior of factories during the first 6 h. The low factory staining may be due to the early promoter driving L2 expression before DNA replication and factory formation and to the cytoplasmic distribution of the ER. It is also possible that the factories are less permeable to the ER and L2 antibodies than the surrounding cytoplasm. The association of L2 with the entire ER network, however, may signify a role in addition to that of viral membrane formation. L2 did not specifically colocalize with markers for the ERGIC or the Golgi apparatus, although there could be some association with these membranes. Expression of L2 in transfected cells indicated that other viral proteins were not needed for ER localization. L2 has two hydrophobic domains near the C terminus, one or both of which likely serves as a transmembrane domain but lacks a KDEL-ER retention motif. The topology of L2 was determined by specifically permeabilizing the plasma membrane with digitonin to allow entry of the MAb into the cytoplasm. The ability of the MAb to label L2 indicated that the N terminus was cytoplasmic, allowing for the possibility of L2 interactions with other viral and cellular proteins. Thus far, however, we have not been able to identify binding partners by immunoprecipitation following detergent lysis of infected cells.
The HA MAb was also used in conjunction with protein A-gold spheres to localize L2 in thin sections of infected cells by transmission electron microscopy. L2 was present in tubular membranes including the perinuclear envelope, confirming the ER distribution determined by confocal microscopy. Within the factory, the tubules were near crescents, invaded the interior of crescents and in some instances were so close to the edge or rim of the crescents that it was not possible to demonstrate discontinuity even by tilting. A similar tubular labeling pattern was found for the luminal ER protein PDI. Other viral transmembrane proteins required for crescent formation, i.e., A17 and A14 localize strongly to viral factories when viewed by confocal microscopy and along the entire crescent, suggesting that they are structural components of the membrane (24, 37, 54, 56). The proteins that are required for viral membrane formation can be divided into two groups based on conditional lethal phenotypes: (i) repression of A17 or A14 leads to the accumulation of large, electron-dense masses of viroplasm resembling the interior of IVs and numerous small vesicles, and (ii) repression of A11, F10, H7, and L2 also results in accumulation of electron dense masses of viroplasm but without vesicles. These results suggest that the second group of proteins are involved with the recruitment of ER membrane and the first group with modeling the membrane into the crescents. The phenotype of a conditional lethal L2 mutant and the localization of L2 during a normal infection suggest that it participates in elongation of crescents by the addition of ER membrane to the growing edge.
We had previously reported that L2 was associated with purified virions and suggested it was present in the viral membrane based on extraction with NP-40 detergent (25). Although we confirmed the association of L2 with virions by electron microscopy of thin sections of infected cells, the immunogold labeling was sparse and predominantly in the interior of virions rather than on the external membrane. Furthermore, the number of gold particles associated with virions was far less than we had previously seen by immunogold labeling of HA-tagged A9 protein, which was localized on the virion membrane (59). We confirmed the much lower amounts of L2 compared to A9 by analyzing equivalent numbers of virions containing HA-tagged L2 and virions containing HA-tagged A9 by Western blotting with the same anti-HA monoclonal antibody. We suspect that the presence of L2 in immature and mature virions is due to trapping during assembly and that it can be released by the NP-40 treatment.
In our previous report (27), we had noted a reduction in the processing of the A17 protein and were curious as to the fate of other viral membrane proteins in the absence of L2. Here, we found that a subset of viral membrane proteins that are components of or are associated with the entry-fusion complex were not detected or greatly reduced in amount when L2 expression was repressed. These entry proteins included A21, A28, L1, and F9. Using the L1 and F9 proteins as models, we demonstrated by pulse-chase experiments that the reduction was due to instability rather than decreased synthesis. Interestingly, another MV membrane protein D8 was not destabilized, nor were the core proteins A3 and A4, the IV scaffold protein D13, or the extracellular envelope protein F13. However, two additional proteins A11 and A26 were greatly reduced. A11 is predicted to have two transmembrane domains near the C terminus like L2 but is not associated with virus particles, and its precise localization within the virus factory has not been reported (35). A26 is not a transmembrane protein but interacts with the A27 protein (19), which is anchored to the MV membrane through interaction with A17, which is not processed when L2 was repressed. A26 was previously reported to be unstable when A27 was deleted (19), raising the possibility that A27 is also unstable when L2 is repressed.
