

Abstract
Alphaviruses such as Semliki Forest virus (SFV) are enveloped viruses that infect cells through a low-pH-triggered membrane fusion reaction mediated by the transmembrane fusion protein E1. E1 drives fusion by insertion of its hydrophobic fusion loop into the cell membrane and refolding to a stable trimeric hairpin. In this postfusion conformation, the immunoglobulin-like domain III (DIII) and the stem region pack against the central core of the trimer. Membrane fusion and infection can be specifically inhibited by exogenous DIII, which binds to an intermediate in the E1 refolding pathway. Here we characterized the properties of the E1 target for interaction with exogenous DIII. The earliest target for DIII binding was an extended membrane-inserted E1 trimer, which was not detectable by assays for the stable postfusion hairpin. DIII binding provided a tool to detect this extended trimer and to define a series of SFV fusion-block mutants. DIII binding studies showed that the mutants were blocked in distinct steps in fusion protein refolding. Our results suggested that formation of the initial extended trimer was reversible and that it was stabilized by the progressive fold-back of the DIII and stem regions.
INTRODUCTION
The alphaviruses are members of a genus of small spherical enveloped viruses with positive-sense RNA genomes (reviewed in reference 23). Alphaviruses include a number of medically important pathogens such as Eastern equine encephalitis virus and the emerging pathogen chikungunya virus, which has caused recent epidemics in India (10, 41, 43). Although human infections by pathogenic alphaviruses are increasing, to date there are no vaccines or antiviral therapies available for use in treatment of patients. Well-characterized alphaviruses such as Semliki Forest virus (SFV) and Sindbis virus have been used extensively to study the structure, entry, replication, and biogenesis of this important group of viruses (23).
The alphavirus particle contains an inner core of the viral RNA in a complex with the capsid protein (23). This is surrounded by a lipid membrane containing the transmembrane E2 and E1 proteins, organized as trimers of E2 and E1 (E2/E1) heterodimers and arranged with T = 4 icosahedral symmetry. Alphaviruses infect host cells by binding to receptors at the plasma membrane followed by uptake via clathrin-mediated endocytosis (reviewed in reference 18). The low-pH environment of the endosome then triggers the fusion of the viral and endosome membranes to deliver the nucleocapsid into the cytosol. Endocytic uptake and virus infection are blocked by expression of dominant-negative versions of host proteins involved in endocytosis (e.g., see references 7 and 42), whereas fusion and virus infection are inhibited by neutralizing the low pH of endocytic vesicles (e.g., see references 9 and 16). During entry, the E2 protein binds the virus receptor(s) while E1 mediates membrane fusion.
The structures of the E2/E1 heterodimer and the prefusion and postfusion structures of the E1 protein provide important information about the alphavirus membrane fusion reaction (14, 24, 26, 37, 39, 46). E1 and E2 are both elongated molecules composed primarily of β sheets. E1 contains a central domain, domain I (DI), that connects on one side to domain II (DII), which has the hydrophobic fusion loop at its distal tip. On the other side, DI connects via a linker region to domain III (DIII), an immunoglobulin-like domain that is followed by the stem region and C-terminal transmembrane domain. On the surface of the virus, E1 is arranged tangential to the virus membrane and is largely covered by E2. Upon exposure to low pH, the E2/E1 heterodimer dissociates (47), exposing the E1 fusion loop, which then inserts into the target membrane (12). Monomers of E1 then trimerize and refold to form the stable postfusion homotrimer (48). The structure of the final homotrimer reveals a central core trimer composed of DI and DII (14). DIII folds back to pack against this core trimer, moving toward the target membrane-inserted fusion loop to generate a hairpin-like structure with the fusion loops and transmembrane domains on the same side of the trimer. The conversion of E1 from the metastable prefusion conformation to the final postfusion homotrimer drives the fusion reaction. Flaviviruses such as dengue virus (DV) have a structurally similar membrane fusion protein E, which mediates fusion through a comparable conversion to a membrane-inserted trimeric hairpin (e.g., see references 33 and 34).
Given the important movement and packing of DIII during E1's rearrangement to the final homotrimer, we explored the use of exogenous DIII as a fusion inhibitor (27). We found that alphavirus or dengue virus DIII proteins can specifically bind to E1 or E during the low-pH-triggered fusion reaction. The bound DIII protein acts as a dominant-negative inhibitor of virus fusion and infection. No cross-inhibition of alphaviruses by dengue DIII (or vice versa) is observed. Using an in vitro reconstitution approach, we showed that a truncated form of E1 containing domains I and II and the linker region (DI/II) could form stable trimers on target membranes at low pH (40). These core trimers act as an efficient target for DIII binding, whereas monomeric DI/II does not stably bind DIII. Together, these data suggest that exogenous DIII inhibits fusion by binding to unoccupied sites on a trimeric E1 fusion intermediate, thus inhibiting the fold-back of endogenous DIII.
Here we set out to determine the properties of the viral target for exogenous DIII. We showed that DIII binds to a membrane-inserted E1 intermediate formed at a very early stage of the fusion reaction in a process independent of target membrane receptors. This target was not detectable by the biochemical assays used to monitor the final postfusion homotrimer; thus, the interaction with exogenous DIII represents a novel tool to assay this fusion intermediate. We then used DIII binding to characterize the fusion blocks caused by mutations in the E1 fusion loop, the adjacent ij loop, and the E1 stem region. While all three mutations allowed the formation of the DIII target, they define three distinct stages in the pathway of E1 rearrangement to its final postfusion conformation.
MATERIALS AND METHODS
Cells and viruses.
BHK-21 cells were propagated in complete BHK medium (Dulbecco's modified Eagle's medium containing 5% fetal calf serum, 10% tryptose phosphate broth, 100 U penicillin/ml, and 100 μg streptomycin/ml) at 37 or 28°C. Cholesterol-containing C6/36 mosquito cells and cholesterol-depleted C6/36 cells were cultivated as described for previous studies (45).
The wild-type (WT) SFV used in the study was a well-characterized plaque-purified stock (15) or was derived by in vitro RNA transcription of the WT SFV infectious clone pSP6-SFV4 (30). The SFV E1 point mutants H230A (3) and G91D (8) and the E1 stem deletion mutant Δ20 (28) were constructed by mutagenesis of pSP6-SFV4 as previously described.
Preparation of pyrene and radiolabeled viruses.
Pyrene-labeled WT SFV was prepared essentially as described previously (5). In brief, BHK cells were cultured for 25 h in medium containing 1-pyrenehexadecanoic acid (Invitrogen, Carlsbad, CA) (10 μg/ml). The cells were then infected at multiplicity of infection (MOI) of 5 PFU/cell for 20 h. The labeled virus was purified by banding on a discontinuous sucrose gradient (22) and stored in aliquots at −80°C until use. Radiolabeled WT SFV was prepared by infecting BHK cells at high MOI and overnight incubation in methionine-free medium containing Express labeling mix (Perkin Elmer Life and Analytical Sciences) (83 μCi/ml). Labeled virus was purified and stored as described above. Radiolabeled H230A and G91D viruses were prepared similarly, but BHK-21 cells were infected by electroporation with in vitro-transcribed virus RNA and radiolabeled overnight at 28°C using 133 μCi/ml (3, 8).
Expression, refolding, and purification of recombinant proteins.
The proteins used in this study were N-terminally hexahistidine-tagged SFV domain III with the stem region and N-terminally hexahistidine-tagged DV domain III, previously termed SFV HDIIIS and DV HDIII (27). For simplicity, these proteins are referred to here as SFV DIII and DV DIII, respectively. The proteins were generated by expression in Escherichia coli strain BL21(DE3), refolding in vitro, and purification on a Superdex G-75 gel filtration column (GE Healthcare) (27).
Liposomes.
