

Abstract
The biology of cerebral white matter injury is woefully understudied, in part due to the difficulty to reliably model this type of injury in rodents. Periventricular leukomalacia (PVL) is the predominant form of brain injury and the most common cause of cerebral palsy in premature infants. PVL is characterized by predominant white matter injury. No specific therapy for PVL is presently available because the pathogenesis is not well understood. Here we report that two types of mouse PVL models have been created by hypoxia-ischemia with or without systemic co-administration of lipopolysaccharide (LPS). LPS co-administration exacerbated hypoxic-ischemic white matter injury and led to enhanced microglial activation and astrogliosis. Drug trials with the anti-inflammatory agent minocycline, the anti-excitotoxic agent NBQX and the antioxidant agent edaravone showed various degrees of protection in the two models, indicating that excitotoxic, oxidative and inflammatory forms of injury are involved in the pathogenesis of injury to immature white matter. We then applied immune-electron microscopy to reveal fine structural changes in the injured white matter, and found that synapses between axons and oligodendroglial precursor cells (OPCs) are quickly and profoundly damaged. Hypoxia-ischemia caused a drastic decrease in the number of postsynaptic densities associated with the glutamatergic axon-OPC synapses defined by the expression of vesicular glutamate transporters, vGluT1 and vGluT2, on axon terminals that formed contacts with OPCs in the periventricular white matter, resulted in selective shrinkage of the postsynaptic OPCs contacted by vGluT2 labeled synapses, and led to excitotoxicity mediated by GluR2-lacking, Ca2+-permeable AMPA receptors. Taken together, the present study provides novel mechanistic insights into the pathogenesis of PVL, and reveals that axon-glia synapses are highly vulnerable to white matter injury in the developing brain. More broadly, the study of white matter development and injury has general implications for a variety of neurological diseases including PVL, stroke, spinal cord injury and multiple sclerosis.
Keywords: White matter injury, Axon-glia synapse, Oligodendrocyte, Hypoxia-ischemia, Lipopolysaccharide, Periventricular leukomalacia
Introduction
In the United States, approximately 63000 infants are born yearly with a very low birth weight < 1500 g (Martin et al., 2008). Nearly 90% of such infants survive the neonatal period, but 5–10% of the survivors subsequently exhibit cerebral palsy and approximately 40–50% have cognitive and behavioral deficits. The predominant form of brain injury underlying this neurological morbidity in premature infants is periventricular leukomalacia (PVL), which is characterized by selective cerebral white matter injury with prominent oligodendroglial loss (Volpe, 2001). Currently, there is no specific therapy for PVL because the pathogenesis is not well understood. Although the etiology of PVL is multifactorial, brain hypoxia-ischemia associated with prematurity and maternal-fetal infection and/or inflammation are thought to be the primary causes (Volpe, 2001, 2009; Pleasure et al., 2006; Deng, 2010). A number of animal models of PVL have been developed in various species, such as rabbits, dogs, cats and sheep, by ischemia or administration of infectious agents (Hagberg et al., 2002). Considering cost efficiency and easier access to antibodies/sequence and molecular genetics data, it is necessary to develop rodent models of PVL. A hypoxic-ischemic model of PVL in the rat at P7 has been previously established (Follet et al., 2000, 2004). This unique model resembles to a great degree the PVL pathology seen in premature infants. The hallmark of this model is selective white matter pathology, in contrast to the majority of the stroke models that are characterized by gray matter infarction. However, the limitations of this hypoxic-ischemic model in the rat should not be overlooked. First, a major shortcoming with a rat model is the difficulty in access to transgenic studies as compared to a mouse model; the optimal proof-of-concept experimental approach to establish a causative role for a molecule of interest often requires the use of transgenic mice. Second, combined hypoxia-ischemia and infection/inflammation would likely create more clinically relevant PVL models that would more accurately reflect the physiological underpinnings of the human disease. The rodent age that best correlates with the human developmental period of greatest risk for PVL lesions (between the 24th and 32nd gestational weeks) is postnatal (P6-7), and oligodendroglial differentiation is predominately a postnatal event in rodents. As it is impossible to produce a maternal-fetal infection/inflammation in postnatal ages, it would be reasonable to simulate an infection at a relevant postnatal developmental stage to create a rodent model of PVL. Thus, combining LPS injection with hypoxia-ischemia at P6-7 in rodents may produce more PVL-like lesions in the white matter. In the present study, we sought to convert the experimental model of PVL from the rat to mouse, and used combined hypoxia-ischemia and infection/inflammation at P6. The consensus of our current understanding of the pathogenesis of PVL is that multiple forms of injury including excitotoxic, oxidative, and inflammatory mechanisms are involved (Volpe, 2001; Deng et al., 2008, 2010). Thus, building upon previous pharmacological studies (Follett et al., 2004; Lechpammer et al., 2008; Fan et al., 2005; Cai et al., 2006), here we conducted drug trials using the anti-inflammatory agent minocycline, anti-excitotoxic agent NBQX and the antioxidant agent edaravone in our mouse models of PVL.
Recent studies have surprisingly revealed that oligodendroglial precursor cells (OPCs) form synaptic contacts with axons in cerebral white matter in neonatal rodents (Kukley et al., 2007; Ziskin et al., 2007), and that the axon-OPC synapses appear to present only during development (De Biase et al., 2010). At axon-glia synapses, white matter axons derived from cortical pyramidal cells release excitatory neurotransmitter glutamate and activate AMPA subtype glutamate receptors on OPCs and mediate excitatory responses (Kukley et al., 2007; Ziskin et al., 2007). Under pathological conditions, AMPA receptors expressed on OPCs mediate excitotoxicity at excitatory synapses and cause oligodendrocyte injury and ultimately lead to hypomyelination (Deng et al., 2003; McDonald et al., 1998). The vesicular release of glutamate via axon-OPC synapses may be an important mechanism underlying Ca2+-permeable AMPA receptor mediated oligodendrocyte injury. We hypothesize that hypoxic-ischemic injury may alter ultrastructural features of the glutamatergic synapses that can be defined by immunostaining of vesicular glutamate transporters (vGluTs). In the present study, axon-OPC synapses were localized by immune-electron microscopy in the external capsule of the neonatal mouse subjected to hypoxia-ischemia. vGluT1 and vGluT2 were used as markers for glutamatergic synapses to determine whether the axon-OPC synapses are sensitive targets in white matter injury using our hypoxic-ischemic mouse model of PVL.
Materials and Methods
Animals
P6 C57BL/6 mice (The Jackson Laboratories, Bar Harbor, ME) were used to characterize neuropathological features under different treatments. P6 NG2-DsRed BAC transgenic mice [The Jackson Laboratories, stock #: Tg(Cspg4-DsRed.T1)1Akik/J] were used to study the identity of postsynaptic profiles in white matter. Animal research was approved by the University of California Davis Committee on Animal Research. All procedures were carried out by following the guidelines set by the institutional animal care and use committee.