We considered that the effect of L2 repression on protein stability could be a specific property of L2 or a more general effect due to the inhibition of viral membrane formation. To address this question, we determined the stability of proteins when F9, A11 and A17 were repressed. F9 is a viral membrane protein that is not involved in viral membrane biogenesis and predictably did not alter the stability of any of the proteins tested. In contrast, repressing A11 and A17, which are required for crescent membrane formation, had a destabilizing effect similar to that of repressing L2. However, L2 was not destabilized by repression of A11 or A17, which is consistent with the synthesis of L2 before either of those proteins. In another study, it was found that L1 was greatly reduced in amount when H7, another protein required for viral membrane formation was repressed (39). It seems likely that a subset of proteins associated with the viral membrane is dependent on the latter for stability. These proteins may be degraded after ER insertion unless rapidly shunted to the viral membrane or perhaps need to directly insert into the viral membrane, which is absent when L2, A11, A17, or H7 is repressed.
ACKNOWLEDGMENTS
We thank Catherine Cotter and Jeffrey Americo of our laboratory for preparation of cells, P. S. Satheskumar of our laboratory for sharing data, Geoffrey L. Smith (Imperial College), and Gary Cohen (University of Pennsylvania) for antibodies.
The research was supported by the Division of Intramural Research, National Institute of Allergy and Infectious Diseases, National Institutes of Health.
Footnotes
Published ahead of print on 14 September 2011.
REFERENCES
- 1. Alexander W. A., Moss B., Fuerst T. R. 1992. Regulated expression of foreign genes in vaccinia virus under the control of bacteriophage T7 RNA polymerase and the Escherichia coli lac repressor. J. Virol. 66:2934–2942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Betakova T., Wolffe E. J., Moss B. 1999. Membrane topology of the vaccinia virus A17L envelope protein. Virology 261:347–356 [DOI] [PubMed] [Google Scholar]
- 3. Bisht H., Brown E., Moss B. 2010. Kinetics and intracellular location of intramolecular disulfide bond formation mediated by the cytoplasmic redox system encoded by vaccinia virus. Virology 398:187–193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bisht H., Weisberg A. S., Moss B. 2008. Vaccinia virus L1 protein is required for cell entry and membrane fusion. J. Virol. 82:8687–8694 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bisht H., Weisberg A. S., Szajner P., Moss B. 2009. Assembly and disassembly of the capsid-like external scaffold of immature virions during vaccinia virus morphogenesis. J. Virol. 83:9140–9150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Blasco R., Moss B. 1992. Role of cell-associated enveloped vaccinia virus in cell-to-cell spread. J. Virol. 66:4170–4179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Brown E., Senkevich T. G., Moss B. 2006. Vaccinia virus F9 virion membrane protein is required for entry but not virus assembly, in contrast to the related l1 protein. J. Virol. 80:9455–9464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Condit R. C., Moussatche N., Traktman P. 2006. In a nutshell: structure and assembly of the vaccinia virion. Adv. Virus Res. 66:31–124 [DOI] [PubMed] [Google Scholar]
- 9. Da Fonseca F. G., Weisberg A. S., Caeiro M. F., Moss B. 2004. Vaccinia virus mutants with alanine substitutions in the conserved G5R gene fail to initiate morphogenesis at the nonpermissive temperature. J. Virol. 78:10238–10248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dales S., Mosbach E. H. 1968. Vaccinia as a model for membrane biogenesis. Virology 35:564–583 [DOI] [PubMed] [Google Scholar]
- 11. Dales S., Siminovitch L. 1961. The development of vaccinia virus in Earle's L strain cells as examined by electron microscopy. J. Biophys. Biochem. Cytol. 10:475–503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. DeMasi J., Traktman P. 2000. Clustered charge-to-alanine mutagenesis of the vaccinia virus H5 gene: isolation of a dominant, temperature-sensitive mutant with a profound defect in morphogenesis. J. Virol. 74:2393–2405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Demkowicz W. E., Maa J. S., Esteban M. 1992. Identification and characterization of vaccinia virus genes encoding proteins that are highly antigenic in animals and are immunodominant in vaccinated humans. J. Virol. 66:386–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Earl P. L., Cooper N., Wyatt L. S., Moss B., Carroll M. W. 1998. Preparation of cell cultures and vaccinia virus stocks, p. 16.16.1–16.16.3 In Ausubel F. M., et al. (ed.), Current protocols in molecular biology, vol. 2 John Wiley & Sons, Inc., New York, NY [Google Scholar]
- 15. Earl P. L., Moss B. 1998. Characterization of recombinant vaccinia viruses and their products, p. 16.18.1–16.18.11 In Ausubel F. M., et al. (ed.), Current protocols in molecular biology, vol. 2 Greene Publishing Associates/Wiley Interscience, New York, NY [Google Scholar]