Liposomes were prepared by freezing-thawing and extrusion of the lipid mixture through two stacked 200-nm-pore-size polycarbonate filters using a Liposofast mini-extruder (Avestin, Ottawa, Canada) as described previously (5). Complete liposomes contained a 1:1:1:1.5 molar ratio of 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoethanolamine (POPE), bovine brain sphingomyelin (Avanti Polar Lipids, Alabaster, AL), and cholesterol (Steraloids, Inc., Wilton, NH). Liposomes without cholesterol contained a POPC/POPE/sphingomyelin molar ratio of 1:1:1. The liposomes were prepared in HNE buffer (5 mM HEPES, 150 mM NaCl, 0.1 mM EDTA, pH 7.0) and stored in N2 at 4°C for up to 2 weeks.
Virus-liposome fusion assay.
Virus fusion with target liposomes was monitored using a lipid mixing assay that follows the decrease in the excimer signal of pyrene-labeled virus (2, 5). Pyrene-labeled virus was mixed with liposomes at a final liposomal lipid concentration of 200 μM and the indicated final concentration of SFV or DV DIII protein. The mixture was incubated for 5 min in a stirred thermostated cuvette at 20°C and then adjusted to pH 5.5 by the addition of a pretitrated volume of 0.3 M morpholineethanesulfonic acid (MES), pH 4.85. The pyrene excimer signal was monitored by an Aminco-Bowman AB-2 fluorometer (Spectronic Unicam, Rochester, NY), using excitation at 343 nm, emission at 480 nm, and a 470-nm-pore-size cutoff filter in the emission beam. After low-pH treatment, the reaction was neutralized with a pretitrated amount of 0.11 M triethanolamine. The detergent octaethylene glycol monododecyl ether (C12E8) was added to a final concentration of 22 mM to produce complete dilution of the pyrene excimer. By measuring the excimer fluorescence, the extent of fusion was calculated before the low-pH treatment and after detergent addition as 0 and 100% fusion, respectively.
Virus-liposome association.
Radiolabeled virus was incubated with 200 μM liposomes at 20°C for 1 min at pH 5.5 in the presence of either SFV DIII or DV DIII. The samples were adjusted to pH 8 and 40% sucrose in a final volume of 0.6 ml. The mixture was layered over a 60% sucrose cushion and overlaid with 1 ml (wt/vol) of 25% sucrose and 0.3 ml (wt/vol) of 5% sucrose prepared in 50 mM Tris (pH 7.4)–100 mM NaCl. The gradients were centrifuged in a TL-55 rotor at 54,000 rpm for 3 h at 4°C and collected in 7 fractions. The percentage of total virus radioactivity associated with the liposomes in the top three fractions was determined as previously described (38).
Interaction of DIII with SFV E1.
To measure the interaction of SFV DIII with cell-bound virus (27), radiolabeled WT SFV or G91D or H230A mutant viruses were bound to cells for 90 min on ice with shaking and treated at pH 5.5 for 1 min at 37°C in the presence of 2 μM SFV or DV DIII. Low-pH treatment was performed on ice for 0, 5, 10, or 30 s (see Fig. 2). Cells were then washed at neutral pH and solubilized in lysis buffer (50 mM Tris [pH 7.4], 100 mM NaCl, 1% Triton X-100, 1 mM EDTA, 1 μg/ml pepstatin A, 50 μg/ml leupeptin, 0.1% bovine serum albumin [BSA], 100 μg/ml aprotinin, 1 mM phenylmethylsulfonyl fluoride [PMSF]).
Fig. 2.
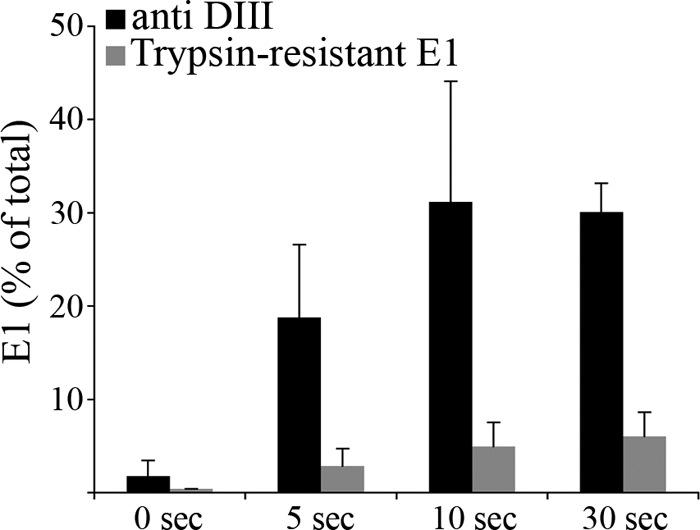
The target for exogenous domain III binding is formed at an early stage of the fusion reaction. BHK cells were incubated with radiolabeled SFV for 90 min on ice. The cells and bound virus were then treated for the indicated times on ice at pH 5.5 in the presence of 2 μM SFV DIII. The cells were washed at neutral pH and lysed. Aliquots of the lysates were immunoprecipitated with antibody to the hexahistidine tag on DIII or with polyclonal antiserum against the E1-E2 proteins, and the retrieved E1 was quantitated by SDS-PAGE and phosphorimaging. The amount of E1 retrieved by exogenous DIII is expressed as a percentage of the total amount of E1 retrieved with the polyclonal serum (black bars). The formation of trypsin-resistant E1 (gray bars) was determined as described in Materials and Methods, using parallel samples treated for the indicated times on ice at pH 5.5 in the absence of exogenous DIII. Data shown represent the mean and standard deviation of the results of 4 independent experiments (5-, 10-, and 30-s time points) or the average and range of the results of 2 independent experiments (0-s time point).
To measure the interaction of DIII with cell surface-expressed WT or Δ20 E1, BHK-21 cells were electroporated with viral RNA and incubated at 37°C for 6 h. The cells were starved for 15 min at 37°C in medium that was free of methionine and cysteine, pulse-labeled for 20 min with 150 μCi of [35S]methionine-cysteine/ml, and chased for 1 h in medium containing excess methionine and cysteine. The cells were treated for 1 min at 37°C and the indicated pH in the presence of SFV or DV DIII under additional conditions (see Fig. 6). Cells were washed and lysed as described above.
Fig. 6.
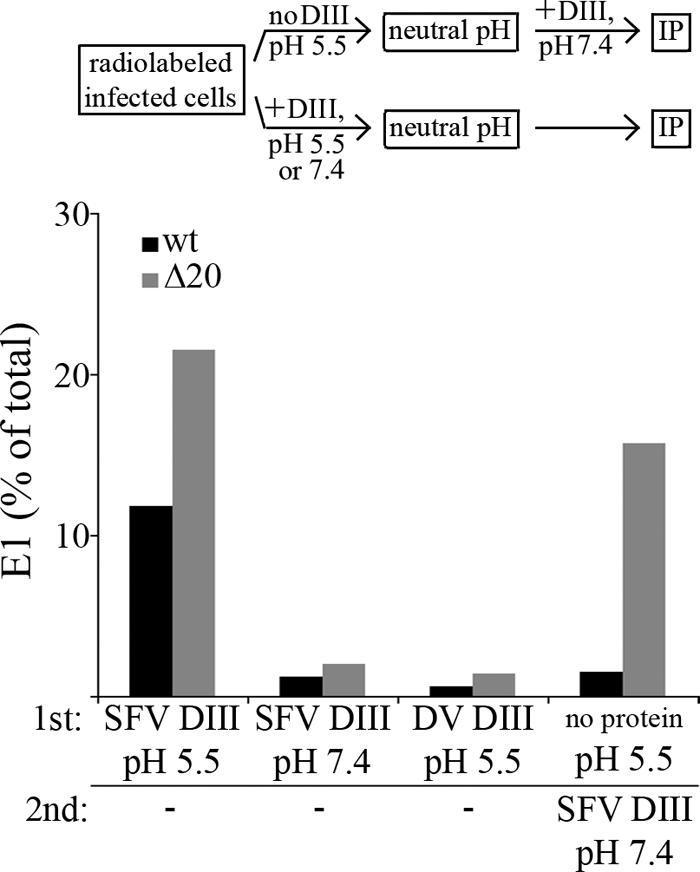
A deletion in the E1 stem region inhibits the packing of endogenous domain III. BHK-21 cells were electroporated with RNA from WT (black bars) or Δ20 (gray bars) SFV and incubated at 37°C for 6 h. The cells were pulse-labeled for 20 min and chased for 60 min to allow transport of radiolabeled envelope proteins to the cell surface. The cells were then treated at neutral or low pH for 1 min at 37°C in the presence of 2 μM SFV or DV DIII. One set of samples was treated at low pH in the absence of DIII and then subjected to a second treatment at neutral pH in the presence of SFV DIII (see schematic). Following these treatments, cells were lysed and E1 was retrieved by the use of DIII-specific antibody as described for Fig. 2. Note that the total E1 value includes both cell surface and intracellular E1, thus accounting for the reduced retrieval compared to experiments performed with purified virus. The data shown are a representative example of the results of two independent experiments.