Unilateral carotid ligation (UCL) plus hypoxia
Animals underwent a permanent ligation of the right common carotid artery under ice anesthesia, followed by 1-h recovery on a thermal blanket, maintaining body temperature at 33–34°C and with a 1-h feeding period with the dam. Animals were then placed in a sealed chamber infused with nitrogen to maintain a level of 6.0% O2. Different durations of hypoxia were applied. The body temperature of mice was maintained at 33–34°C by leaving animals on thermal blanket throughout hypoxia. After a 1-h period of recovery, mice were returned to their dam. Some animals also received LPS co-injection intraperitoneally immediately after hypoxia.
LPS adminitration
To evaluate the effect of LPS on brain injury, LPS (0111:B4, Sigma) was injected i.p. immediately after hypoxia-ischemia at various doses (0.1, 1 or 3 mg/kg).
Histology and immunohistochemistry
After hypoxia-ischemia with or without LPS injection, animals were sacrificed at different time points: 24, 48, 72 or 96 h. The animals were anesthetized and then perfused with 4% paraformaldehyde in 0.1 M PBS (pH=7.4), and cryoprotected in 30% sucrose in 0.1 M PBS. Coronal brain sections were cut by cryostat from the anterior extent of the lateral ventricles through the posterior extent of the dorsal hippocampus. Representative sections were stained with the following antibodies: MBP (1:1000, Sternberg) as a protein marker for myelin; O1 (1:100, Millipore) as a lipid marker for oligodendrocytes (Follett et al., 2000); CD68 (1:100, Serotec) as a marker for activated microglia; GFAP (1:1000, Millipore) as a marker for astrocytes.
To evaluate white matter injury, we used a previously established semi-quantitative scoring scale (0–5) based on the absence of MBP staining: 0 = no MBP loss; 1= some loss of processes perpendicular to capsule; 2 = moderate loss of processes; 3 = complete loss of processes with intact capsule; 4 = loss of processes with thinning or breaks in capsule; 5 = loss of processes with complete loss of capsule (Follett et al., 2000, 2004; Shen et al., 2010). O1 was used as a complementary marker for evaluating white matter injury, and also scored on a 0–5 scale, with 0 being no loss, and 5 being virtually complete loss (Follett et al., 2000, 2004; Shen et al., 2010). Cell counting was performed to evaluate changes in other immunocytochemical markers, including CD68 and GFAP. We counted all positive cells present within the field of view at 200 × magnification in the white matter track overlying the hippocampus for six fields of each view/section and six evenly spaced coronal sections per animal. Cell counting was performed in hemispheres ipsilateral and contralateral to the ligation to take into account developmental variations between mice, and all tests were performed by an observer blinded to treatment groups or the side of carotid artery ligation.
Drug administration
The drug treatment paradigms were previously well-studied for each agent and used to achieve the maximum efficacy with minimum toxicity, while considering the pharmacokinetic and pharmacodynamic properties of each agent. Minocycline (Sigma) dissolved in saline was injected daily intraperitoneally at the dose of 15 mg/kg at 96 h after hypoxia. NBQX (Tocris) sodium salt dissolved in water was injected intraperitoneally every 12 h at the dose of 10 mg/kg at 48 h after hypoxia. Edaravone dissolved in 10% DMSO in saline (BioMol) was injected twice intraperitoneally at the dose of 10 mg/kg immediately after the artery occlusion and immediately after hypoxia.
Electron microscopy (EM)
Mice subjected to UCL/hypoxia at P6 were perfused with 4% paraformaldehyde + 0.2% glutaraldehyde in 0.1 M PBS, and brains were cut with a vibratome (Leica) at 60 μm. Sections were stained for vGluT1 and vGluT2 antibodies (Millipore, Billerica, MA) using the immunoperoxidase method. The stained sections were processed for electron microscopy as described in a previous study (Graziano et al., 2008). Sections from the ipsilateral (injury) side and the contralateral (control) side were first examined in the light microscope at 10x and 20x, the white matter was identified and photographed and the core of white matter regions (approximately 2 × 0.5 mm) was dissected under a dissecting microscope and glued to blank blocks. Thin sections were cut at 70 nm using an ultramicrotome (Leica Ultracut) and collected on single-slot (2 × 1 mm) Nickel grids and stained with uranyl acetate and lead citrate, and examined in a Philips CM120 electron microscope. The regions examined were carefully scanned and exclude the regions containing neuronal somata or dendrites, and the orientation of the region was guided by light microscopic images taken before EM processing. Much effort was made to ensure that the regions were from the white matter. Images were taken by a 2K × 2 K CCD camera (Gatan, Inc., Pleasanton, CA) and processed by using the software provided by Gatan, Inc (DigitalMicrograph). Images were composed in Photoshop CS (Adobe System, Mountain View, CA).
Western blot analysis
Immunoblots of membrane preparations of white matter tissue were performed using protocols and anti-GluR antibodies as previously published (Deng et al., 2006).
Electrophysiology
Recordings were performed using conventional patch-clamp techniques in whole-cell mode as previously published (Follett et al., 2004; Rosenberg et al., 2003). Experiments were performed with application of AMPA (100 μM) and the desensitization blocker cyclothiazide (50 μM).
Statistical analysis
All data were presented as mean ± SEM. Statistical differences were assessed by one-way ANOVA with Tukey post hoc analysis for multiple comparisons. Student’s t test was used when only two independent groups were compared. Significance was considered for p values ≤ 0.05.
Results
Systemic co-administration of LPS exacerbates hypoxic-ischemic white matter injury in the developing brain
To determine the appropriate hypoxia condition for use in order to create hypoxic-ischemic mouse model of PVL, we first evaluated the effect of different durations of hypoxia (6.0% O2) on white matter injury in mice after unilateral carotid ligation (UCL) at P6. Animals were sacrificed and brains were collected 96 h (at P10) after UCL/hypoxia. MBP and O1 immunostaining for myelin and oligodendrocytes showed the lack of white matter injury in animals that received only UCL or UCL/hypoxia for 20 min. Myelin and oligodendrocyte loss ipsilateral to the ligation started to show with UCL/hypoxia for 30 min (6% O2, 30 min). Increasing the hypoxia time to 45 min enhanced the white matter injury (Fig. 1A, C). Semi-quantitative white matter injury score based on MBP and O1 staining density demonstrated that white matter injury ipsilateral to the ligation significantly increased in animals that received UCL/hypoxia for 30 min or 45 min as compared to UCL only or UCL/hypoxia for 20 min. No injury contralateral to the ligation in animals subjected to UCL plus hypoxia for any duration (20, 30, or 45 min) (Fig. 1B, D). These results showed that UCL/hypoxia for 30 min created appropriate white matter injury ipsilateral to the ligation, and the injury increased with a longer duration of hypoxia with UCL.
Fig. 1.