- 16. Heuser J. 2005. Deep-etch EM reveals that the early poxvirus envelope is a single membrane bilayer stabilized by a geodetic “honeycomb” surface coat. J. Cell Biol. 169:269–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hiller G., Weber K. 1985. Golgi-derived membranes that contain an acylated viral polypeptide are used for vaccinia virus envelopment. J. Virol. 55:651–659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hollinshead M., Vanderplasschen A., Smith G. L., Vaux D. J. 1999. Vaccinia virus intracellular mature virions contain only one lipid membrane. J. Virol. 73:1503–1517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Howard A. R., Senkevich T. G., Moss B. 2008. Vaccinia virus A26 and A27 proteins form a stable complex tethered to mature virions by association with the A17 transmembrane protein. J. Virol. 82:12384–12391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hsiao J. C., Chung C. S., Chang W. 1999. Vaccinia virus envelope D8L protein binds to cell surface chondroitin sulfate and mediates the adsorption of intracellular mature virions to cells. J. Virol. 73:8750–8761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Husain M., Moss B. 2003. Evidence against an essential role of COPII-mediated cargo transport to the endoplasmic reticulum-Golgi intermediate compartment in the formation of the primary membrane of vaccinia virus. J. Virol. 77:11754–11766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Husain M., Weisberg A. S., Moss B. 2006. Existence of an operative pathway from the endoplasmic reticulum to the immature poxvirus membrane. Proc. Natl. Acad. Sci. U. S. A. 103:19506–19511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Husain M., Weisberg A. S., Moss B. 2007. Sequence-independent targeting of transmembrane proteins synthesized within vaccinia virus factories to nascent viral membranes. J. Virol. 81:2646–2655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krijnse-Locker J., et al. 1996. The role of a 21-kDa viral membrane protein in the assembly of vaccinia virus from the intermediate compartment. J. Biol. Chem. 271:14950–14958 [DOI] [PubMed] [Google Scholar]
- 25. Laliberte J. P., Moss B. 2009. Appraising the apoptotic mimicry model and the role of phospholipids for poxvirus entry. Proc. Natl. Acad. Sci. U. S. A. 106:17517–17521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Lustig S., et al. 2005. Combinations of polyclonal or monoclonal antibodies to proteins of the outer membranes of the two infectious forms of vaccinia virus protect mice against a lethal respiratory challenge. J. Virol. 79:13454–13462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maruri-Avidal L., Domi A., Weisberg A. S., Moss B. 2011. Participation of vaccinia virus L2 protein in the formation of crescent membranes and immature virions. J. Virol. 85:2504–2511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morgan C. 1976. The insertion of DNA into vaccinia virus. Science 193:591–592 [DOI] [PubMed] [Google Scholar]
- 29. Moss B., Rosenblum E. N. 1973. Protein cleavage and poxvirus morphogenesis: tryptic peptide analysis of core precursors accumulated by blocking assembly with rifampicin. J. Mol. Biol. 81:267–269 [DOI] [PubMed] [Google Scholar]
- 30. Moss B., Rosenblum E. N., Katz E., Grimley P. M. 1969. Rifampicin: a specific inhibitor of vaccinia virus assembly. Nature 224:1280–1284 [DOI] [PubMed] [Google Scholar]
- 31. Nagayama A., Pogo B. G. T., Dales S. 1970. Biogenesis of vaccinia: separation of early stages from maturation by means of rifampicin. Virology 40:1039–1051 [DOI] [PubMed] [Google Scholar]
- 32. Nelson G. E., Sisler J. R., Chandran D., Moss B. 2008. Vaccinia virus entry/fusion complex subunit A28 is a target of neutralizing and protective antibodies. Virology 380:394–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Parkinson J. E., Smith G. L. 1994. Vaccinia virus gene A36R encodes a Mr 43–50 K protein on the surface of extracellular enveloped virus. Virology 204:376–390 [DOI] [PubMed] [Google Scholar]
- 34. Punjabi A., Traktman P. 2005. Cell biological and functional characterization of the vaccinia virus F10 kinase: implications for the mechanism of virion morphogenesis. J. Virol. 79:2171–2190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Resch W., Weisberg A. S., Moss B. 2005. Vaccinia virus nonstructural protein encoded by the A11R gene is required for formation of the virion membrane. J. Virol. 79:6598–6609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Risco C., et al. 2002. Endoplasmic reticulum-Golgi intermediate compartment membranes and vimentin filaments participate in vaccinia virus assembly. J. Virol. 76:1839–1855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rodriguez J. R., Risco C., Carrascosa J. L., Esteban M., Rodriguez D. 1997. Characterization of early stages in vaccinia virus membrane biogenesis: implications of the 21-kilodalton protein and a newly identified 15-kilodalton envelope protein. J. Virol. 71:1821–1833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rodriguez J. R., Risco C., Carrascosa J. L., Esteban M., Rodriguez D. 1998. Vaccinia virus 15-kilodalton (A14L) protein is essential for assembly and attachment of viral crescents to virosomes. J. Virol. 