

Abstract
West Nile virus (WNV) is similar to other RNA viruses in that it forms genetically complex populations within hosts. The virus is maintained in nature in mosquitoes and birds, with each host type exerting distinct influences on virus populations. We previously observed that prolonged replication in mosquitoes led to increases in WNV genetic diversity and diminished pathogenesis in mice without remarkable changes to the consensus genome sequence. We therefore sought to evaluate the relationships between individual and group phenotypes in WNV and to discover novel viral determinants of pathogenesis in mice and fitness in mosquitoes and birds. Individual plaque size variants were isolated from a genetically complex population, and mutations conferring a small-plaque and mouse-attenuated phenotype were localized to the RNA helicase domain of the NS3 protein by reverse genetics. The mutation, an Asp deletion, did not alter type I interferon production in the host but rendered mutant viruses more susceptible to interferon compared to wild type (WT) WNV. Finally, we used an in vivo fitness assay in Culex quinquefasciatus mosquitoes and chickens to determine whether the mutation in NS3 influenced fitness. The fitness of the NS3 mutant was dramatically lower in chickens and moderately lower in mosquitoes, indicating that RNA helicase is a major fitness determinant of WNV and that the effect on fitness is host specific. Overall, this work highlights the complex relationships that exist between individual and group phenotypes in RNA viruses and identifies RNA helicase as an attenuation and fitness determinant in WNV.
INTRODUCTION
West Nile virus (WNV; Flaviviridae: Flavivirus) is a positive-sense, single-stranded RNA (ssRNA) virus belonging to the Japanese encephalitis virus (JEV) serological complex of the flaviviruses. WNV perpetuates in nature in enzootic transmission cycles through alternating replication in (mainly avian) vertebrates and mosquitoes. The specific mosquito and avian hosts that are most important in a particular locality differ but tend to include Culex species mosquitoes and passerine birds (2, 10). WNV infection of mammals, including humans and horses, occurs via spillover from this enzootic cycle. Since its introduction into North America in 1999 (20), molecular epidemiologic studies have clearly demonstrated that the virus has evolved to maximize its transmission potential within local transmission cycles (8, 11, 23). This finding stimulated subsequent efforts to understand the details of the underlying evolutionary mechanisms that lead to population-level genetic and phenotypic changes in the virus. In brief, these studies demonstrated that WNV populations are genetically diverse within hosts (15), that genetic diversity may be shared between hosts (15), and that mosquito infection drives genetic diversification of the virus population both through relaxation of purifying selection and through selection of rare genotypes resulting from RNA interference (RNAi) (4, 16, 17). Thus, in the WNV transmission cycle, different host types differentially influence the virus population. Whereas infection of mosquitoes leads to high levels of population variation and consequent adaptive plasticity, vertebrate infection maintains high fitness through strong purifying selection.
The WNV genome is approximately 11 kb in length and encodes a single polyprotein that is co- and posttranslationally cleaved by viral and host proteases into three structural and seven nonstructural proteins. The capsid (C), premembrane (prM), and envelope (E) structural proteins are encoded at the 5′ portion of the genome and, along with viral RNA and a host-derived lipid membrane, comprise the WNV virion. The nonstructural proteins, encoded at the 3′ portion of the genome, include NS1, NS2A, NS2B, NS3, NS4A, NS4B, and NS5. Translated nonstructural WNV proteins are multifunctional. During the course of infection, they assemble on host cell membranes to replicate the viral RNA and interfere with host antiviral responses. Several functions have been assigned to individual proteins. The flavivirus NS5 protein functions as the viral RNA-dependent RNA polymerase, has methyltransferase activity (36), and antagonizes JAK-STAT signaling in host cells (3). The viral NS3 protein has RNA helicase, serine protease, and NTPase functions (35). To date, most studies of virulence determinants of viruses, including arboviruses, have focused on the influence of specific genetic changes to the virus genome. For example, loss of an NYS glycosylation motif in the envelope protein significantly reduces WNV neuroinvasion in mice, and mutations in the active site of the NS5 methyltransferase are highly attenuating (1, 36). A mutation in the NS3 helicase domain of WNV confers virulence for American crows (5). Several mutations to the NS4B coding sequences that result in an attenuated phenotype have been identified (24, 26, 33), and several live attenuated flavivirus vaccines have been developed, with the mutations conferring attenuation as previously described (reviewed in reference 25). It is increasingly apparent, however, that virus determinants at the population level influence pathogenesis. Studies of mumps virus and poliovirus demonstrated that increases in the genetic heterogeneity of the virus population led to increases in pathogenesis (28, 32). In contrast, a high level of variation in the population of hepatitis C virus and WNV has been associated with decreases in pathogenesis (16, 31). In addition, studies using a mouse model of foot and mouth disease virus pathogenesis suggested that the overall pathogenic potential of a virus population arises through a complex interplay of viral genomes that differ in their individual potential to cause disease (27). This work supported previous seminal observations on the ability of vesicular stomatitis virus quasispecies to suppress more rapidly replicating (and thus, presumably, higher-fitness) mutant genomes (9). The relationships between the phenotypic variability of individual virus genomes within a population and the overall population fitness and pathogenic potential are thus clearly complex and generally underappreciated.
We therefore sought to determine whether the attenuation we observed in a genetically complex WNV population (16) can be attributed to individual attenuated genotypes present within the population. Since the attenuation we observed occurred after prolonged replication in mosquitoes, we also assessed whether the attenuated phenotype was associated with loss of fitness in vertebrates and/or gain of fitness in mosquitoes. To accomplish this, we used WNV in a model two-host virus system and took advantage of a mouse-attenuated WNV strain that was generated by serial passage in mosquitoes. This WNV strain was previously characterized with respect to both overall consensus sequence and mutational diversity (16). Specifically, we isolated WNV mutants from the attenuated population, determined their genome sequences, and evaluated pathogenesis in mice. We then used reverse genetics to localize attenuating mutations to the RNA helicase domain of the NS3 protein. Finally, the relative levels of fitness of the NS3 mutant in mosquitoes and chickens were evaluated. Our results revealed that attenuated genotypes within a viral mutant swarm might profoundly influence pathogenesis and suggest that these genotypes suppress genotypes of greater virulence. Further, our findings demonstrate the presence of virulence determinants within the RNA helicase domain and suggest that these determinants influence virus fitness in a species-specific manner.
MATERIALS AND METHODS
Cells.