Immunoprecipitation.
Retrieval of E1 by the use of bound DIII was determined by immunoprecipitation as previously described (27). In brief, aliquots of the lysates were immunoprecipitated with rabbit polyclonal serum to E2/E1 (19), the acid conformation-specific E1 monoclonal antibody (MAb) E1a-1 (1, 19), an MAb to the hexahistidine tag on DIII (Sigma, St. Louis, MO), or a preimmune rabbit serum. Samples were analyzed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and quantitated by phosphorimaging (29). The level of retrieved E1 was expressed as a percentage of the total E1, defined as the E1 immunoprecipitated with the rabbit polyclonal serum to E2/E1.
Assays of E1 homotrimer formation.
Radiolabeled virus was bound to BHK-21 cells on ice and treated at the indicated pH under the indicated conditions. The cells were lysed (1% Triton X-100 in phosphate-buffered saline [PBS]), and the cellular debris was removed by centrifugation. The SDS resistance of the E1 homotrimer was assayed by treating aliquots of the lysates with 4% SDS at 30°C for 3 min. Parallel aliquots were treated at 95°C to disrupt the homotrimer and used to determine the total E1 level. Samples were analyzed by SDS-PAGE and phosphorimaging. The amount of E1 migrating at the homotrimer position at 30°C was compared to the total E1. Trypsin-resistant E1 homotrimers were quantitated by treatment of aliquots of the cell lysates with 100 μg tosylamino-phenylethyl chloromethyl ketone (TPCK)-trypsin/ml at 37°C for 10 min, followed by quenching with a 3-fold excess of soybean trypsin inhibitor. The samples were then analyzed by SDS-PAGE and phosphorimaging. The amount of trypsin-resistant E1 was expressed as a percentage of the amount of E1 recovered from the cell lysate incubated in the absence of trypsin.
Analysis of E1 stem packing.
The packing of the E1 stem in the postfusion homotrimer was assayed using an anti-peptide antibody generated against the N-terminal half of the stem as previously described (29). Radiolabeled virus was prebound to BHK-21 cells on ice and treated at pH 5.5 for 1 min at 37°C. The cells were solubilized in 1% Triton X-100 in PBS, and the monomeric E1 was removed by digestion with 100 μg/ml TPCK-trypsin for 15 min at 37°C. As a control, an aliquot of each sample was denatured with 1% SDS to expose the stem, using 3 cycles of incubation for 3 min at 95°C, followed by dilution in 1% Triton X-100–PBS to a final SDS concentration of 0.05%. The samples were immunoprecipitated with a polyclonal serum against E2-E1 proteins, affinity-purified antibody to the stem (29), or preimmune serum and analyzed by SDS-PAGE and phosphorimaging.
Statistical analysis.
The Prism program (GraphPad Software, Inc.) was used for statistical evaluation of data as indicated, using t tests to generate two-tailed P values. A P value (p) of <0.05 was considered to indicate a significant difference.
RESULTS
Effect of DIII proteins on SFV membrane association and liposome fusion.
Our previous studies of DIII inhibition were performed using virus prebound to cells on ice and then treated at low pH in the presence or absence of DIII (27). We observed inhibition of fusion using assays of either virus infection (full fusion) or the dilution of the lipid probe pyrene from the virus membrane (lipid mixing). Exogenous DIII also inhibited infection when present during alphavirus endocytic entry. In all of these assays, the virus interacts with the cellular receptor, a condition that may itself alter the arrangement of the envelope proteins and potentiate their interaction with exogenous DIII (11). We therefore used liposome target membranes to test whether DIII could inhibit fusion in the absence of virus-receptor binding.
Liposomes were mixed with pyrene-labeled SFV and domain III proteins. Note that throughout this paper, “SFV DIII” is used to designate the most inhibitory form of DIII containing the N-terminal His tag and C-terminal stem region (see Materials and Methods for details). The fluorescence emission of the pyrene excimer was recorded during incubation at 20°C (Fig. 1A). Lipid mixing occurred rapidly when the control sample was adjusted to pH 5.5 (curve a), in keeping with the reported results of previous studies (2, 5). Exogenous SFV DIII caused a dose-dependent inhibition of virus fusion (curves c to f). Maximal inhibition was observed at a concentration of 20 μM SFV DIII, with lipid mixing reduced by ∼60% compared to buffer control results (curve f). Higher concentrations of SFV DIII did not cause further inhibition (data not shown). Inhibition was specific, since fusion in the presence of 20 μM DV DIII was similar to that seen in the presence of buffer alone (curve b versus curve a). SFV DIII inhibition of virus-liposome fusion was less complete than inhibition of virus-cell fusion (27), which could reflect differences in the assays, target membrane concentration and composition, kinetics, and/or the accessibility or geometry of the fusion site.
Fig. 1.
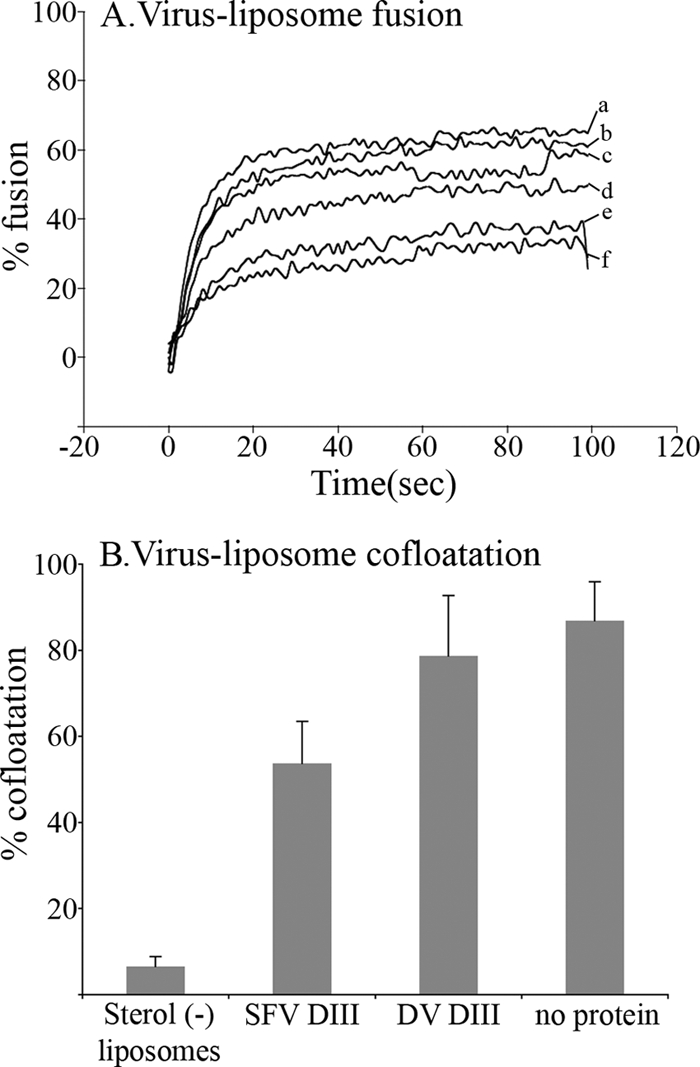
Exogenous domain III impairs SFV fusion without significantly affecting virus-membrane association. (A) Virus-liposome fusion activity in the presence of increasing concentrations of DIII. Pyrene-labeled SFV was mixed with complete liposomes plus buffer alone (a), 20 μM DV DIII (b), or SFV DIII at a concentration of 5 μM (c), 10 μM (d), 15 μM (e), or 20 μM (f). The fusion reaction was triggered at pH 5.5 at time zero, and the fluorescence signal was recorded in real time during incubation at 20°C. (B) Effect of DIII on virus-liposome association. Radiolabeled SFV was mixed with complete liposomes in the presence of control buffer, 20 μM SFV DIII, or 20 μM DV DIII or with cholesterol-deficient liposomes, treated for 1 min at pH 5.5 and 20°C, and adjusted to neutral pH. The virus-liposome association was analyzed by cofloatation on a discontinuous sucrose gradient; data are expressed as the percentage of the total virus in the gradient present in the top 3 fractions. Data shown in panel B represent the average of the results of two independent experiments; bars indicate the range.