72:1287–1296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Satheshkumar P. S., Weisberg A., Moss B. 2009. Vaccinia virus H7 protein contributes to the formation of crescent membrane precursors of immature virions. J. Virol. 83:8439–8450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Schmelz M., et al. 1994. Assembly of vaccinia virus: the second wrapping cisterna is derived from the trans-Golgi network. J. Virol. 68:130–147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Senkevich T. G., White C. L., Koonin E. V., Moss B. 2002. Complete pathway for protein disulfide bond formation encoded by poxviruses. Proc. Natl. Acad. Sci. U. S. A. 99:6667–6672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Senkevich T. G., Wyatt L. S., Weisberg A. S., Koonin E. V., Moss B. 2008. A conserved poxvirus NlpC/P60 superfamily protein contributes to vaccinia virus virulence in mice but not to replication in cell culture. Virology 374:506–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Smith G. L., Law M. 2004. The exit of vaccinia virus from infected cells. Virus Res. 106:189–197 [DOI] [PubMed] [Google Scholar]
- 44. Sodeik B., et al. 1993. Assembly of vaccinia virus: role of the intermediate compartment between the endoplasmic reticulum and the Golgi stacks. J. Cell Biol. 121:521–541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Sodeik B., Griffiths G., Ericsson M., Moss B., Doms R. W. 1994. Assembly of vaccinia virus: effects of rifampin on the intracellular distribution of viral protein p65. J. Virol. 68:1103–1114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Szajner P., Jaffe H., Weisberg A. S., Moss B. 2003. Vaccinia virus G7L protein interacts with the A30L protein and is required for association of viral membranes with dense viroplasm to form immature virions. J. Virol. 77:3418–3429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Szajner P., Weisberg A. S., Lebowitz J., Heuser J., Moss B. 2005. External scaffold of spherical immature poxvirus particles is made of protein trimers, forming a honeycomb lattice. J. Cell Biol. 170:971–981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Szajner P., Weisberg A. S., Moss B. 2004. Evidence for an essential catalytic role of the F10 protein kinase in vaccinia virus morphogenesis. J. Virol. 78:257–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Szajner P., Weisberg A. S., Wolffe E. J., Moss B. 2001. Vaccinia virus A30L protein is required for association of viral membranes with dense viroplasm to form immature virions. J. Virol. 75:5752–5761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Tolonen N., Doglio L., Schleich S., Locker J. K. 2001. Vaccinia virus DNA replication occurs in endoplasmic reticulum-enclosed cytoplasmic mini-nuclei. Mol. Biol. Cell 12:2031–2046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Tooze J., Hollinshead M., Reis B., Radsak K., Kern H. 1993. Progeny vaccinia and human cytomegalovirus particles utilize early endosomal cisternae for their envelopes. Eur. J. Cell Biol. 60:163–178 [PubMed] [Google Scholar]
- 52. Townsley A., Senkevich T. G., Moss B. 2005. Vaccinia virus A21 virion membrane protein is required for cell entry and fusion. J. Virol. 79:9458–9469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Traktman P., Caligiuri A., Jesty S. A., Sankar U. 1995. Temperature-sensitive mutants with lesions in the vaccinia virus F10 kinase undergo arrest at the earliest stage of morphogenesis. J. Virol. 69:6581–6587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Traktman P., et al. 2000. Elucidating the essential role of the A14 phosphoprotein in vaccinia virus morphogenesis: construction and characterization of a tetracycline-inducible recombinant. J. Virol. 74:3682–3695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ulaeto D., Grosenbach D., Hruby D. E. 1995. Brefeldin A inhibits vaccinia virus envelopment but does not prevent normal processing and localization of the putative envelopment receptor P37. J. Gen. Virol. 76:103–111 [DOI] [PubMed] [Google Scholar]
- 56. Wolffe E. J., Moore D. M., Peters P. J., Moss B. 1996. Vaccinia virus A17L open reading frame encodes an essential component of nascent viral membranes that is required to initiate morphogenesis. J. Virol. 70:2797–2808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Yang Z., Bruno D. P., Martens C. A., Porcella S. F., Moss B. 2011. Genome-wide analysis of the 5′ and 3′ ends of vaccinia virus early mRNAs delineates regulatory sequences of annotated and anomalous transcripts. J. Virol. 85:5897–5909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yang Z., et al. 2011. Expression profiling of the intermediate and late stages of poxvirus replication. J. Virol. 85:9899–9808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yeh W. W., Moss B., Wolffe E. J. 2000. The vaccinia virus A9L gene encodes a membrane protein required for an early step in virion morphogenesis. J. Virol. 74:9701–9711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang Y., Moss B. 1992. Immature viral envelope formation is interrupted at the same stage by lac operator-mediated repression of the vaccinia virus D13L gene and by the drug rifampicin. Virology 187:643–653 [DOI] [PubMed] [Google Scholar]