African green monkey kidney cells (Vero; ATCC CCL-81), baby hamster kidney cells (BHK-21; ATCC CCL-10), and mouse fibroblasts (L929; ATCC CCL-1) were grown at 37°C with 5% CO2 in minimal essential medium (MEM; Gibco Invitrogen, Carlsbad, CA) supplemented with 10% heat-inactivated fetal bovine serum (HI-FBS), 2 mM l-glutamine, and sodium bicarbonate (1.5 g/liter) (complete medium). Aedes albopictus mosquito cells (C6/36; ATCC CRL-1660) were grown at 28°C with 5% CO2 in MEM supplemented with 10% HI-FBS, sodium bicarbonate (1.1 g/liter), 2 mM l-glutamine, 0.1 mM nonessential amino acids, penicillin (100 U/ml), and streptomycin (100 μg/ml). Plaque assays were performed using Vero cells in complete medium with the addition of 0.6% oxoid agar, penicillin (100 U/ml), and streptomycin (100 μg/ml).
Virus strains.
The parental WNV strain used for these studies was derived from an infectious cDNA clone (30) that had been passaged 20 times in mosquitoes (mp20c) as described elsewhere (16). Individual strains were isolated from this genetically diverse mosquito-passaged strain (mp20c) by plaque isolation from monolayers of Vero cells by the use of a sterile pipette tip. The pipette tip was then inserted into 1 ml of BA-1 diluent (M199; 1% bovine serum albumin, 0.05 M Tris [pH 7.6], sodium bicarbonate [0.35 g/liter], penicillin [100 μg/ml], streptomycin [100 μg/ml], fungizone [1 μg/ml]), and virus was amplified once using Vero cells grown in T25 tissue culture flasks. After 4 days of replication, the medium was harvested and brought to a concentration of 20% HI-FBS and stored at −80°C until use. Viral titers for all stocks of WNV were determined on Vero cells.
Encephalomyocarditis virus (EMCV) was a generous gift from Laura White at University of North Carolina at Chapel Hill. The virus was produced in BHK-21 cells, and viral titers were determined by a plaque assay using L929 cells.
Sequencing and construction of WNV mutants.
The large-plaque (lp) (LP1v1) and small-plaque (sp) (SP3v1) biological isolates were sequenced according to methods described elsewhere (19). Briefly, RNA was extracted from amplified virus stocks by the use of a QIAamp Viral RNA kit (Qiagen, Valencia, CA) and resuspended in 100 μl of RNase-free water. The WNV genome was amplified as six overlapping fragments of approximately 2 kb each using a Titan One Tube reverse transcription-PCR (RT-PCR) kit (Roche, Indianapolis, IN) according to the manufacturer's instructions. Amplicons were then sequenced in both directions using WNV-specific primers (available from the authors on request) designed to bind at approximately 400- to 500-nucleotide (nt) intervals on each strand, such that a primer binding site was located approximately every 200 nt. Sequences were assembled using the SeqMan module within DNAStar (LaserGene, Madison, WI), with a requirement of at least 2× coverage for the sequence to be considered complete. A modified full-length WNV infectious clone (pFLWNV) (30) was used to engineer the Asp deletion at NS3 483 (NS3Δ483), the Met-to Thr substitution at NS4a 64 (NS4aM64T), or double mutations (NS3Δ483+NS4aM64T). A standard fusion PCR protocol was used to prepare cDNA fragments (spanning nucleotide positions 5000 to 8000 of the WNV genome; GenBank accession no. AF404756) that contained the NS3Δ483, NS4aM64T, or NS3Δ483+NS4aM64T mutations. The PCR products were digested with restriction enzyme KpnI (KpnI cleaves at nucleotide positions 5340 and 7762 of the WNV sequence). The cleaved fragments were swapped into the pFLWNV plasmid, resulting in pFLWNV(NS3Δ483), pFLWNV(NS4aM64T), and pFLWNV(NS3Δ483+NS4aM64T). All plasmid constructs were verified by DNA sequencing. For generation of recombinant viruses, wild-type and mutant pFLWNV plasmids were linearized with XbaI. Genome-length RNAs were subjected to in vitro transcription using a T7 mMessage mMachine kit (Ambion, Austin, TX) and transfected into BHK-21 cells as described previously (30). Culture fluids from the transfected cells were collected on day 5 posttransfection.
Plaque size determination and replication kinetics.
The plaque phenotype of isolated and engineered viruses was verified by a plaque assay using Vero cells. The sizes of at least 40 plaques per strain were measured using Zeiss Axioscope software.
Replication kinetics of engineered viruses were evaluated in vitro in Vero and L929 cells according to standard methods. Briefly, confluent monolayers were infected at a multiplicity of infection (MOI) of 1, and supernatant was removed at various time points postinfection. Infectious virus was quantified by a plaque assay using Vero cells. The original mp20c stock was not used in these studies. Since mp20c was derived from a single infected mosquito that had been homogenized in 1 ml of buffer, we were severely limited in our ability to study it directly in vitro at a specific MOI.
Mouse studies.
All work with mice was conducted in strict accordance with protocols approved by the Wadsworth Center's Institutional Animal Care and Use Committee and followed criteria established by the National Institutes of Health. Five-week-old female C3H/HeN (C3H) mice were obtained from Taconic Farms (Hudson, NY) and allowed to acclimate in the BSL-3 vivarium for 1 week. Mice were inoculated subcutaneously (s.c.) in the left rear footpad with diluent (mock treatment) or virus as previously described (6). For morbidity and mortality studies, groups of eight mice were inoculated with biological isolates of WNV at 103 PFU or with clone-derived parental or mutant WNV at 10, 103, or 105 PFU. Control groups of four mice were subjected to mock inoculation. Mice were weighed and clinically assessed daily. Clinical signs included ruffled fur, hunching, ataxia, and weakness. A mouse was considered to have clinical West Nile disease when at least one of the following criteria was met: (i) ≥10% weight loss or (ii) clinical signs for at least 2 days. Mice that exhibited severe disease were euthanized. Serum was harvested from surviving mice at the termination of the study, and infection was confirmed by an enzyme-linked immunosorbent assay (ELISA) detecting antibodies against WNV, essentially as described (12). For tissue tropism and viral load studies, mice were sacrificed at various times postinoculation (p.i.) and transcardially perfused with 60 ml of phosphate-buffered saline (PBS) plus 1% fetal bovine serum (FBS). Tissues were harvested and processed as previously described (6).