The effect of different hypoxia durations on white matter injury in the developing brain. UCL/hypoxia (30 min) caused selective white matter injury. MBP staining showed myelin loss ipsilateral to the ligation in animals subjected to UCL/hypoxia (30 min). UCL/hypoxia (45 min) caused severe myelin loss (A). Semi-quantitative score of MBP staining showed that UCL/hypoxia (30 or 45 min) caused significant myelin loss ipsilateral to the ligation (B) (I, ischemia; I/H, ischemia-hypoxia; n=9–11; * p <0.001). O1 staining showed oligodendrocyte loss ipsilateral to the ligation with hypoxia for 30 min or 45 min (C). Semi-quantitative score of O1 staining showed that UCL/hypoxia (30 or 45 min) caused significant myelin loss ipsilateral to the ligation (D) (I, ischemia; I/H, ischemia-hypoxia; n=9–11; * p <0.001).
The etiology of PVL is often multifactorial; in addition to hypoxia-ischemia, maternal-fetal infection/inflammation often is strongly associated with PVL (O’Shea et al., 1998; Leviton et al., 1999). To model multifactorial PVL, we next examined co-administration of different doses of LPS (0.1, 1 or 3 mg/kg, i.p.) in the animals after UCL/hypoxia (6.0% O2, 30 min). Intraperitoneal injection of LPS at 0.1 mg/kg did not exacerbate white matter injury caused by UCL/hypoxia. LPS at 1 or 3 mg/kg significantly increased white matter injury caused by UCL/hypoxia, interestingly, not only in ipsilateral white matter but also in contralateral white matter (Fig. 2).
Fig. 2.

Co-administration of LPS injection with hypoxia-ischemia (H/I) exacerbated white matter injury. MBP staining of animals killed at 96 h after H/I with vehicle (PBS) or different doses of LPS showed that 1 or 3 mg/kg LPS, but not 0.1 mg/kg LPS, combined with UCL/hypoxia (6% O2, 30 min), caused myelin loss both contralateral and ipsilateral to the ligation (A). O1 staining of same animals showed similar results on oligodendrocyte loss both contralateral and ipsilateral to the ligation (C). Statistical analysis of the semi-quatitative scores of MBP and O1 staining showed that LPS co-administration (1 or 3 mg/kg) with UCL/hypoxia (30 min) significantly increased the white matter injury bilaterally, as compared to UCL/hypoxia (30 min) (n=6–9, *p<0.01 ** p<0.001) (B, D).
In addition, upon neuropathological examination of coronal sections stained with hematoxylin-and-eosin (H&E) through the entire forebrain, we also noted that some degree of gray matter injury in these mice (Fig. 3). Different injury patterns were observed under different conditions. UCL/hypoxia (30 min) with or without LPS injection (1 mg/kg) caused selective white matter injury with minor injury in the hippocampus and cortex. UCL/hypoxia (45 min) or UCL/hypoxia (30 min) plus LPS injection (3 mg/kg) caused infarctions in the hippocampus and cortex. These pathological features that are present in our mouse models resemble human PVL, which also does not occur in complete isolation from accompanying gray matter lesions; the hallmark of PVL is the relative, although not complete, sparing of the cerebral cortex overlying the periventricular focal and diffuse white matter injury.
Fig. 3.

H&E staining revealed different patterns of injury under different conditions. UCL/hypoxia (30 min) with or without LPS injection (1 mg/kg) caused selective white matter injury with minor injury in the hippocampus and cortex. UCL/hypoxia (45 min) or UCL/hypoxia (30 min) plus LPS injection (3 mg/kg) caused stroke-like injury. Note the infarcts in the hippocampus and cortex, as pointed by arrows.
Systemic co-administration of LPS leads to enhanced microglial activation and astrogliosis
Next, microglial activation and astrogliosis were studied by CD68 and GFAP immunostaining on animals killed at 24, 48, 72, or 96 h after hypoxia-ischemia with or without LPS injection. Quantification of CD68-positive cells demonstrated that microglial activation peaked at 48 h after insult in both the H/I model and the H/I/LPS model (Fig. 4A). More CD68-immunostained cells were seen in both contralateral and ipsilateral white matter of animals subjected to H/I/LPS as compared to H/I (Fig. 4B). Thus, LPS co-administration with hypoxia-ischemia significantly increased the number of activated microglia in contralateral and ipsilateral white matter as compared to hypoxia-ischemia only (Fig. 4B).
Fig. 4.

Co-administration of LPS with hypoxia-ischemia (H/I) increased microglial activation. Quantification of CD68-positive cells on sections from animals killed at different time points after H/I or H/I/LPS showed that the peak of microglial activation in both H/I and H/I/LPS models was at 48 h after treatment. Co-administration of LPS with H/I significantly increased microglial activation in contralateral and ipsilateral white matter at 48 h after H/I/LPS as compared to H/I (A) (n=6/group, * P <0.001). CD68-staining showed that LPS injection increased microglial activation in contralateral and ipsilateral white matter (B).
Quantification of GFAP-positive cells demonstrated that astroglial activation continued to increase 96 h after insult in both the H/I model and the H/I/LPS model (Fig. 5A). More prominent GFAP staining was shown in contralateral and ipsilateral white matter in animals subjected to H/I/LPS as compared to H/I (Fig. 5B). Thus, LPS co-administration with hypoxia-ischemia significantly increased astroglial activation in contralateral and ipsilateral white matter as compared to hypoxia-ischemia only (Fig. 5B).
Fig. 5.

Co-administration of LPS with hypoxia-ischemia (H/I) increased astrogliosis. Quantification of GFAP-positive cells on sections from animals killed at different time points after H/I or H/I/LPS showed that astroglial activation in both H/I and H/I/LPS models kept increasing during 96 h after treatment. LPS injection with H/I significantly increased astrogliosis in contralateral and ipsilateral white matter at 96 h after H/I/LPS injection, as compared to H/I (A) (n=6/group, * P <0.001). GFAP staining showed that LPS injection increased astrocyte activation in contralateral and ipsilateral white matter (B).
Minocycline attenuates white matter injury caused by hypoxia-ischemia plus LPS injection
We next conducted three drug trials on our mouse models of PVL, aiming to use well-studied pharmacological agents to target excitotoxic, oxidative, and inflammatory mechanisms of injury by using previously published treatment paradigms for each agent. The times selected for administration of each agent and the concentrations of each agent we used were based on previous studies in other animal models, where these particular treatment paradigms were used to achieve the maximum efficacy with minimum toxicity, while considering the pharmacokinetic and pharmacodynamic properties of each agent.
First, minocycline was administrated daily at the dose of 15 mg/kg 96 h after UCL/hypoxia (6% O2, 30 min) with or without LPS co-administration (1 mg/kg). While MBP and O1 staining (Fig. 6A, B) and semi-quantitative injury score with MBP or O1 staining (Fig. 6C, D) showed no difference between minocycline group and vehicle group after UCL/hypoxia, minocycline treatment significantly attenuated white matter injury in both contralateral and ipsilateral white matter as compare to vehicle group after UCL/hypoxia/LPS injection (Fig. 6E, F, G, H).
Fig. 6.