Based on our previous results, we hypothesized that the DIII target is a trimeric membrane-inserted intermediate generated during the E1 low-pH conformational change (27). However, we were unable to directly test the effect of DIII on E1 membrane insertion by the use of our previous cell-based virus fusion assays. We here evaluated membrane insertion by the use of a virus-liposome cofloatation assay. Radiolabeled SFV was mixed with liposomes and domain III proteins and incubated at pH 5.5 for 1 min to trigger E1-membrane interaction. The liposomes and bound virus particles were separated from unbound virus by floatation on sucrose gradients. As shown in Fig. 1B, SFV efficiently associated with complete liposomes, which support E1-membrane insertion and fusion, but did not associate with cholesterol-deficient liposomes, which do not support insertion and fusion. The presence of SFV DIII versus control DV DIII did not significantly affect the interaction of SFV with complete target liposomes (P = 0.0997), even though SFV DIII strongly inhibits virus-liposome fusion. The small reduction in the interaction that was observed in the presence of SFV DIII was similar to that observed with sphingolipid-deficient liposomes that are fusion deficient but support E1 insertion (6). Our data thus indicate that SFV DIII inhibits the initial lipid-mixing step of fusion without having a substantial effect on E1-membrane interaction and support the hypothesis that the target of DIII is a membrane-inserted trimeric intermediate.
Formation of the target for exogenous domain III.
Brief low-pH treatment of SFV at 4°C in the presence of target membranes causes the dissociation of the E1/E2 dimer without triggering the fusion of the virus membrane (2, 17). Under these conditions, the trypsin-and-SDS-resistant E1 homotrimer does not form, in keeping with the absence of the final postfusion trimer in the cold-arrested intermediate. To further define the intermediate, we assayed for the formation of the DIII target under the described conditions.
Radiolabeled SFV was prebound to cells and treated on ice at pH 5.5 in the presence or absence of SFV DIII. The cells were lysed, and the percentage of E1 interacting with DIII was determined by coimmunoprecipitation. Parallel samples treated in the absence of DIII were tested for trypsin resistance. The E1 interaction with exogenous DIII was clearly detectable after 5 to 10 s of low-pH treatment, time points at which the amount of trypsin-resistant E1 was negligible (Fig. 2).
Alphavirus membrane fusion is strongly promoted by the presence of cholesterol in the liposome or cellular target membrane (21, 36, 44). In the absence of cholesterol, E1 converts to a trypsin-and-SDS-resistant form at low pH, indicating formation of the E1 homotrimer, but complete stem fold-back and packing do not occur (29). This might reflect a change in the core trimer when it is generated in the absence of cholesterol. We therefore tested the cholesterol dependence of the target for DIII binding. Radiolabeled SFV was prebound to control and cholesterol-depleted C6/36 mosquito cells and treated at pH 5.5 for 1 min at 37°C in the presence of either SFV DIII or DV DIII (Fig. 3). There was no significant difference in the amounts of E1 retrieved by SFV DIII in control versus sterol-depleted cells (P = 0.4872). Together, the results of these experiments thus demonstrate that the target for DIII binding is formed at a very early stage in the fusion reaction but does not require cholesterol in the target membrane.
Fig. 3.
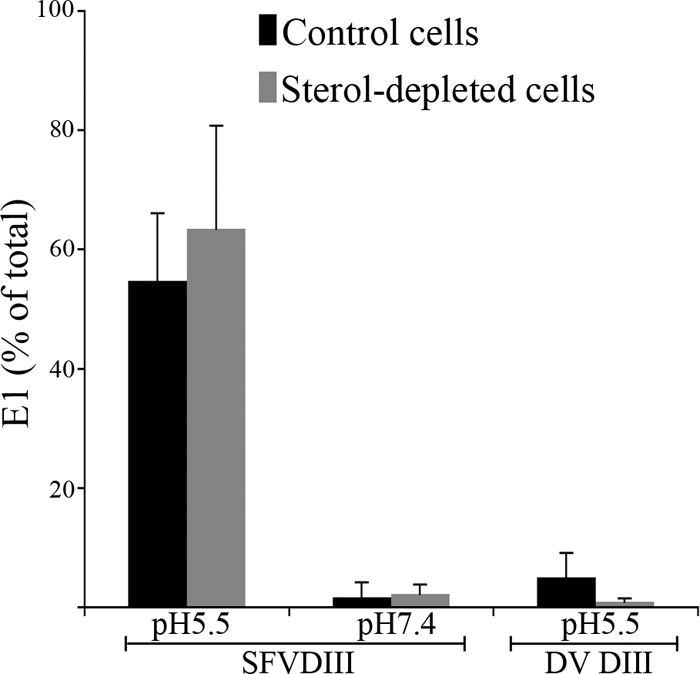
Formation of the core trimer is independent of target membrane cholesterol. Monolayers of control (black bars) and cholesterol-depleted (gray bars) C6/36 mosquito cells were incubated with radiolabeled SFV for 90 min on ice. The cells and bound virus were treated at pH 5.5 or 7.4 for 1 min at 37°C in the presence of either SFV DIII or DV DIII. The cells were lysed, and the percentage of E1 interacting with SFV DIII was assessed by immunoprecipitation as described for Fig. 2. Data shown represent the average and range of the results of 2 independent experiments.
Exogenous DIII interaction destabilizes the E1 trimer.
Our data to date suggest that the target of DIII protein interaction is a trimeric membrane-inserted intermediate. While our previous data indicated that E1 retrieved by DIII binding was in a trypsin-resistant conformation, this retrieval was performed using relatively low concentrations of DIII, conditions under which some fold-back of endogenous DIII might have occurred (27). We hypothesized that full occupancy of the DIII binding sites on E1 completely blocks the fold-back of all three E1 subunits, thus blocking formation of the SDS-and-trypsin-resistant E1 trimer.
We therefore tested the effect of increasing concentrations of DIII on the stability of the E1 trimer (Fig. 4). The proportion of E1 in the SDS-and-trypsin-resistant conformation showed a dose-dependent decrease when low-pH treatment was carried out in the presence of SFV DIII. DV DIII had no effect. At the highest concentration of SFV DIII, most of the E1 trimer was destabilized and no longer showed either SDS or trypsin resistance. This suggests that DIII was interacting with extended E1 trimers in which none of the endogenous DIII had packed against the core trimer. The interaction of exogenous DIII with viral E1 thus defines a distinct membrane-inserted fusion intermediate that, in its earliest form, is not detectable by the biochemical methods used to analyze the stable postfusion E1 homotrimer.