Vector competence of C. pipiens quinquefasciatus.
Colonized C. pipiens quinquefasciatus mosquitoes were maintained in a humidified chamber at a constant temperature of 27°C with 16-h/8-h light/dark photoperiods. At 6 to 7 days postemergence, adult females were deprived of sucrose for 24 h and then fed on artificial blood meals of defibrinated goose blood containing WNV strains (approximately 2 × 108 PFU/ml) by the use of a membrane feeding apparatus (Hemotek, Accrington, United Kingdom). Fully engorged females were separated under conditions of cold anesthesia and held for 7 or 14 days under the conditions described above.
At 7 and 14 days postfeeding, mosquitoes were anesthetized with triethylamine (Sigma, St. Louis, MO). Mosquito legs were removed and inserted into a microcentrifuge tube containing 1 ml of mosquito diluent (20% FBS–PBS plus penicillin and streptomycin [50 μg/ml], 50 μg/ml of gentamicin, and 2.5 μg/ml of fungizone). Salivary secretions were collected by inserting mosquito mouthparts into a capillary tube containing a 1:1 mixture of FBS and 50% sucrose for approximately 30 min; the mixture was then added to 0.3 ml of mosquito diluent, and the body was placed in a tube containing 1 ml of mosquito diluent. Bodies and legs were homogenized in a mixer mill (Retsch, Germany) at 24 cycles/s for 1 min and clarified by centrifugation. Bodies, legs, and salivary secretions were assayed for infection by a plaque assay using Vero cells. Infection, dissemination, and transmission values are defined as the proportions of mosquitoes with infected bodies, legs, and salivary secretions, respectively.
Competition fitness assays.
In vivo viral fitness in mosquitoes and chickens was measured as described elsewhere (13). Briefly, test and reference viruses were mixed in approximately 1:1 PFU ratios and either fed to mosquitoes by the use of a membrane feeding apparatus or inoculated s.c. into day-old chickens. The reference virus contains five consecutive noncoding mutations that are readily detectable by sequencing and do not influence virus phenotype (13). The starting proportions of test and reference viruses were determined by analysis of the chick inoculum or of samples taken from four mosquitoes harvested immediately after feeding. Fitness was assessed in mosquitoes at 7 and 14 days postfeeding and in chickens at 48 h p.i. RNA was extracted using spin columns (QIAamp Viral RNA kit, Qiagen) according to the manufacturer's instructions. RT-PCR was performed using a One Step RT-PCR kit (Qiagen) with primers designed to amplify the region containing the genetic marker (forward primer, 5′-GCTCTGCCCCTACATGCCGAAAGT-3′; reverse primer, 5′-TACTTCACTCCTTCTGGCGGTTCA-3′) under the following thermocycling conditions: 50°C for 30 min; 95°C for 15 min; 40 cycles of 94°C for 30 min, 60°C for 30 s, and 72°C for 1 min; and 72°C for 10 min. Products were purified using a StrataPrep PCR purification kit (Stratagene, La Jolla, CA). Capillary sequencing was performed at the University of New Mexico (UNM) Health Sciences Center DNA Research Services core facility. Sequence chromatograms were analyzed using polySNP software (14) (http://staging.nybg.org/polySNP.html). Briefly, polySNP calculated the proportion of each genotype at the five nucleotide sites mutated in the marked reference virus by determining the area under the curve for each fluorescent dye at each variant chromatogram position. Thus, proportions of unmarked and marked reference nucleotides were quantitatively determined for each of the 5 sites that marked the reference. The median value of these five estimates was adopted as the genotype proportion for the sample. In cases in which only one genotype was detected, we fixed the value at either 0.05 or 0.95 to account for uncertainty in the measurement. Statistical analyses were performed using the GraphPad software package.
Alpha/beta interferon (IFN-α/β) bioassays.
Functional type I IFN in sera was measured as described previously (22). Briefly, serum samples and standards (recombinant mouse IFN-β; Chemicon International, Temecula, CA) were diluted in complete medium, treated with 2 N HCl, and incubated at 4°C overnight. The next day, samples and standards were neutralized with 2 N NaOH, added to triplicate wells of L929 cells, serially diluted 2-fold, and incubated at 37°C overnight. EMCV was added to the cells treated with standards and samples, and plates were incubated at 37°C. After 24 h, cell viability was measured using a CellTiter 96 AQueous Non-Radioactive Cell Proliferation assay (Promega, Madison, WI) per the manufacturer's instructions. The amounts of functional IFN-α/β in the serum were extrapolated from the standard curve. The values plotted for individual mice represent averages of the results from experiments performed using triplicate wells.
Effect of IFN on virus replication.
Vero cells (3 × 105 per well) were seeded in 12-well plates and incubated at 37°C overnight. Cells were incubated with complete medium alone (no treatment) or with recombinant human IFN-β (Calbiochem, San Diego, CA) at 100 or 1,000 U/ml in complete medium for 24 h at 37°C. The medium was removed, and the cells were inoculated with WNV (MOI of 1) at 37°C. At 1 h p.i., cells were washed and incubated in complete medium at 37°C. At various times p.i., an aliquot of the culture supernatant was harvested and stored at −70°C prior to titration by a plaque assay using Vero cells.
Statistics.
Statistical analyses were performed using GraphPad Prism (GraphPad Software, San Diego, CA). A two-tailed Fisher's exact test was used to compare percentages of morbidity and mortality. A two-tailed Mann-Whitney U test was used to test for differences between tissue titers. A P value of less than 0.05 was considered significant.
RESULTS
Isolation and characterization of mutants from a mixed WNV population.
Visual inspection of the plaques formed by the mosquito-passaged, mouse-attenuated WNV population mp20c (16) indicated that both small and large plaques were present in the virus population (Fig. 1A). Individual plaque size variants were isolated by plaque picking and amplified once on Vero cells to obtain working virus stocks. Isolates were designated LP1v1 (large plaque) and SP3v1 (small plaque). The sizes of the plaques formed by these isolates (Fig. 1A and B) indicated that their plaque size phenotypes were retained after a single passage in cell culture. Mouse survival studies were then conducted to assess the virulence of LP1v1 and SP3v1. The parental mixed WNV population caused 25% mortality, SP3v1 caused 0% mortality, and LP1v1 caused 50% mortality in mice (Fig. 1C). The complete genome sequence of SP1v1 was then determined to define genetic changes associated with attenuation in mice (Table 1). Four coding changes were identified, including substitutions at positions 244 and 308 of the E glycoprotein, deletion of an aspartic acid residue at position 483 in the RNA helicase domain of the NS3 protein, and a substitution at position 64 of the NS4A protein. Comparison of the sequence of SP3v1 to the consensus sequence of mp20c in the mixed population revealed that, in contrast to the substitutions in the E and NS4A coding sequences, which were apparent as either consensus or ambiguous base calls in both, the Asp deletion was not detectable by consensus sequence analysis of mp20c (Table 1).