Minocycline was only protective on the H/I/LPS model. There was no effect on the MBP (A) or O1 (B) staining after minocycline treatment in animals after UCL/hypoxia (30 min). Statistical analysis of white matter injury score with MBP and O1 staining showed no significant difference between vehicle and minocycline treatment (C, D). MBP (E) and O1 (F) immunostaining showed that minocycline treatment increased MBP staining and O1 staining contralateral and ipsilateral to the ligation in the animals after H/I/LPS injection. Statistical analysis of white matter injury score with MBP and O1 staining showed that minocycline treatment significantly decreased white matter injury score by MBP and O1 contralateral and ipsilateral to the ligation in the H/I/LPS model (G, H). (n=6/group, * p <0.01, ** p <0.001).
NBQX attenuates white matter injury caused by hypoxia-ischemia with or without LPS co-administration
Second, NBQX was administrated every 12 h at the dose of 10 mg/kg 48 h after UCL/hypoxia (6% O2, 30 min) with or without LPS co-administration (1 mg/kg). The results showed that NBQX was protective against white matter injury in the H/I model (Fig. 7A, B, C, D). NBQX treatment increased MBP and O1 staining in ipsilateral white matter but not in contralateral white matter as compared to vehicle after hypoxia/ischemia/LPS injection (Fig. 7E, F). Semi-quantitative injury score with MBP and O1 staining demonstrated that NBQX significantly reduced the white matter injury in ipsilateral white matter in the H/I/LPS model (Fig. 7G, H).
Fig. 7.

NBQX was protective on both the H/I model and the H/I/LPS model. MBP (A) and O1 (B) staining as well as white matter injury score with MBP and O1 staining showed significant differences between vehicle and NBQX treatment (C, D) on the H/I model. MBP (E) and O1 (F) staining as well as white matter injury score with MBP and O1 staining showed that NBQX treatment significantly decreased white matter injury score by MBP and O1 ipsilateral to the ligation in the H/I/LPS model (G, H). (n=6/group, * p <0.01).
Edaravon attenuates white matter injury caused by hypoxia-ischemia with or without LPS co-administration
Third, edaravon was injected twice intraperitoneally at the dose of 10 mg/kg immediately after the artery occlusion and immediately after hypoxia. Similarly to NBQX treatment, edaravon treatment reduced white matter injury in the H/I model and ipsilateral white matter injury in the H/I/LPS model (Fig. 8).
Fig. 8.

Edaravone was protective on both the H/I model and the H/I/LPS model. There was increase of MBP (A) and O1 (B) staining in animals received edaravone treatment as compared to vehicle following UCL/hypoxia (30 min). Statistical analysis of white matter injury score with MBP and O1 staining showed significant differences between vehicle (saline containing 10% DMSO) and edaravone group (C, D). MBP (E) and O1 (F) staining showed that edaravone treatment increased MBP staining and O1 staining ipsilateral to the ligation as compared to vehicle in the H/I/LPS model. Statistical analysis of white matter injury score with MBP and O1 staining showed that edaravone treatment significantly decreased white matter injury score by MBP and O1 ipsilateral to the ligation in the H/I/LPS model (G, H). (n=6/group, * p <0.01).
Axon-OPC synapses are highly vulnerable to hypoxic-ischemic white matter injury
To reveal fine structural changes during white matter injury, we used electron microscopy to examine the general ultrastructure using vGluT1 and vGluT2 immunostained sections from mice subjected to hypoxia-ischemia (Fig. 9). Samples taken from the ipsilateral side and contralateral side were examined separately. In the contralateral white matter, ultrastructure of the tissues appeared intact; notably, organized, orientated axon bundles and oligodendrocytes or other cellular components were found normal; more electron-light and -intermediate oligodendroctes were found (Mori and Leblond, 1970; Back et al., 2007); and more importantly, some myelinated axons were identified in these samples (Fig. 9). In the injury side, degenerated elements, such as electron dense organelles, accumulated profiles, and degenerating unmyelinated axons and watery vascular processes were commonly found near the oligodendrocyte somata. In general, the nucleus of the oligodendrocyte showing multiple chromatin aggregated patches appeared normal and contained rough endoplasmic reticulum and mitochondria. However, in many cases, their processes were heavily vacuolated, showing signs of losing their structure. The cellular components including axon bundles were in general intact, but no myelinated axons were found.
Fig. 9.
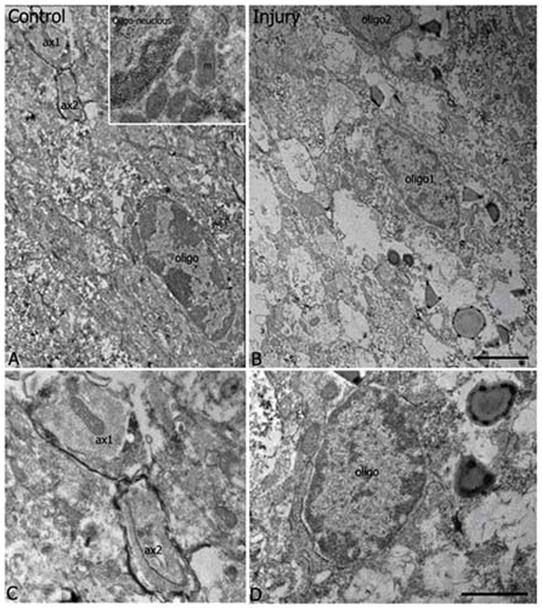
General ultrastructure. Electron microscopic examination showed ultrastructural integrity of contralateral (A, C) as compared to ipsilateral (B, D) white matter in mice subjected to hypoxia-ischemia. (A) Low power EM taken from contralateral side showed an intermediate dense oligodendrocyte soma (oligo) which contained a characteristic elongated nucleus with multiple chromatin patches and had a large amount of mitochondria (m) accumulated in cytoplasm (the area enlarged in the inset). A portion of its process extended toward the up left side where two axon profiles (ax1, ax2) were myelinated. No sign of degeneration was identified in cellular components and in the surrounding tissue. (B) Low power EM taken from the white matter of ipsilateral injury side showed two intermediate dense oligodendrocytes somata (oligo1, partial oligo2) which contained patched chromatins in their nucleus and exhibited some signs of alteration in the fine structure, such as vacuolated processes, accumulated electron dense materials in some vacuolar structures and some large membrane-delineated profiles surrounding the somata and the processes. No myelinated axons were identified in the surrounding region. (C) High magnification EM showed two myelinated axons (ax1, ax2) as indicated in (A). Several lamina of myelin sheath were obvious to wrap the axolemma. (D) High power EM showed the detailed ultrastructure of another oligodendrocyte soma (oligo), dense vacuolar structures; and some vacuolated profiles were seen in the surrounding region near the nucleus. (Bar in C= 2 μm; Bar in D=0.5 μm).
Previous studies have shown that OPCs can form synapses with axons in the white matter (Kukley et al., 2007; Ziskin et al., 2007). We thus sought to identify the postsynaptic densities (PSDs) under electron microscope (EM) as the synaptic features (Fig. 10). While PSDs associated with vGluT1 or vGluT2 immunolabeled terminals in the contralateral white matter were prominent (Fig. 10A, B), profound loss of PSDs in the labeled synapses was detected in the ipsilateral white matter at 24 h after hypoxia-ischemia (Fig. 10C, D).