Fig. 4.
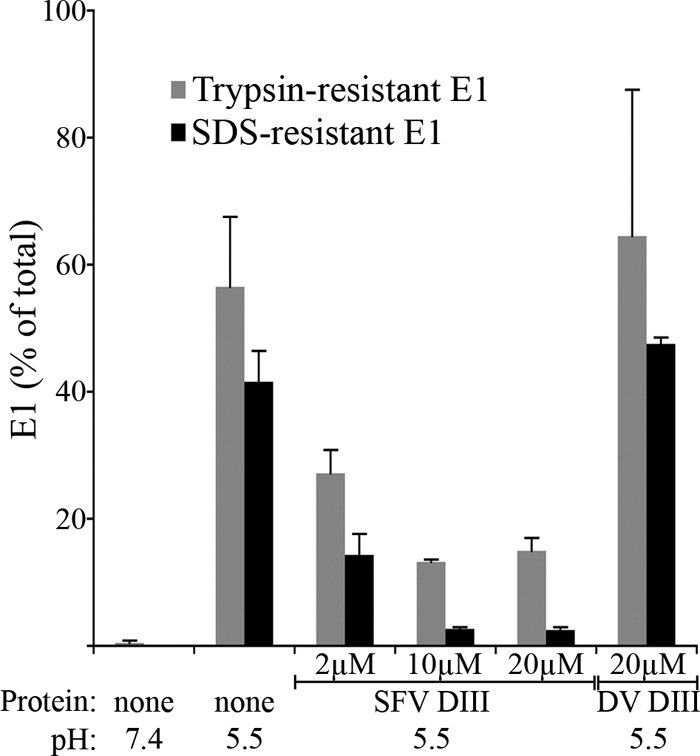
Binding of exogenous domain III reduces the stability of the E1 homotrimer. BHK cells were incubated with radiolabeled SFV for 90 min on ice. The cells were then washed, and the cell-bound virus was treated at neutral or low pH in the presence of control buffer or the indicated concentrations of SFV or DV DIII. The cells were washed at neutral pH and lysed, and aliquots of the lysates were assayed for E1 trypsin resistance (gray bars) as described in Materials and Methods. Parallel aliquots were used to determine the resistance of the E1 homotrimer to treatment with SDS sample buffer at 30°C (black bars). Data shown represent the average and range of the results of 2 independent experiments.
DIII interaction with the G91D fusion loop mutant.
Given the properties of the DIII target described above, the DIII-E1 interaction can be used as an important and novel tool to analyze the conformation of E1 during the fusion pathway. We therefore used DIII binding to analyze three previously characterized SFV fusion mutants, as described in detail in this and the following sections. All three mutants are completely blocked with respect to membrane fusion, including the initial lipid mixing, but they progressed to different points in the E1 rearrangement.
The SFV E1 G91D mutant contains a lethal glycine-to-aspartate substitution at position 91 in the E1 fusion loop at the tip of domain II (20, 25). This mutation does not inhibit low-pH-triggered E1 insertion into the target membrane or reactivity with the acid conformation-specific MAb E1a-1 but blocks formation of the stable E1 trimer, as assayed by sucrose gradient sedimentation and SDS and trypsin resistance.
We used DIII binding to investigate whether the G91D mutant was able to form the trimeric DIII target. Radiolabeled WT or G91D SFV was prebound to cells, treated for 1 min at pH 5.5 in the presence of SFV DIII, and analyzed for DIII-E1 binding by coimmunoprecipitation (Fig. 5A). Both WT E1 and the G91D mutant E1 were efficiently bound by exogenous SFV DIII, with the G91D mutant showing a slight increase in the binding level compared to the WT. The interaction required low-pH treatment and was not observed for the DV DIII.
Fig. 5.
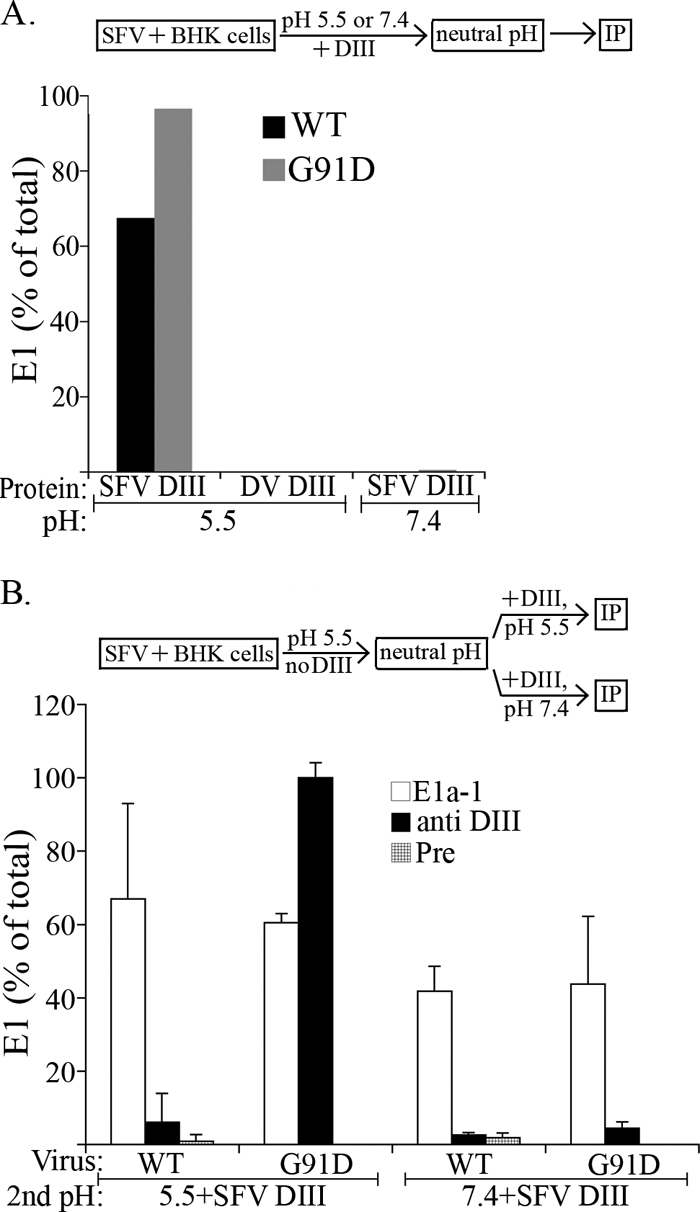
Formation of the core trimer is reversible in the G91D fusion loop mutant. (A) Formation of the target of SFV DIII binding in WT and G91D SFV. BHK cells were incubated with either radiolabeled WT SFV (black bars) or the G91D mutant (gray bars) on ice for 90 min, treated at the indicated pH for 1 min at 37°C in the presence of 2 μM SFV or DV DIII, washed in neutral pH buffer, and lysed. E1 retrieval with antibody to exogenous DIII was assayed and quantitated as described for Fig. 2. Data shown are a representative example of the results of two independent experiments. IP, immunoprecipitation. (B) Reversibility of core trimer formation in the G91D mutant. BHK cells were incubated on ice with WT or mutant SFV as described for panel A, treated at pH 5.5 for 1 min at 37°C in the absence of DIII, and returned to neutral pH (see schematic). Cells were then incubated at 37°C in the presence of exogenous SFV DIII for 1 min at the indicated pH, washed in neutral pH buffer, and lysed. Parallel aliquots of the lysates were precipitated with an E1 acid conformation-specific MAb (E1a-1; white bars), an antibody that recognizes exogenous SFV DIII (anti DIII; black bars), and a control preimmune serum (Pre; crosshatched bars). The percentage of E1 retrieved with each antibody was quantitated by comparison with retrieval by polyclonal antiserum to E1/E2, as described for Fig. 2. Data shown represent the average and range of the results of 2 independent experiments.