Fig. 1.

Minority components of the WNV mutant spectrum arising during passage in mosquitoes differ in plaque morphology and pathogenic potential in mice. Passage of WNV derived from an infectious cDNA (WT) clone in mosquitoes resulted in a population that contained different plaque sizes. (A and B) Unpassaged, clone-derived WNV (WT) had large plaques approximately 3 mm in diameter (plaque size measured for at least 20 individual plaques by the use of Zeiss Axioscop software). After 20 passages in mosquitoes, the population (mp20c) contained both small- and large-plaque-producing variants that were isolated and passaged once in Vero cells. The resulting strains (larger plaque, LP1v1; smaller plaque, sp3v1) were isolated, and pathogenic potential in mice was determined. (C) Adult C3H mice were inoculated s.c. with one of the WNV strains at 103 PFU (n = 8 per group). Mice with severe disease were euthanized. All surviving mice were seropositive for WNV at 28 days p.i.
Table 1.
Coding changes in WNV strains
| Nucleotide position(s) | Unpassaged nucleotide | mp20c nucleotide | sp3v1 nucleotide | Coding region | Amino acid changea |
|---|---|---|---|---|---|
| 1697 | A | G | G | E | Glu to Gly (244) |
| 1889 | C | Y | T | E | Ala to Val (308) |
| 6058–6060 | GAC | GAC | —b | NS3 | Asp to —b (483) |
| 6662 | T | Y | C | NS4A | Met to Thr (65) |
Numbers in parentheses represent codon positions numbered from the start of the protein.
Dashes indicate that the residue is deleted.
Identification of the SP3v1 attenuating mutation.
We reasoned that defects in RNA replication were most likely to produce the small-plaque (sp), mouse-attenuated phenotype of SP3v1. Therefore, the Asp deletion at NS3 483 (NS3Δ483) and the Met-to-Thr substitution at NS4A 64 (NS4aM64T) were engineered alone and in tandem into a wild-type WNV NY99 backbone by reverse genetics. Viral production of the mutants was not significantly different than wild-type production in Vero cells (Fig. 2A); however, growth curves in mouse L929 cells and plaque size measurements clearly identified the deletion in NS3 as the determinant of slower replication in mouse cells and the sp phenotype (Fig. 2B and C). In mice, NS3Δ483 and double-mutant viruses were similarly attenuated across a range of doses (inoculations of 101 to 105 PFU), but the NS4A substitution mutant was not distinguishable from WT WNV in its ability to produce morbidity and mortality (Table 2). The tissue tropism and viral loads of the NS3Δ483 were then determined in mice at various time points after inoculation. In the initial sites of WNV replication, the skin at the inoculation site and the draining lymph node (6, 21), viral loads in mice inoculated with NS3Δ483 were indistinguishable from those seen with the WT (Fig. 3A and B). In addition, NS3Δ483 produced adequate viremia and spread efficiently to other tissues, as evidenced by determination of titers in the spleen and distant lymph node early during infection that were equal to or greater than those seen with the WT virus (Fig. 3C, D, and E). Later during infection, NS3Δ483 replicated to lower titers in the spleen (2 to 4 days p.i.) and distal lymph node (4 days p.i.) and resulted in lower viremia (4 days p.i.) compared to WT virus. Furthermore, viral loads in the brain and spinal cord were lower in mice inoculated with NS3Δ483 (3 to 5 days p.i.) (Fig. 3F) and the proportion of animals with infectious virus in either the brain or spinal cord (i.e., neuroinvasion) was lower for NS3Δ483 than for the WT (Fig. 3G).
Fig. 2.
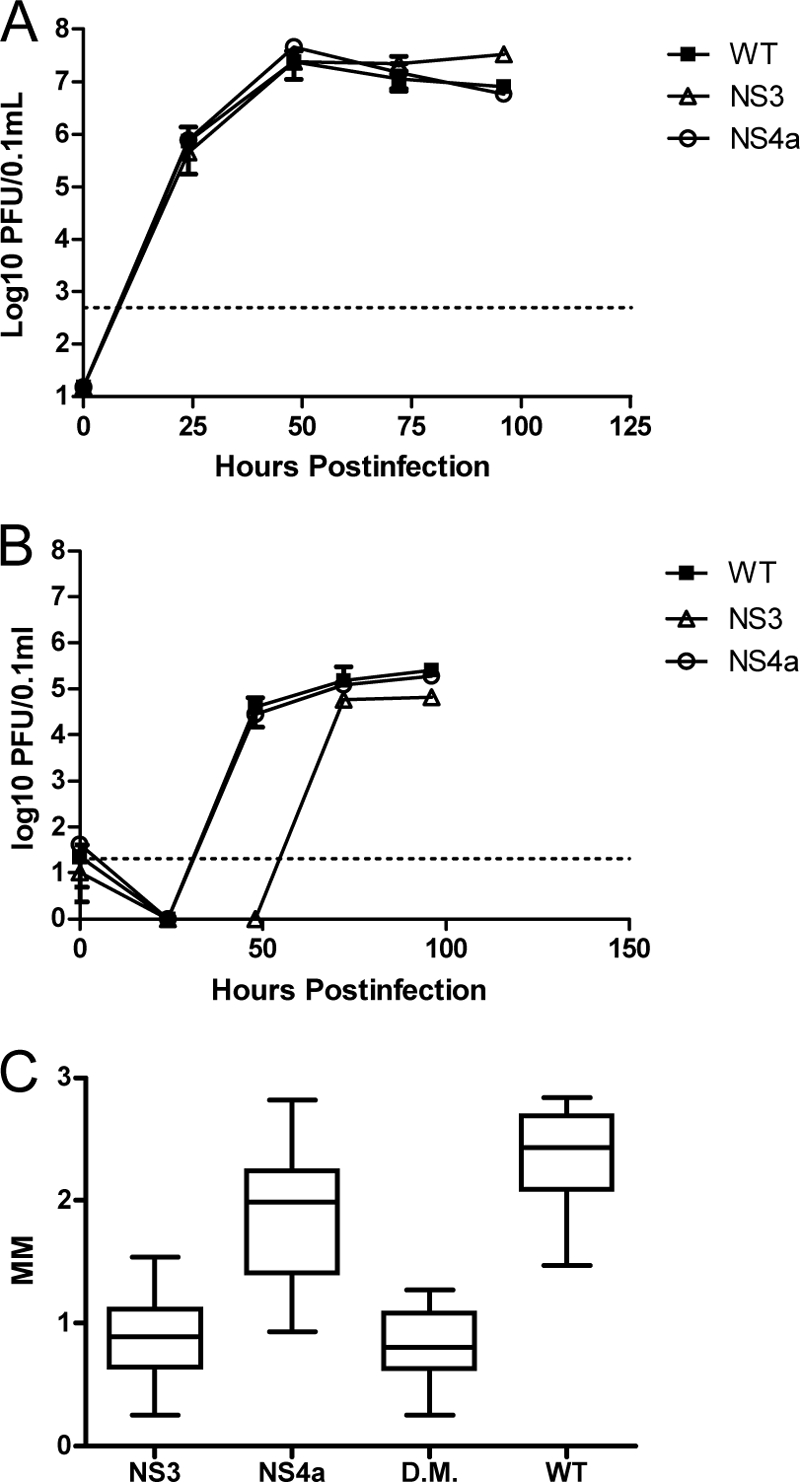
Asp deletion from the RNA helicase domain of WNV NS3 confers a small-plaque phenotype and diminished replication in L929 cells but not in Vero cells. Growth of infectious-clone-derived WNV was determined on Vero (A) and L929 (B) cells. Virus was applied to six-well plates containing confluent monolayers of each cell type at an MOI of 1.0 and allowed to adsorb for 1 h. A 0.1-ml volume of medium was removed at indicated times, and virus content was quantified by a plaque assay using Vero cells. Dotted lines on growth curves indicate the limit of detection of the assay. Plaque sizes for each single mutant (NS3 and NS4a) and the double mutant (DM) were measured as described in the text.