Fig. 10.

Loss of postsynaptic densities (PSDs) in vGluT1 and vGluT2 labeled synapses. Electron microscopic examination showed that loss of PSDs in vGluT1 and vGluT2 labeled synapses was detected in the ipsilateral, injured white matter at 24 h after hypoxia-ischemia (Higher magnification images of PSDs in contralateral white matter are shown in the insets). PSDs associated with the labeled terminals in ipsilateral white matter were not prominent or absent altogether (arrow heads).
To ascertain that the location of the synapses was on NG2+ OPCs, we next used the NG2-DsRed transgenic mice to study the synaptic interaction of OPC with axons in white matter. Immunostaining of vGluT1 and vGluT2 on sections from NG2-DsRed mice showed that vGluT1 or vGluT2 positive vesicles were on DsRed-labeled NG2 cell bodies or processes (Fig. 11A, B). In the vGluT1 and vGluT2 immunostained tissues, electron-dense immunoperoxidase labeled presynaptic terminals were found in the white matter of contralateral side (Fig. 11C, D), however, the number of immunolabeled profiles was drastically reduced in the white matter compared to gray matter. They usually contained synaptic vesicles and mitochrondria, significant portion of these terminals formed asymmetrical synapses exhibiting prominent PSDs with postsynaptic profiles which showed variable sizes and contained microtubules and mitochrondria. Some of the profiles were in proximity to the somata of oligodendrocytes, but few labeled terminals contacted the somata. In the ipsilateral side, the number of vGluT1 and vGluT2 labeled terminals were drastically decreased (Fig. 11E, F). Although in many occasions presynaptic structures such as synaptic vesicles and mitochrondria could be identified, few labeled synapses had recognizable PSDs to associate with postsynaptic profiles. The PSD number of both vGluT1 and vGluT2 labeled synapses decreased approximately 4 times in ipsilateral white matter compared to synapses in contralateral side (Fig. 11G, H), whereas the decreases in PSDs were prevented or reversed in NBQX-treated mice (Fig. 11G, H).
Fig. 11.
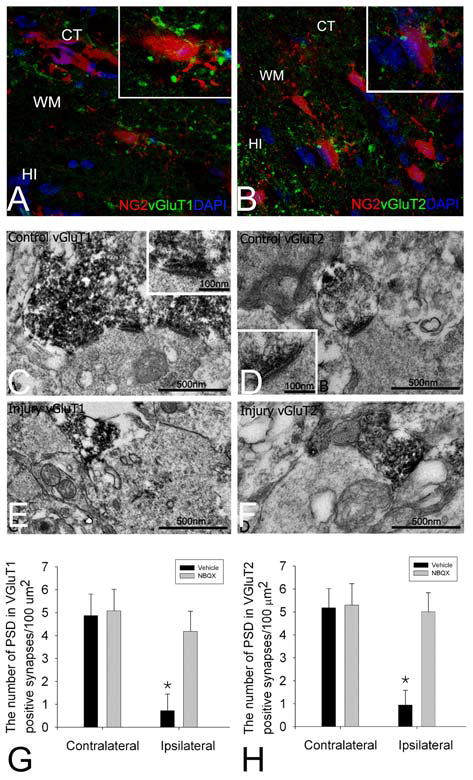
Analysis of the location of synapses on NG2-labeled cells, profound loss of these synapses induced by hypoxic-ischemic injury, and the prevention of synaptic loss by NBQX treatment. Immunostaining of vGluT1 and vGluT2 on sections from NG2-DsRed mice showed that vGluT1 or vGluT2 positive terminals were on DsRed-labeled NG2 cell bodies or processes (A and B). CT: cortex; WM, white matter; HI: hippocampus. Electron microscopic examination showed more vGluT1 (C) or vGluT2 (D) immunolabeled synapses in contralateral side compared to ipsilateral side stained for vGluT1 (E) or vGluT2 (F) in animals subjected to hypoxia-ischemia. In the contralateral side, postsynaptic densities (PSDs) which associated with vGluT1 (C) and vGluT2 labeled terminals (D) were prominent and clearly exhibited asymmetrical feature (higher magnification images of PSDs were shown in the insets). Although vGluT1 (E) or vGluT2 labeled presynaptic terminals (F) were identified in the injured side, PSDs associated with the labeled terminals were not prominent or absent altogether. Quantification of the number of PSDs counted in vGluT1 (G) and vGluT2 (H) labeled synapses showed a significant decrease in the ipsilateral white matter as compared to the contralateral side, whereas the decreases in PSDs were reversed in NBQX-treated mice (G and H) (n=8, *<0.05).
Next, we examined whether the loss of PSDs was related to the change in the sizes of presynaptic terminals or postsynaptic profiles (Fig. 12). The measurement of presynptic and postsynaptic area showed no significant change in the sizes of vGluT1 and vGluT2 labeled presynaptic terminals in both ipsilateral and contralateral sides, and no change in postsynaptic profiles contacted by vGluT1 labeled terminals in both sides. However, a drastic decrease in the sizes of postsynaptic profiles contacted by vGluT2 labeled synapse was found in the ipsilateral injured white matter (p<0.001) (Fig. 12). Collectively, these results indicate that hypoxia-ischemia causes alterations in glutamatergic axon-OPC synapses, the prominent effect is on PSDs, and hypoxia-ischemia selectively damage subgroups of OPC contacted by specific glutamatergic cells that synthesize vGluT2 protein.
Fig. 12.
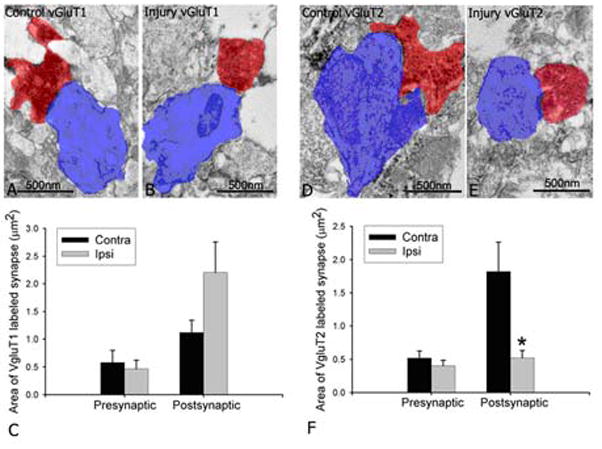
Selective loss in postsynaptic profiles of vGluT2 labeled synapses. Electron microscopic examination showed areas of vGluT1 labeled presynaptic terminals (red) and their postsynaptic profiles (blue) in the contralateral side (A) and ipsilateral side (B), and vGluT2 labeled presynaptic terminals (red) and their postsynaptic profiles (blue) in contralateral side (D) and ipsilateral side (E) of immature white matter of the mice after hypoxia-ischemia. For vGluT1, no difference was found in presynaptic and postsynaptic areas in both sides by quantitative analysis (C) (p=0.934 for presynaptic; p=0.083 for postsynaptic). For vGluT2, no difference was found in presynaptic areas, however, a significant decrease in postsynaptic areas was found in the injured side compared to contralateral side (F) (n=8, * p<0.05).