This result indicated that, although the G91D mutant was incapable of forming the final E1 homotrimer, it was able to form an extended trimeric intermediate that can bind exogenous DIII. We then asked whether this G91D trimer could be generated at low pH and remain stable for subsequent DIII binding. Radiolabeled WT or G91D SFV was prebound to cells, treated for 1 min at pH 5.5 in the absence of DIII, and returned to neutral pH. The cells and bound virus were then incubated with exogenous DIII for 1 min at either neutral pH or pH 5.5. The cells were lysed and immunoprecipitated with the acid conformation-specific MAb E1a-1 or with an antibody specific for exogenous DIII (Fig. 5B). As expected, during the first treatment at low pH, WT SFV efficiently formed the E1 homotrimer, as shown by its reactivity with MAb E1a-1. However, this end-stage E1 homotrimer was no longer accessible for exogenous DIII binding during a second incubation at either neutral or low pH, in keeping with our previous results (27). In contrast, the G91D mutant E1 was able to bind exogenous DIII during a second low-pH incubation. Mutant E1-DIII binding required incubation at low pH. Our previous studies conducted with truncated E1 proteins showed that the DIII binding step does not require low pH per se (40) (see also Fig. 6 below). Thus, the results suggest that G91D E1 can form an extended prehairpin homotrimer but that this intermediate is reversible upon return to neutral pH. It can be captured and stabilized by binding exogenous DIII.
DIII interaction with the Δ20 stem deletion mutant.
We previously showed that a specific sequence of the SFV E1 stem region is not required for membrane fusion but that a minimal stem length is required (28). A 20-amino-acid deletion within the stem (Δ20) completely blocked the ability of E1 to mediate cell-cell fusion, and fusion was rescued by the addition of 11 alanine residues. Although it is inactive with respect to fusion, Δ20 E1 can still form an E1 homotrimer that is resistant to trypsin and SDS (28). Here, we used exogenous DIII to characterize the Δ20 core trimer and the extent of E1 fold-back.
Since the Δ20 deletion affects virion assembly (28), these experiments were performed using BHK cells electroporated with WT or Δ20 infectious viral RNA. Cells were pulse-labeled and chased to allow envelope protein transport to the plasma membrane (28). Cells were then incubated for 1 min at pH 5.5 in the presence of exogenous DIII proteins, and E1-DIII interaction was assessed by coimmunoprecipitation. The Δ20 E1 protein reproducibly showed more efficient binding to exogenous DIII than did the WT E1, while no significant retrieval was observed using neutral pH treatment or DV DIII (Fig. 6). We also tested the efficiency of fold-back of the endogenous DIII, which might have been compromised by the stem deletion. Pulse-labeled cells expressing the WT or Δ20 proteins were treated at low pH in the absence of exogenous DIII to trigger homotrimer formation. The cells were then incubated with SFV DIII at neutral pH, and the amount of E1 interacting with exogenous DIII was determined. The WT E1 showed no interaction with DIII under these conditions, in agreement with the full occupancy of the binding sites by endogenous DIII during final hairpin formation. In contrast, the Δ20 mutant showed retrieval levels that were ∼75% of those observed when DIII was included in the initial treatment at low pH. These results indicated that the stem deletion allowed efficient formation of a stable core trimer but greatly decreased the folding back of the endogenous DIII.
DIII interaction with the H230A ij loop mutant.
The SFV H230A mutant contains an alanine substitution of a highly conserved histidine residue at E1 position 230 in the ij loop, which lies adjacent to the fusion loop at the tip of domain II. The H230A mutant is blocked in fusion and infection, but its interaction with target membranes and the formation of the SDS-and-trypsin-resistant homotrimer are comparable to those seen with WT SFV (3). Thus, this mutant is blocked in a late stage of the fusion pathway, which we hypothesize involves a direct or indirect effect of H230A on the packing of the outer trimer layer (DIII and stem) against the core trimer (3, 4). We therefore set out to define these late steps in E1 H230A trimerization.
We used DIII binding to evaluate the ability of the H230A mutant to form the extended intermediate. Radiolabeled WT and H230A mutant virus were prebound to cells as described above and treated at low pH in the presence of SFV DIII. Retrieval of H230A E1 by DIII was comparable to that of WT SFV, was low pH dependent, and did not occur with DV DIII (Fig. 7A). Thus, the fusion block in H230A was not due to a detectable defect in the formation of the DIII target.
Fig. 7.

The fusion-block H230A mutation does not affect core trimer formation or proximal stem packing. (A) Formation of the target of SFV DIII binding in WT and H230A SFV. BHK cells were incubated with radiolabeled WT SFV or H230A mutant on ice and treated at low pH in the presence of exogenous DIII, and E1 was retrieved by the use of DIII-specific antibody, all as described for Fig. 5A. The data shown are a representative example of the results of two independent experiments. (B) Packing of the proximal stem region in WT and H230A SFV. WT and H230A mutant viruses were prebound to BHK cells on ice as described for panel A, treated for 1 min at pH 5.5 at 37°C, and adjusted to neutral pH. The cells were lysed and digested with trypsin to remove monomeric E1 as described in Materials and Methods. The resultant E1 homotrimers were denatured with SDS where indicated. Aliquots of the samples were precipitated with polyclonal serum to E2/E1, antibody to the proximal stem region (29), and control preimmune serum. The amount of retrieved E1 was expressed as a percentage of the total amount of E1 retrieved using the polyclonal antiserum. Data shown represent the average and range of the results of 2 independent experiments.
We then used stem3, an antibody directed against the N-terminal third of the stem region (29), to test the packing of the stem to the core trimer of WT versus H230A E1 (Fig. 7B). Viruses were prebound to cells and treated at low pH. Trypsin-resistant E1 homotrimers were isolated and tested for reactivity with the stem3 antibody versus a polyclonal antibody to the SFV envelope proteins. Both the WT and H230A mutant homotrimers were poorly recognized by the stem3 antibody (white bars), in keeping with this region of the stem being packed tightly against the core trimer (29). The stem3 epitope was present on both homotrimers, as shown by efficient stem3 recognition following SDS denaturation of the homotrimers (black bars). Taken together, the current and previous data suggest that the late-stage fusion block in H230A occurs subsequent to formation of the core trimer, fold-back of E1 domain III, and packing of the N-terminal region of the stem.
DISCUSSION
Here we describe the properties of the E1 target for exogenous DIII. This fusion intermediate was rapidly generated after low-pH-induced dissociation of the E2/E1 heterodimer but without a detectable requirement for the virus-receptor interaction or target membrane cholesterol. Binding of DIII blocked lipid mixing of the virus and target membranes but did not inhibit E1-membrane insertion. In the presence of excess DIII, the E1 intermediate exhibited neither SDS nor trypsin resistance, presumably reflecting a complete block in fold-back of endogenous DIII. Previous in vitro experiments showed that DIII binds to trimers of E1 DI/II proteins but not to monomers (40). This agrees with the postfusion trimer structure, which demonstrates packing of DIII into the groove formed between two E1 proteins in the trimer (14). While it is possible that dimers are generated during the core trimer assembly process, our electron microscopy studies of DI/DII membrane interactions have not detected dimers (40, 49). Thus, our current and previous data strongly support the idea that the initial DIII target is a membrane-inserted extended E1 trimer. As E1 refolds to the hairpin, exogenous DIII can bind to available sites until formation of the postfusion trimer is complete. The E1 extended intermediate was not detectable by assays for completely or partially folded-back trimers, which rely on the increased stability of these conformations. Thus, our results are also important in demonstrating that binding of exogenous DIII can be used to follow this early fusion intermediate.