Table 2.
Mutation in NS3 confers attenuation in micea
| Virus inoculum | Dose (PFU) | Disease outcome |
|||
|---|---|---|---|---|---|
| Morbidity (no. of sick mice/total no. of mice) | Mortality (no. of mice that died/total no. of mice) | Avg day of disease onset (SD)b | Avg survival time (SD)c | ||
| Diluent | NA | 0/4 | 0/4 | NA | NA |
| WNV-NS3:Δ483D | 10 | 2/7d** | 1/7d** | 8.5 (0.7) | 11 |
| 103 | 3/8* | 2/8* | 8.3 (2.3) | 6.5 (0.7) | |
| 105 | 1/8*** | 1/8 | 7 | 9 | |
| WNV-NS4A:M65T | 10 | 8/8 | 7/8 | 8.0 (0.5) | 9.6 (1.3) |
| 103 | 7/8 | 5/8 | 8.7 (2.6) | 12.8 (6.9) | |
| 105 | 8/8 | 4/8 | 7.1 (0.4) | 7.3 (0.5) | |
| WNV-NS3:Δ483D + NS4A:M65T | 10 | 1/8*** | 0/8*** | 8 | NA |
| 103 | 1/8*** | 1/8*** | 7 | 7 | |
| 105 | 4/8 | 2/8 | 8.3 (1.0) | 7.5 (2.1) | |
| WNV-wild type | 10 | 8/8 | 7/8 | 8.6 (0.7) | 10.1 (1.7) |
| 103 | 8/8 | 7/8 | 8.1 (0.4) | 9.7 (1.1) | |
| 105 | 8/8 | 5/8 | 7.4 (0.5) | 8.6 (0.5) | |
Six-week-old female C3H mice were inoculated subcutaneously in the left rear footpad with 10 PFU, 103 PFU, or 105 PFU of virus or with diluent alone. Mice were monitored for weight loss and clinical signs. Fisher's exact test was used to compare mutants to WT groups at the same doses; P values are designated as follows: *, 0.01 to 0.05; **, 0.001 to 0.01; ***, <0.001. SD, standard deviation. NA, not applicable.
Data represent average day of disease onset for the mice that were sick but did not die.
Data represent average survival time in days for the mice that died.
All surviving mice were tested by ELISA at 28 days p.i. for seroconversion. One mouse in the WNV-NS3:Δ483D 10 PFU group did not seroconvert. All other survivors inoculated with WNV seroconverted.
Fig. 3.

NS3Δ483 replicates and spreads efficiently early during infection but produces lower titers in secondary tissue targets later during infection. (A to G) Adult C3H mice were inoculated s.c. with 103 PFU of WT or NS3Δ483 WNV. Viral loads in tissues were determined by a plaque assay for the left rear footpad skin at the inoculation site (A), left popliteal lymph node (B), serum (C), spleen (D), right popliteal lymph node (E), brain (F), and spinal cord (G). (H) Data from panels F and G are shown as percentages of mice with WNV in the brain and/or spinal cord. Each data point represents an individual mouse; data represent the results of two independent studies with four mice per group for each study. The solid horizontal lines represent geometric means for each group. Significant P values are indicated as follows: *, P = 0.01 to 0.05; **, P = 0.001 to 0.01. The dotted horizontal lines indicate the limits of detection at 500 PFU/g (A, B, D, and E), 50 PFU/ml (C), and 250 PFU/g (F and G). CNS, central nervous system.
Overall, the tissue tropism and kinetics of replication in mice for NS3Δ483 suggested that the mutant virus replicates in initial tissue targets and spreads to distal lymphoid tissues similarly to WT virus but that NS3Δ483 is defective in viral production later in infection at distal sites, including the central nervous system. We then hypothesized that NS3Δ483 induced more and/or was more sensitive to type I IFN than the WT virus. We compared the amount of IFN-α/β induced by NS3Δ483 and WT viruses in sera of mice; there were no significant differences between the two viruses (Fig. 4A). We modeled sensitivity to type I IFN by pretreating Vero cells with IFN-β and measuring viral production. Vero cells do not produce type I IFN but can respond to exogenous IFN-α/β. After treatment with 1,000 U/ml, NS3Δ483 was 10-fold more sensitive to inhibition by IFN-β than WT virus (10,000-fold versus 1,000-fold inhibition) at 24 h p.i. (Fig. 4B). By 48 h p.i., NS3Δ483 still exhibited 50-fold inhibition compared to 3-fold inhibition for WT virus. These results are consistent with the in vitro growth curves. There was a 24-h delay in viral production for NS3Δ483 in L929 cells (Fig. 2B), which are competent with respect to the type I IFN response, but equivalent growth kinetics were seen in Vero cells (Fig. 2A), which are type I IFN incompetent. Thus, our results strongly suggest that attenuation of NS3Δ483 is due in part to greater sensitivity to IFN-α/β.