Ca2+-permeable AMPA receptors have been shown to mediate both the function of axon-OPC synapses (Ge et al., 2006) and excitotoxicity to OPCs (Deng et al., 2006). We performed immunoblots of AMPA receptor subunits (GluR1, 2, 3, 4) in the membrane preparations of white matter tissue, and the results showed that GluR2 levels were selectively downregulated 24 h after hypoxia-ischemia in mice at P6 (Fig. 13A). We also performed patch-clamp recording of NG2-DsRed cells in cerebral white matter slices from mice after hypoxia-ischemia. The I-V relationship showed that AMPA receptor function was altered following hypoxia-ischemia, showing more inward rectification as compared to slices from controls (Fig. 13B). Consistent with the previous demonstration that GluR2-lacking, Ca2+-permeable AMPA receptors critically regulate excitotoxicity to OPCs (Deng et al., 2003, 2006), these results indicate that axon-OPC synapses represent a crucial subcellular region for regulating AMPA receptor subunits and controlling GluR2 levels and Ca2+ permeability during white matter injury.
Fig. 13.
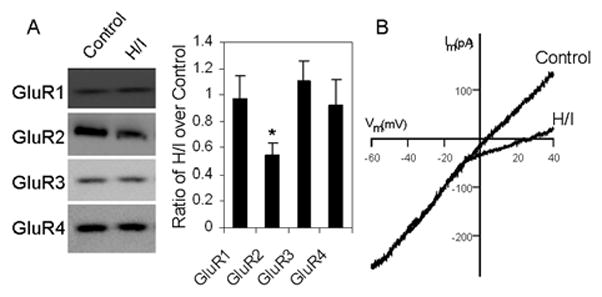
Immunoblotting of AMPA receptor subunits GluR1-4 in membrane preparations of white matter tissue 24 h after hypoxia-ischemia (H/I) showed that GluR2 levels were selectively downregulated (by western blots followed by densitometrical measurements for calculating the ratio of H/I over control tissue, * p<0.001) (A). In parallel, the I-V relationship from electrophysiological recording revealed that AMPA receptor function was altered in slices from mice 24 h after hypoxia-ischemia, showing more inward rectification consistent with the presence of GluR2-laking AMPA receptors (B).
We performed morphologic studies in both the H/I model and the H/I/LPS model. As the injury was more severe in the H/I/LPS model than that in the H/I model, the vulnerability of axon-OPC synapses revealed by the EM analysis in the H/I model expectedly was also seen in the H/I/LPS model. In addition, the decreases in the postsynaptic densities were prevented when neuroprotective strategies were used to decrease the white matter injury. Taken together, the findings concerning the vulnerability of axon-OPC synapses are the principal novel data of this report, linking the potential relevance of injury to axon-OPC synapses to the subsequent impairment of oligodendroglial differentiation and myelination revealed by the previous work (Back et al., 2001). This connection is of key importance of this work.
Discussion
In the present study, we not only have created and characterized clinically relevant mouse models of white matter injury, thus allowing for investigations into the disease pathogenesis using powerful mouse genetics (Wang et al., 2009; Nijboer et al., 2010) that confirmed major findings and principles obtained from previous rat models (Follett et al., 2000; 2004), but also have shown that axon-OPC synapses are quickly and profoundly disrupted via a Ca2+-permeable AMPA receptor dependent mechanism, thus identifying the axon-OPC synapses as a novel, highly sensitive target site during white matter damage. Therefore, the reported findings represent a significant conceptual advance in our understanding of white matter disease.
Although there has been a growing recognition of the importance of white matter pathology in human disease, the biology of white matter damage is woefully understudied, in part due to the difficulty to reliably model this type of injury in rodents. The traditional view from rodent studies is that gray matter is more vulnerable than white matter to common neurological insults such as ischemia. This view may be an artifact of how ischemic brain injury has been studied. Most experimental work has used rodents, in which white matter constitutes only about 15% of total brainvolume. In humans, white matter is about 50% of brain volume (Zhang et al., 2000). The study of white matter development and injury indeed has general implications for a variety of CNS diseases. In the present study, we have created age-specific models of cerebral white matter injury in the immature brain, a disorder known as periventricular leukomalacia. We first sought to convert the experimental model of PVL from the rat to mouse, and used combined hypoxia-ischemia and infection/inflammation at P6 to create clinically relevant models of PVL. It is expected that once such mouse models can replicate the critical features of PVL from phenotype to molecular and cellular events, we should have established the foundation to define disease biology and pathogenesis using available transgenic mouse strains. In the present study, the availability of the NG2-DsRed transgenic mice enabled us to analyze the synapses better than would be possible in rats. In addition, a defined murine model will also allow for the conduction of drug trials to discover candidate agents with therapeutic potential in a relatively high throughput manner, and for the testing of the utility of stem cells using available immunodeficienct mouse strains.
The pattern of hypoxic-ischemic brain injury is highly age-specific and also depends on the severity of the insult (Follett et al., 2002, 2004). In our hypoxic-ischemic mouse model, hypoxia for 30 min at P6 appeared to cause PVL-like white matter injury in ipsilateral side to the ligation with relative sparing of neurons in the overlying cerebral cortex compared to the contralateral hemisphere. Prolonged hypoxia (45 min) caused more profound gray matter injury or even infarcts in the cerebral cortex. P6 in mice corresponds to the human developmental period of greatest risk for PVL lesions, i.e. 24–32 gestational weeks in humans with regard to white matter maturation (Follett et al., 2000; Marret et al., 1995; Muse et al., 2001). Therefore, hypoxic-ischemic insult at this developmental stage causes relatively selective white matter injury in mice. More severe hypoxia conditions could lead to more profound gray matter injury (Rice et al., 1981). The vulnerability of immature white matter to hypoxic-ischemic injury is at least in part due to the intrinsic, maturation-dependent susceptibility of premyelinating oligodendrocytes that populate the immature white matter, and these immature cells have been shown to be a key cellular substrate in PVL (Rivkin et al., 1995; Kinney et al., 1998; Back et al., 1997, 2001, 2002, 2007).
Meanwhile, inflammation is one of major causes of PVL. LPS has been used as a type of endotoxin to induce white matter injury in various species including the cat, sheep, dog and rat (Gilles et al., 1977; Young et al., 1983; Duncan et al., 2002; Dean et al., 2009; Cai et al., 2003). To study multifactorial pathology of PVL, we combined LPS systemic administration with hypoxia-ischemia to create a new mouse model of PVL. The mechanisms by which LPS causes white matter injury include LPS induced hypoperfusion and inflammatory effects (Kopp et al., 1999; Aderem et al., 2001; Eklind et al., 2001; Young et al., 1982; Ando et al., 1988; Lehnardt et al., 2002, 2006). Consistent with previous studies (Degos et al., 2010; Verney et al., 2010; Hagberg et al., 2002; Eklind et al., 2005; Wang et al., 2006), our results demonstrate that LPS systemic administration increases microglia activation and astrogliosis and exacerbates hypoxic-ischemic white matter injury, suggesting that the inflammatory effects of LPS play an important role in our H/I/LPS model of PVL.