G91D fusion loop mutation.
We used exogenous DIII as a tool to define the E1 conformational changes in several previously described fusion-negative E1 mutants (summarized in Fig. 8). The G91D mutation in the fusion loop blocks E1 at a point following protein-membrane insertion (20, 25). While the mutant protein responds to low pH, as evinced by its reactivity with MAb E1a-1, no E1 trimerization is observed using SDS resistance, trypsin resistance, or sucrose gradient sedimentation assays (20). Here we showed that the G91D mutant G91D E1 formed an extended prehairpin homotrimer that bound exogenous DIII. In keeping with the lack of a biochemically detectable postfusion homotrimer, G91D E1 did not complete hairpin formation and remained an extended intermediate available for interaction with exogenous DIII. Unexpectedly, however, this G91D E1 intermediate was reversible, as shown by its capture when DIII was added during a second low-pH pulse but not during a second incubation at neutral pH. This appears to reflect the disassociation of the G91D extended trimer during neutral pH incubation. Once clamped by exogenous DIII, the trimer did not dissociate.
Fig. 8.

Model for intermediates in alphavirus fusion. This working model summarizes the intermediates in E1-mediated membrane fusion, the properties of the intermediates, and the point of inhibition for several E1 fusion mutants. The reversibility of the WT extended trimer is hypothesized based on the results seen with the G91D mutant (indicated by a question mark). It is not known whether H230A is blocked in packing of the C-terminal stem (question mark) and/or in possible intertrimer interactions (not diagrammed). See the text for further discussion.
How might the G91D mutation destabilize or prevent the progression of the extended trimer intermediate? Several key contacts appear to promote this conformation and trimer formation. In the center of the core trimer, residues D188 and K176 on DII form a ring of salt bridges that link the E1 proteins (31, 32). At the membrane-distal end of the core trimer, a network of interactions connects DI and the DI-DIII linker (49). Indeed, in the absence of the linker, the core trimer is unstable. Studies of a synthetic flavivirus fusion peptide suggested that residues in the fusion loop can affect trimer stability (35). Although G91D E1 inserts into target membranes and forms the extended trimer, intratrimer interactions of the fusion loops may be disrupted. It is also possible that cooperative interfusion loop interactions normally help to stabilize the core trimer and are disrupted in the mutant (40). While we cannot distinguish among these possibilities at this point, our data demonstrate an important role of the fusion loops in trimer stability and the reversibility of the initial extended E1 trimer in the G91D mutant.
Δ20 stem mutation.
The Δ20 stem deletion mutant inhibits fusion but allows the formation of a trypsin-and-SDS-resistant E1 homotrimer (28). Here we show that Δ20 E1 efficiently produced the DIII-binding core trimer and that it was blocked in a subsequent “partial hairpin” state. In this intermediate, some DI-DIII linker and endogenous DIII had folded back and stabilized the trimer, but DIII-binding sites remain open. We could detect this intermediate because, following the low-pH-induced refolding step, Δ20 E1 efficiently bound exogenous DIII at neutral pH. This neutral pH binding of DIII to a preformed core trimer target is also in agreement with our in vitro studies (40). The Δ20 mutant helps to corroborate our model for DIII inhibition, in which we proposed that exogenous DIII binds to a mixed population of trimers with various degrees of endogenous DIII packing and stability (27).
H230A ij loop mutation.
The E1 H230A mutant refolds to produce the SDS-and-trypsin-resistant homotrimer but is blocked in a late and unidentified stage in the fusion reaction (3). We showed here that E1 H230A was efficiently retrieved by exogenous DIII and thus that formation of the core trimer was similar to that seen with WT SFV. We then tested for alterations in the packing of the stem against the core trimer. Using a site-directed antibody (29), we showed that the N-terminal stem region of the H230A mutant packed as efficiently as that of WT E1. Taken together, our results suggest that the H230A mutation may inhibit packing of the C-terminal stem-transmembrane region in the final stage of refolding of the E1 homotrimer. This would be in keeping with H230A second-site resuscitating mutations, which are in positions that could affect stem packing (4). Alternatively, it is possible that H230A affects the intertrimer interactions observed in electron microscopy studies of membrane-inserted E1 proteins (13, 40, 49). To date, there is no direct evidence that these interactions play a role in fusion. Further studies of the H230A mutant may be an important tool in defining the roles of stem packing and cooperative trimer interactions in the late stages of membrane fusion.
Ordering alphavirus fusion intermediates.
Exogenous DIII binding provided a tool to define the fusion block in a series of E1 mutants (this study and reference 32). These intermediates can now be ordered to generate a refined model for the alphavirus fusion pathway (Fig. 8). Following dissociation of the E2/E1 dimer at low pH, E1 monomers insert into the target membrane. In support of this, the E1 D188K fusion block mutant inserts into the target membrane but does not form the core trimer and is unable to bind exogenous DIII (32). E1 monomers then associate to form an extended E1 trimer that is initially reversible and represents the intermediate identified by the G91D mutant. Fold-back of the linker and DIII regions then occurs through the partial hairpin intermediate identified by the Δ20 mutant. Hairpin formation continues with completion of packing of DIII and the stem region. Together with possible intertrimer cooperative interactions, this refolding drives the fusion reaction and completes formation of the final postfusion conformation. While a number of mechanistic questions remain, this working model is a starting point for their study.
ACKNOWLEDGMENTS
We thank all of the members of our laboratory for helpful discussions and Yan Zheng and Claudia Sánchez-San Martín for their thoughtful comments on the manuscript. We thank Maofu Liao for his insightful comments on the manuscript and for his contributions to Fig. 8. We also thank Kartik Chandran and the members of his laboratory for helpful discussions and Sonu Nanda for excellent technical assistance.
This work was supported by a grant to M.K. from the National Institute of Allergy and Infectious Diseases (R01-AI075647) and by Cancer Center Core Support grant NIH/NCI P30-CA13330.
The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy and Infectious Diseases or the National Institutes of Health.
Footnotes
Published ahead of print on 21 September 2011.