Fig. 4.

NS3Δ483 induces levels of type I IFN similar to those induced by the WT but is more sensitive to its effects. (A) Functional type I IFN levels were measured by a bioassay using sera from adult C3H mice that were inoculated s.c. with 103 PFU of WNV WT or NS3Δ483 and sacrificed at various times p.i. (B) Vero cells were pretreated with IFN-β (100 or 1,000 U/ml) or with medium alone (untreated) for 24 h prior to inoculation with WT or NS3Δ483 at an MOI of 1. Viral production was measured by a plaque assay at 24 and 48 h p.i.; the ratios of viral titers for IFN-β-treated cells versus untreated cells are shown. Tx, treatment.
Influence of Asp deletion in NS3 on vector competence.
The ability of NS3Δ483 to infect, disseminate throughout, and be transmitted by mosquitoes was determined using colonized mosquitoes. At both 7 and 14 days postfeeding on an infectious blood meal containing approximately 107 PFU/0.1 ml, no differences in vector competence were detected (Table 3).
Table 3.
Impact of NS3 deletion on mosquito vector competence
| Virus strain | % (n) of virus positive in indicated category at postfeeding day: |
|||||
|---|---|---|---|---|---|---|
| 7a |
14b |
|||||
| Infected | Disseminated | Transmitted | Infected | Disseminated | Transmitted | |
| WT | 43 (40) | 0 | 0 | 57 (28) | 14 | 7 |
| NS3Δ483 | 60 (40) | 10 | 0 | 52 (23) | 13 | 0 |
For strains in the infected, disseminated, and transmitted categories, P = 0.1793, 0.1156, and 1.000, respectively (Fisher's exact test).
For strains in the infected, disseminated, and transmitted categories, P = 0.7823, 1.000, and 0.4949, respectively (Fisher's exact test).
Influence of Asp deletion in NS3 on virus fitness in vivo.
The relative fitness of NS3Δ483 was then assessed in vivo in mosquitoes and chickens. After replication for 2 days in chickens, NS3Δ483 was displaced by a genetically marked control reference virus in the circulating blood (Fig. 5A). After 7 days of replication in mosquitoes, the fitness of NS3Δ483 was approximately equal to that of the WT WNV (Fig. 5B), but by day 14, NS3Δ483 showed significantly less fitness in mosquitoes (Fig. 5C[Mann Whitney; P = 0.0063]).
Fig. 5.

Deletion of Asp483 in WNV NS3 decreases fitness in chickens and mosquitoes. (A) Day-old chickens were inoculated s.c. in the cervical region with 2 × 103 PFU of WNV containing equal proportions of each test virus (NS3 and WT) and a genetically marked reference virus that has been previously described. Serum was withdrawn by brachial venipuncture at the time of peak viremia (48 h p.i.) and RNA extracted according to standard methods. The proportions of test and reference viruses were estimated by quantitative sequencing. (B and C) To assess fitness in mosquitoes, colonized C. quinquefasciatus mosquitoes were offered an infectious blood meal containing 107 PFU of equal proportions of test and reference viruses and held for 7 (B) or 14 (C) days of extrinsic incubation (EI). After EI, mosquitoes were sacrificed and homogenized in RNA lysis buffer (RNEasy) according to standard methods. Proportions of test and reference viruses were determined as described above. Dotted lines indicate approximate linear ranges of quantitative sequencing. For chicks, the mean fitness levels of NS3 and WT viruses were compared by a one-sample t test using the observed WT virus-reference virus proportion as a predicted mean. For mosquitoes, fitness of NS3 and that of the WT were compared using a Mann-Whitney test and a Gaussian approximation.
DISCUSSION
RNA viruses form genetically complex populations of closely related variants within hosts; such populations are frequently referred to as a mutant swarm or as a quasispecies. In WNV, as with other RNA viruses (29), the amount of genetic variation present in the mutant swarm is host dependent (15, 16): replication in mosquitoes tends to promote viral genetic diversity, and replication in vertebrates tends to limit this diversity. In this study, we examined mp20c, a WNV population that had replicated in mosquitoes for 20 consecutive passages and was highly genetically diverse and attenuated in mice. Consensus genome sequence data revealed that the nucleotide substitutions that accumulated during 20 mosquito passages were unremarkable (16). However, a range of plaque sizes was observed when the titer of mp20c was determined on Vero cells. This range of plaque sizes led us to question whether minor genomic variants might be present within the mutant swarm and whether these might be capable of influencing the population phenotypes. Therefore, we examined two variants from the attenuated mp20c population that differed in plaque size and evaluated several phenotypes in order to gain insight into the relationships between individual and group phenotypes in WNV.
First, we isolated biological clones representing small-plaque and large-plaque phenotypes from the mixed population and characterized several phenotypes. Plaque sizes were consistent after a single Vero cell passage, indicating the stability of the phenotype and the suitability of the passaged stocks for further experiments. We found that the mouse mortality rate was variable following peripheral inoculation with a constant virus dose. Specifically, whereas the small-plaque variant produced no mortality, the large-plaque variant produced 50% mortality. The mortality rate of the mixed population was 25%. These results establish that the SP3v1 small-plaque variant is attenuated in mice. This finding was not particularly surprising: previous work has shown that the sp phenotype in WNV is associated with attenuation (7, 18). Slightly more surprising was the finding that mp20c caused an intermediate (25%) degree of mortality compared to sp3v1 (0%) and lp1v1 (50%). We had expected that the pathogenic potential of a virus population such as mp20c would be dominated by that of its most pathogenic component. This clearly was not the case, suggesting that, as has been reported for vesicular stomatitis virus (9), high-fitness WNV variants (here we directly equate fitness with pathogenesis in mice) can be “suppressed” through replication along with a genetically diverse mutant swarm containing lower-fitness variants.