In our drug trials, we used NBQX, edaravone and minocycline in the H/I model and the H/I/LPS model. NBQX and edaravone protected the white matter after either H/I or H/I/LPS, indicating that both excitotoxicity and oxidative damage contribute importantly to the disease pathogenesis in the two models (Follett et al., 2000; 2004). Minocycline was protective against white matter injury in the H/I/LPS model but not the H/I model, presumably because inflammation is more critical in the H/I/LPS model. Minocycline is a clinically available antibiotic and anti-inflammatory drug that also demonstrates remarkable neuroprotective properties and seemingly acts as “A Jack of All Trades” in a variety of experimental models of neurological disease. Initial studies conducted on the neuroprotective effects of minocycline in experimental models of neurological diseases gave promising results. However, more recently, minocycline has been shown to have variable effects in different species and models of neurological disorders (Lechpammer et al., 2008; Fan et al., 2005; Cai et al., 2006). Results from clinical trials have shown beneficial or detrimental effects on different neurological diseases. Our results appear to highlight the anti-inflammatory function of minocycline against white matter injury. Although abundant evidence points to a convergent action of minocycline in suppression of both apoptosis and neural inflammation by preventing neuronal cell death and by inhibiting microglial activation, the exact primary targets of minocycline have yet to be identified and characterized, and its clinical potential for neuroprotection remains elusive. On the other hand, our data indicate that the protective effects of anti-excitotoxic and antioxidant agents on white matter injury are impressive. Previous studies have revealed a critical role of glutamate receptors in white matter development and injury, and hypoxic-ischemic white matter injury in rats could be prevented by the AMPA/KA subtype glutamate receptor antagonist NBQX (Follett et al., 2000). A subsequent study examined the efficacy of topiramate, a clinically safe anticonvulsant drug with AMPA/KA receptor blocking properties, in in vitro and in vivo models, and demonstrated similar protective efficacy of this agent (Follett et al., 2004). Edaravone, a free radical scavenger that is clinically available in Europe, has been shown to protect neurons in H/I models (Otomo et al., 2003; Zhang et al., 2005). Our demonstration of its efficacy in white matter injury provides a critical proof of evidence for its translational potential in such human diseases as PVL.
Overriding a long-standing rule in cellular neuroscience that the fast neurotransmission in the CNS only occurs at synaptic junctions between neurons, recent studies have unexpectedly revealed that OPCs can form synaptic contacts with neurons in the hippocampus (Bergles et al., 2000) and in the white matter (Kukley et al., 2007; Ziskin et al., 2007). Upon electrical stimulation, axons can release glutamate through synaptic contact to activate AMPA receptors on postsynaptic OPCs. The electrical activity in axons plays an important role in guiding oligodendrocyte development (Barres et al., 1993; Demerens et al., 1996), and glutamate release at axon-OPC synapses could be a key molecular signal used by OPCs to locate and myelinate axons. Under ischemic conditions, AMPA receptor mediated excitotoxicity is one of the main factors to cause white matter injury (Follett et al., 2000, 2004; Deng et al., 2003, 2006). Axon-OPC synapses could be a crucial subcellular region for recruiting and composing AMPA receptors under pathological conditions such as PVL. We found that PSD structures between axons and OPCs in cerebral white matter are selectively damaged as early as 24 h after hypoxia-ischemia in our mouse model of PVL. Although electron dense immunoperoxidase reaction products labeled for vGluT1 and vGluT2 obstruct ultrastructural details of the axonal terminals on postsynaptic processes, synaptic vesicles and some mitochrondria were still visible and no obvious signs of degeneration in these labeled terminals. Interestingly, there was no difference in the sizes of vGluT1 and vGluT2 labeled presynaptic axon terminals between ipsilateral side and contralateral side to the ligation, indicating that presynaptic axons could be stable during this time period. However, the significant decrease in PSD number on postsynaptic processes was demonstrated in both vGluT1 and vGluT2 labeled synapses in ipsilateral side. The disappearance of PSD suggests that a reorganization or disassociation of proteins or other postsynaptic components could be going on in this dynamic structure that is sensitive to pathological insult. We are confident that the postsynaptic processes were derived from OPCs and this is based on the following considerations: (i) the regions selected for EM analysis were verified first at light microscopic level and only the core of the cerebral white matter was chosen (ii) guided by LM, only regions showing axon bundles but no neuronal dendritic profiles or somata were subjected to EM analysis, and (iii) the tissues for our analysis on DsRed-labeled OPCs were also taken from the same regions used for EM analysis.
The postsynaptic processes contacted by vGluT2 positive terminals exhibited a significant shrinkage in the sizes indicating dramatic changes in their processes, suggesting that a portion of OPCs targeted by vGluT2 axons undergo a target-specific structural change such that their processes become thinner or retracted after the insult. The consequence of this structural alteration could lead to failures in myelinating the surrounding axon bundles. Our observation is consistent with previous findings showing that OPCs in areas of demyelinated lesions display a degenerative morphology (Reynolds et al., 2002; Cenci di Bello et al., 1999), and that vGluT1 and vGluT2 have differential distribution and function in the brain (Fremeau et al., 2004a, 2004b; Graziano et al., 2008). Our results show that the decrease of postsynaptic area selectively in vGluT2 positive axon-OPC synapse but not vGluT1 positive synapse, indicating that OPCs respond differently to vGluT1 or vGluT2 mediated synaptic transmissions during white matter injury. In addition, the axon-OPC synapses could be a crucial site for regulating AMPA receptor subunits, Ca2+ permeability and excitotoxicity (Liu et al., 2006; Follett et al., 2004; Talos et al., 2006a; 2006b). Further investigations into the underlying molecular and cellular mechanisms will shed light on pathogenesis of white matter injury that underlies such devastating human diseases as PVL.
Acknowledgments
We thank Edward G. Jones for his support and advice and for his critical comments on this manuscript, and Laird Miers for technical assistance. This study was supported by grants to W.D. from the NIH (R01 NS059043 and R01 ES015988), National Multiple Sclerosis Society, Feldstein Medical Foundation, and Shriners Hospitals for Children. We declare no conflicts of interest related to this article.