REFERENCES
- 1. Ahn A., Klimjack M. R., Chatterjee P. K., Kielian M. 1999. An epitope of the Semliki Forest virus fusion protein exposed during virus-membrane fusion. J. Virol. 73:10029–10039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bron R., Wahlberg J. M., Garoff H., Wilschut J. 1993. Membrane fusion of Semliki Forest virus in a model system: Correlation between fusion kinetics and structural changes in the envelope glycoprotein. EMBO J. 12:693–701 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Chanel-Vos C., Kielian M. 2004. A conserved histidine in the ij loop of the Semliki Forest virus E1 protein plays an important role in membrane fusion. J. Virol. 78:13543–13552 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Chanel-Vos C., Kielian M. 2006. Second-site revertants of a Semliki Forest virus fusion-block mutation reveal the dynamics of a class II membrane fusion protein. J. Virol. 80:6115–6122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chatterjee P. K., Vashishtha M., Kielian M. 2000. Biochemical consequences of a mutation that controls the cholesterol dependence of Semliki Forest virus fusion. J. Virol. 74:1623–1631 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Corver J., et al. 1995. Sphingolipid-dependent fusion of Semliki Forest virus with cholesterol-containing liposomes requires both the 3-hydroxyl group and the double bond of the sphingolipid backbone. J. Virol. 69:3220–3223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. DeTulleo L., Kirchhausen T. 1998. The clathrin endocytic pathway in viral infection. EMBO J. 17:4585–4593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Duffus W. A., Levy-Mintz P., Klimjack M. R., Kielian M. 1995. Mutations in the putative fusion peptide of Semliki Forest virus affect spike protein oligomerization and virus assembly. J. Virol. 69:2471–2479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Earp L. J., Delos S. E., Netter R. C., Bates P., White J. M. 2003. The avian retrovirus avian sarcoma/leukosis virus subtype A reaches the lipid mixing stage of fusion at neutral pH. J. Virol. 77:3058–3066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Enserink M. 2007. Infectious diseases. Chikungunya: no longer a third world disease. Science 318:1860–1861 [DOI] [PubMed] [Google Scholar]
- 11. Flynn D. C., Meyer W. J., Mackenzie J. M., Johnston R. E. 1990. A conformational change in Sindbis virus glycoproteins E1 and E2 is detected at the plasma membrane as a consequence of early virus-cell interaction. J. Virol. 64:3643–3653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gibbons D. L., et al. 2004. Multistep regulation of membrane insertion of the fusion peptide of Semliki Forest virus. J. Virol. 78:3312–3318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gibbons D. L., et al. 2003. Visualization of the target-membrane-inserted fusion protein of Semliki Forest virus by combined electron microscopy and crystallography. Cell 114:573–583 [DOI] [PubMed] [Google Scholar]
- 14. Gibbons D. L., et al. 2004. Conformational change and protein-protein interactions of the fusion protein of Semliki Forest virus. Nature 427:320–325 [DOI] [PubMed] [Google Scholar]
- 15. Glomb-Reinmund S., Kielian M. 1998. fus-1, a pH-shift mutant of Semliki Forest virus, acts by altering spike subunit interactions via a mutation in the E2 subunit. J. Virol. 72:4281–4287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Helenius A., Kartenbeck J., Simons K., Fries E. 1980. On the entry of Semliki Forest virus into BHK-21 cells. J. Cell Biol. 84:404–420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Justman J., Klimjack M. R., Kielian M. 1993. Role of spike protein conformational changes in fusion of Semliki Forest virus. J. Virol. 67:7597–7607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kielian M., Chanel-Vos C., Liao M. 2010. Alphavirus entry and membrane fusion. Viruses 2:796–825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kielian M., Jungerwirth S., Sayad K. U., DeCandido S. 1990. Biosynthesis, maturation, and acid-activation of the Semliki Forest virus fusion protein. J. Virol. 64:4614–4624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kielian M., Klimjack M. R., Ghosh S., Duffus W. A. 1996. Mechanisms of mutations inhibiting fusion and infection by Semliki Forest virus. J. Cell Biol. 134:863–872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kielian M. C., Helenius A. 1984. The role of cholesterol in the fusion of Semliki Forest virus with membranes. J. Virol. 52:281–283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kielian M. C., Keränen S., Kääriäinen L., Helenius A. 1984. Membrane fusion mutants of Semliki Forest virus. J. Cell Biol. 98:139–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kuhn R. J. 2007. Togaviridae: the viruses and their replication, p. 1001–1022 In Knipe D. M. (ed.), Fields virology, fifth ed., vol. 1 Lippincott, Williams and Wilkins, Philadelphia, PA [Google Scholar]
- 24. Lescar J., et al. 2001. The fusion glycoprotein shell of Semliki Forest virus: an icosahedral assembly primed for fusogenic activation at endosomal pH. Cell 105:137–148 [DOI] [PubMed] [Google Scholar]
- 25. Levy-Mintz P., Kielian M. 1991. Mutagenesis of the putative fusion domain of the Semliki Forest virus spike protein. J. Virol. 65:4292–4300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li L., Jose J., Xiang Y., Kuhn R. J., Rossmann M. G. 2010. Structural changes of envelope proteins during alphavirus fusion. Nature 468:705–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liao M., Kielian M. 2005. Domain III from class II fusion proteins functions as a dominant-negative inhibitor of virus-membrane fusion. J. Cell Biol. 171:111–120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liao M., Kielian M. 2006. Functions of the stem region of the Semliki Forest virus fusion protein during virus fusion and assembly. J. Virol. 80:11362–11369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liao M., Kielian M. 2006. Site-directed antibodies against the stem region reveal low pH-induced conformational changes of the Semliki Forest virus fusion protein. J. Virol. 80:9599–9607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liljeström P., Lusa S., Huylebroeck D., Garoff H. 1991. In vitro mutagenesis of a full-length cDNA clone of Semliki Forest virus: the small 6,000-molecular-weight membrane protein modulates virus release. J. Virol. 65:4107–4113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu C. Y., Besanceney C., Song Y., Kielian M. 2010. Pseudorevertants of a Semliki Forest virus fusion-blocking mutation reveal a critical interchain interaction in the core trimer. J. Virol. 84:11624–11633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Liu C. Y., Kielian M. 2009. E1 mutants identify a critical region in the trimer interface of the Semliki Forest virus fusion protein. J. Virol. 83:11298–11306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Modis Y., Ogata S., Clements D., Harrison S. C. 2003. A ligand-binding pocket in the dengue virus envelope glycoprotein. Proc. Natl. Acad. Sci. U. S. A. 100:6986–6991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Modis Y., Ogata S., Clements D., Harrison S. C. 2004. Structure of the dengue virus envelope protein after membrane fusion. Nature 427:313–319 [DOI] [PubMed] [Google Scholar]
- 35. Pan J., Lai C. B., Scott W. R., Straus S. K. 2010. Synthetic fusion peptides of tick-borne encephalitis virus as models for membrane fusion. Biochemistry 49:287–296 [DOI] [PubMed] [Google Scholar]
- 36. Phalen T., Kielian M. 1991. Cholesterol is required for infection by Semliki Forest virus. J. Cell Biol. 112:615–623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pletnev S. V., et al. 2001. Locations of carbohydrate sites on alphavirus glycoproteins show that E1 forms an icosahedral scaffold. Cell 105:127–136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Qin Z. L., Zheng Y., Kielian M. 2009. Role of conserved histidine residues in the low-pH dependence of the Semliki Forest virus fusion protein. J. Virol. 83:4670–4677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Roussel A., et al. 2006. Structure and interactions at the viral surface of the envelope protein E1 of Semliki Forest virus. Structure 14:75–86 [DOI] [PubMed] [Google Scholar]
- 40. Sánchez-San Martín C., Sosa H., Kielian M. 2008. A stable prefusion intermediate of the alphavirus fusion protein reveals critical features of class II membrane fusion. Cell Host Microbe 4:600–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schwartz O., Albert M. L. 2010. Biology and pathogenesis of chikungunya virus. Nat. Rev. Microbiol. 8:491–500 [DOI] [PubMed] [Google Scholar]
- 42. Sieczkarski S. B., Whittaker G. R. 2002. Influenza virus can enter and infect cells in the absence of clathrin-mediated endocytosis. J. Virol. 76:10455–10464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Solignat M., Gay B., Higgs S., Briant L., Devaux C. 2009. Replication cycle of chikungunya: a re-emerging arbovirus. Virology 393:183–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Umashankar M., et al. 2008. Differential cholesterol binding by class II fusion proteins determines membrane fusion properties. J. Virol. 82:9245–9253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vashishtha M., et al. 1998. A single point mutation controls the cholesterol dependence of Semliki Forest virus entry and exit. J. Cell Biol. 140:91–99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Voss J. E., et al. 2010. Glycoprotein organization of Chikungunya virus particles revealed by X-ray crystallography. Nature 468:709–712 [DOI] [PubMed] [Google Scholar]
- 47. Wahlberg J. M., Boere W. A. M., Garoff H. 1989. The heterodimeric association between the membrane proteins of Semliki Forest virus changes its sensitivity to low pH during virus maturation. J. Virol. 63:4991–4997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wahlberg J. M., Bron R., Wilschut J., Garoff H. 1992. Membrane fusion of Semliki Forest virus involves homotrimers of the fusion protein. J. Virol. 66:7309–7318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zheng Y., Sanchez-San Martin C., Qin Z. L., Kielian M. 2011. The domain I-domain III linker plays an important role in the fusogenic conformational change of the alphavirus membrane fusion protein. J. Virol. 85:6334–6342 [DOI] [PMC free article] [PubMed] [Google Scholar]