Molecular clones were then constructed to define the viral genetic determinants of the sp and attenuated phenotype. After sequencing the sp and lp biological clones, two candidate attenuating mutations (substitution of Thr for Met in NS4A [NS4aM64T] and an Asp deletion in NS3 [NS3Δ483]) were identified and engineered into a cDNA clone of WNV. We found that NS3Δ483, but not NS4aM64T, was specifically associated with both the sp and mouse-attenuated phenotypes and with decreased replication in mouse L929 cells. This result suggested that less efficient in vivo replication in mice might lead to attenuation. Accordingly, we evaluated the possibility that replication and/or spread in mice might differ between NS3Δ483 and WT WNV.
Examination of viral loads in mouse sera, lymph nodes, and spleen demonstrated that NS3Δ483 replicated and spread similarly to WT WNV, suggesting that a frank deficit in replication efficiency in vivo is unlikely to produce the attenuation observed in mice. However, NS3Δ483 was cleared from circulation more efficiently than WT WNV, and neuroinvasion was less efficient in mice infected by NS3Δ483 than in those infected by WT WNV. Collectively, these observations led us to conclude that NS3Δ483 was more effectively controlled by the host than was WT WNV.
The deleted residue, Asp483, is located within the RNA helicase domain of the NS3 protein and is highly conserved among the encephalitic flaviviruses WNV, Japanese encephalitis virus (JEV), and tick-borne encephalitis virus (TBEV) but not in dengue virus (DEN) or yellow fever virus (YFV) (34), suggesting that it may be an important determinant of neuroinvasion and/or neurovirulence. Specifically, it may influence the efficiency of unwinding double-stranded RNA (dsRNA) replicative intermediates. The presence of higher concentrations of intracellular dsRNA could lead to enhanced interferon activation through TLR3-dependent signaling. Alternatively, the overall slowed rate of RNA replication might render the virus more susceptible to the antiviral state that develops in vertebrate hosts. Therefore, we measured interferon production in mice that had been infected by mutant and wild-type viruses and assessed the in vitro sensitivity of the viruses to interferon in Vero cells. In mice, NS3Δ483 did not induce significantly different levels of type I interferon compared to WT. Upon in vitro treatment of cells with type I IFN, NS3Δ483 WNV was more sensitive than WT WNV, resulting in 10-fold-lower viral production. Taken together, these results led us to conclude that the NS3Δ483 mutation renders the virus more susceptible to the antiviral state that develops in the animal during the course of WNV infection but does not influence initial replication or interferon induction. Furthermore, we propose a model in which the individual phenotype of greater susceptibility to type I IFN (NS3Δ483) contributes to the phenotype of the quasispecies (mp20c). In this model, all genotypes contribute to the initial antiviral state. Once the antiviral state is induced, the NS3Δ483 mutant is more effectively controlled than the more resistant genotypes, such as the WT genotype. With reduced viral production for the more susceptible genotypes (NS3Δ483), viral infection in the animal as a whole was better controlled, resulting in partial attenuation of the quasispecies (mp20c).
Finally, since the NS3Δ483 mutation arose during prolonged replication in mosquitoes, we tested whether this mutation might be important in balancing WNV fitness between vertebrates, where it appears to be required for interferon resistance, and mosquitoes, which lack an interferon response. To accomplish this, we used a previously reported system for estimating competitive fitness in vivo that takes advantage of a genetically marked WNV competitor virus (13). NS3Δ483 was effectively outcompeted by WT WNV in chickens, demonstrating that the RNA helicase function is a major fitness determinant in these hosts. In mosquitoes, we found that NS3Δ483 competed moderately effectively at 7 days after infection but not at 14 days. These results indicate that this determinant within the RNA helicase domain of NS3 is a fitness determinant in both vertebrate and invertebrate components of the WNV transmission cycle but is more important in vertebrates than in mosquitoes. Interestingly, a major mosquito innate antivirus defense (RNA interference [RNAi]) also targets double-stranded RNA via the RNase III enzyme Dicer. It follows that RNA helicase activity should also be associated with fitness in mosquitoes and that defects in helicase function would lead to fitness declines because intracellular concentrations of viral dsRNA replicative intermediates are higher, which could lead to Dicer-mediated cleavage of greater efficiency. Our data on fitness in mosquitoes support this observation. Moreover, the studies we report highlight the complex relationships that exist between individual and group phenotypes in highly variable populations such as RNA viruses and demonstrate that phenotypically significant variants may be undetectable through conventional approaches to obtaining the virus consensus sequence. Finally, our studies have shown that the WNV RNA helicase is both a fitness determinant of natural hosts (mosquitoes and birds) and a determinant of virus virulence in mice.
ACKNOWLEDGMENTS
We thank Kim Appler, Chrystal Chadwick, and Pamela Chin for technical assistance and the Wadsworth Center Tissue Culture Core providing cell culture support. Sequencing was performed at the Wadsworth Center Molecular Genetics Core facility.
This work was supported in part by funds from the National Institute of Allergy and Infectious Diseases (NIAID), National Institutes of Health (NIH), under grant AI067380 and contract N01-AI25490. Funding for ERD was provided by NIGMS IRACDA award K12GM088021 from the National Institute of General Medical Sciences and the UNM ASERT fellowship. The BSL-3 vivarium at the Wadsworth Center was used, which is funded in part as a core facility by NIAID, NIH (Northeast Biodefense Center) (grant U54-AI057158).
Footnotes
Published ahead of print on 21 September 2011.
REFERENCES
- 1. Beasley D. W., Li L., Suderman M. T., Barrett A. D. 2002. Mouse neuroinvasive phenotype of West Nile virus strains varies depending upon virus genotype. Virology 296:17–23 [DOI] [PubMed] [Google Scholar]
- 2. Bernard K. A., et al. 2001. West Nile virus infection in birds and mosquitoes, New York state, 2000. Emerg. Infect. Dis. 7:679–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Best S. M., et al. 2005. Inhibition of interferon-stimulated JAK-STAT signaling by a tick-borne flavivirus and identification of NS5 as an interferon antagonist. J. Virol. 79:12828–12839 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brackney D. E., Beane J. E., Ebel G. D. 2009. RNAi targeting of West Nile virus in mosquito midguts promotes virus diversification. PLoS Pathog. 5:e1000502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brault A. C., et al. 2007. A single positively selected West Nile viral mutation confers increased virogenesis in American crows. Nat. Genet. 39:1162–1166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Brown A. N., Kent K. A., Bennett C. J., Bernard K. A. 2007. Tissue tropism and neuroinvasion of West Nile virus do not differ for two mouse strains with different survival rates. Virology 368:422–430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Davis C. T., et al. 2004. Emergence of attenuated West Nile virus variants in Texas, 2003. Virology 330:342–350 [DOI] [PubMed] [Google Scholar]
- 8. Davis C. T., et al. 2005. Phylogenetic analysis of North American West Nile virus isolates, 2001–2004: evidence for the emergence of a dominant genotype. Virology 342:252–265 [DOI] [PubMed] [Google Scholar]
- 9. de la Torre J. C., Holland J. J. 1990. RNA virus quasispecies populations can suppress vastly superior mutant progeny. J. Virol. 64:6278–6281 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Dusek R. J., et al. 2009. Prevalence of West Nile virus in migratory birds during spring and fall migration. Am. J. Trop. Med. Hyg. 81:1151–1158 [DOI] [PubMed] [Google Scholar]
- 11. Ebel G. D., Carricaburu J., Young D., Bernard K. A., Kramer L. D. 2004. Genetic and phenotypic variation of West Nile virus in New York, 2000–2003. Am. J. Trop. Med. Hyg. 71:493–500 [PubMed] [Google Scholar]
- 12. Ebel G. D., et al. 2002. Detection by enzyme-linked immunosorbent assay of antibodies to West Nile virus in birds. Emerg. Infect. Dis. 8:979–982 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Fitzpatrick K. A., et al. 2010. Population variation of West Nile virus confers a host-specific fitness benefit in mosquitoes. Virology 404:89–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hall G. S., Little D. P. 2007. Relative quantitation of virus population size in mixed genotype infections using sequencing chromatograms. J. Virol. Methods 146:22–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Jerzak G., Bernard K. A., Kramer L. D., Ebel G. D. 2005. Genetic variation in West Nile virus from naturally infected mosquitoes and birds suggests quasispecies structure and strong purifying selection. J. Gen. Virol. 86:2175–2183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Jerzak G. V. S., Bernard K., Kramer L. D., Shi P. Y., Ebel G. D. 2007. The West Nile virus-mutant spectrum is host-dependant and a determinant of mortality in mice. Virology 360:469–476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jerzak G. V. S., Brown I., Shi P. Y., Kramer L. D., Ebel G. D. 2008. Genetic diversity and purifying selection in West Nile virus populations are maintained during host switching. Virology 374:256–260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Jia Y., et al. 2007. Characterization of a small plaque variant of West Nile virus isolated in New York in 2000. Virology 367:339–347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lanciotti R. S., et al. 2002. Complete genome sequences and phylogenetic analysis of West Nile virus strains isolated from the United States, Europe, and the Middle East. Virology 298:96–105 [DOI] [PubMed] [Google Scholar]
- 20. Lanciotti R. S., et al. 1999. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science 286:2333–2337 [DOI] [PubMed] [Google Scholar]
- 21. Lim P. Y., Behr M. J., Chadwick C. M., Shi P. Y., Bernard K. A. 2011. Keratinocytes are cell targets of West Nile virus in vivo. J. Virol. 85:5197–5201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lim P. Y., Louie K. L., Styer L. M., Shi P. Y., Bernard K. A. 2010. Viral pathogenesis in mice is similar for West Nile virus derived from mosquito and mammalian cells. Virology 400:93–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Moudy R. M., Meola M. A., Morin L. L., Ebel G. D., Kramer L. D. 2007. A newly emergent genotype of West Nile virus is transmitted earlier and more efficiently by Culex mosquitoes. Am. J. Trop. Med. Hyg. 77:365–370 [PubMed] [Google Scholar]
- 24. Muñoz-Jordán J. L., et al. 2005. Inhibition of alpha/beta interferon signaling by the NS4B protein of flaviviruses. J. Virol. 79:8004–8013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pugachev K. V., Guirakhoo F., Trent D. W., Monath T. P. 2003. Traditional and novel approaches to flavivirus vaccines. Int. J. Parasitol. 33:567–582 [DOI] [PubMed] [Google Scholar]
- 26. Puig-Basagoiti F., et al. 2007. A mouse cell-adapted NS4B mutation attenuates West Nile virus RNA synthesis. Virology 361:229–241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Sanz-Ramos M., Díaz-San Segundo F., Escarmis C., Domingo E., Sevilla N. 2008. Hidden virulence determinants in a viral quasispecies in vivo. J. Virol. 82:10465–10476 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sauder C. J., et al. 2006. Changes in mumps virus neurovirulence phenotype associated with quasispecies heterogeneity. Virology 350:48–57 [DOI] [PubMed] [Google Scholar]
- 29. Schneider W. L., Roossinck M. J. 2001. Genetic diversity in RNA virus quasispecies is controlled by host-virus interactions. J. Virol. 75:6566–6571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Shi P. Y., Tilgner M., Lo M. K., Kent K. A., Bernard K. A. 2002. Infectious cDNA clone of the epidemic West Nile virus from New York City. J. Virol. 76:5847–5856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Sullivan D. G., et al. 2007. Hepatitis C virus dynamics during natural infection are associated with long-term histological outcome of chronic hepatitis C disease. J. Infect. Dis. 196:239–248 [DOI] [PubMed] [Google Scholar]
- 32. Vignuzzi M., Stone J. K., Arnold J. J., Cameron C. E., Andino R. 2006. Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439:344–348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wicker J. A., et al. 2006. A single amino acid substitution in the central portion of the West Nile virus NS4B protein confers a highly attenuated phenotype in mice. Virology 349:245–253 [DOI] [PubMed] [Google Scholar]
- 34. Wu J., Bera A. K., Kuhn R. J., Smith J. L. 2005. Structure of the Flavivirus helicase: implications for catalytic activity, protein interactions, and proteolytic processing. J. Virol. 79:10268–10277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Zhou H., Singh N. J., Kim K. S. 2006. Homology modeling and molecular dynamics study of West Nile virus NS3 protease: a molecular basis for the catalytic activity increased by the NS2B cofactor. Proteins 65:692–701 [DOI] [PubMed] [Google Scholar]
- 36. Zhou Y., et al. 2007. Structure and function of flavivirus NS5 methyltransferase. J. Virol. 81:3891–3903 [DOI] [PMC free article] [PubMed] [Google Scholar]