References
- Aderem A, Ulevitch RJ. Toll-like receptors in the induction of the innate immune response. Nature. 2000;406:782–787. doi: 10.1038/35021228. [DOI] [PubMed] [Google Scholar]
- Ando M, Takashima S, Mito T. Endotoxin, cerebral blood flow, amino acids and brain damage in young rabbits. Brain Dev. 1988;10:365–370. doi: 10.1016/s0387-7604(88)80094-9. [DOI] [PubMed] [Google Scholar]
- Back SA, Volpe JJ. Cellular and molecular pathogenesis of periventricular white matter injury. Ment Retard Dev Disabil Res Rev. 1997;3:96–107. [Google Scholar]
- Back SA, Luo NL, Borenstein NS, Levine JM, Volpe JJ, Kinney HC. Late ligodendrocyte progenitors coincide with the developmental window of vulnerability for human perinatal white matter injury. J Neurosci. 2001;21:1302–1312. doi: 10.1523/JNEUROSCI.21-04-01302.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Back SA, Han BH, Luo NL, et al. Selective vulnerability of late oligodendrocyte progenitors to hypoxia-ischemia. J Neurosci. 2002;22:455–463. doi: 10.1523/JNEUROSCI.22-02-00455.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barres BA, Raff MC. Proliferation of oligodendrocyte precursor cells depends on electrical activity in axons. Nature. 1993;361:258–260. doi: 10.1038/361258a0. [DOI] [PubMed] [Google Scholar]
- Bergles DE, Roberts JD, Somogyi P, Jahr CE. Glutamatergic synapses on oligodendrocyte precursor cells in the hippocampus. Nature. 2000;405:187–191. doi: 10.1038/35012083. [DOI] [PubMed] [Google Scholar]
- Cai Z, Pang Y, Lin S, Rhodes PG. Differential roles of tumor necrosis factor-alpha and interleukin-1 beta in lipopolysaccharide-induced brain injury in the neonatal rat. Brain Res. 2003;975:37–47. doi: 10.1016/s0006-8993(03)02545-9. [DOI] [PubMed] [Google Scholar]
- Cai Z, Lin S, Fan LW, Pang Y, Rhodes PG. Minocycline alleviates hypoxic-ischemic injury to developing oligodendrocytes in the neonatal rat brain. Neuroscience. 2006;137:425–435. doi: 10.1016/j.neuroscience.2005.09.023. [DOI] [PubMed] [Google Scholar]
- Cenci di Bello I, Dawson M, Levine J, Reynolds R. Generation of oligodendroglial progenitors in acute inflammatory demyelinating lesions of the rat brain stem is stimulated by demyelination rather than inflammation. J Neurocytol. 1999;28:366–381. doi: 10.1023/a:1007069815302. [DOI] [PubMed] [Google Scholar]
- Dean JM, Farrag D, Zahkouk SM, Zawahry EYEI, Hagberg, Kjellmer L, Mallard C. Cerebellar white matter injury following systemic endotoxemia in preterm fetal sheep. Neuroscience. 2009;160:606–615. doi: 10.1016/j.neuroscience.2009.02.071. [DOI] [PubMed] [Google Scholar]
- De Biase LM, Nishiyama A, Bergles DE. Excitability and synaptic communication within the oligodendrocyte lineage. J Neurosci. 2010;30:3600–3611. doi: 10.1523/JNEUROSCI.6000-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degos V, Favrais G, Kaindl AM, Peineau S, Guerrot AM, Verney C, Gressens P. Inflammation processes in perinatal brain damage. J Neural Transm. 2010;117:1009–1017. doi: 10.1007/s00702-010-0411-x. [DOI] [PubMed] [Google Scholar]
- Demerens C, Stankoff B, Logak M, Anglade P, Allinquant B, Couraud F, Zalc B, Lubetzki C. Induction of myelination in the central nervous system by electrical activity. Proc Natl Acad Sci USA. 1996;93:9887–9892. doi: 10.1073/pnas.93.18.9887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W. Neurobiology of injury to the developing brain. Nature Rev Neurol. 2010;6 (6):328–336. doi: 10.1038/nrneurol.2010.53. [DOI] [PubMed] [Google Scholar]
- Deng W, Pleasure J, Pleasure D. Progress in periventricular leukomalacia. Arch Neurol. 2008;65 (10):1291–1295. doi: 10.1001/archneur.65.10.1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Rosenberg PA, Volpe JJ, Jensen FE. Calcium-permeable AMPA/kainite receptors mediate toxicity and preconditioning by oxygen-glucose deprivation in oligodendrocyte precursors. Proc Natl Acad Sci USA. 2003;100:6801–6806. doi: 10.1073/pnas.1136624100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng W, Neve RL, Rosenberg PA, Volpe JJ, Jensen FE. AMPA receptor composition and CREB regulate oligodendrocyte excitotoxicity. J Biol Chem. 2006;281:36004–36011. doi: 10.1074/jbc.M606459200. [DOI] [PubMed] [Google Scholar]
- Duncan JR, Cock ML, Scheerlinck JP, Westcott KT, McLean C, Harding R, Rees SM. White matter injury after repeated endotoxin exposure in the preterm ovine fetus. Pediatr Res. 2002;52:941–949. doi: 10.1203/00006450-200212000-00021. [DOI] [PubMed] [Google Scholar]
- Eklind S, Mallard C, Arvidsson P, Hagberg H. Lipopolysaccharide induces both a primary and a secondary phase of sensitization in the developing rat brain. Pediatr Res. 2005;58:112–116. doi: 10.1203/01.PDR.0000163513.03619.8D. [DOI] [PubMed] [Google Scholar]
- Eklind S, Mallard C, Leverin AL. Bacterial endotoxin sensitizes the immature brain to hypoxia-ischaemic injury. Eur J Neurosci. 2000;13:1101–1106. doi: 10.1046/j.0953-816x.2001.01474.x. [DOI] [PubMed] [Google Scholar]
- Fan LW, Pang Y, Lin S, Rhodes PG, Cai Z. Minocycline attenuates lipopolysaccharide-induced white matter injury in the neonatal rat brain. Neuroscience. 2005;133:159–168. doi: 10.1016/j.neuroscience.2005.02.016. [DOI] [PubMed] [Google Scholar]
- Follett PL, Rosenberg PA, Volpe JJ, Jensen FE. NBQX attenuates excitotoxic injury in developing white matter. J Neurosci. 2000;20:9235–9241. doi: 10.1523/JNEUROSCI.20-24-09235.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Follett PL, Deng W, Dai W, Talos DM, Massillon LJ, Rosenberg PA, Volpe JJ, Jensen FE. Glutamate receptor-mediated oligodendrocyte toxicity in periventricular leukomalacia: A protective role for topiramate. J Neurosci. 2004;24:4412–4420. doi: 10.1523/JNEUROSCI.0477-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Kam K, Qureshi T, Johnson J, Copenhagen DR, Storm-Mathisen J, Chaudhry FA, Nicoll RA, Edwards RH. Vesicular glutamate transporters 1 and 2 target to functionally distinct synaptic release sites. Science. 2004a;304:1815–1819. doi: 10.1126/science.1097468. [DOI] [PubMed] [Google Scholar]
- Fremeau RT, Jr, Voglmaier S, Seal RP, Edwards RH. vGLUTs define subsets of excitatory neurons and suggest novel roles for glutamate. Trends Neurosci. 2004b;27:98–103. doi: 10.1016/j.tins.2003.11.005. [DOI] [PubMed] [Google Scholar]
- Ge WP, Yang XJ, Zhang Z, Wang HK, Shen W, Deng QD, Duan S. Long-term potentiation of neuron-glia synapses mediated by Ca2+permeable AMPA receptors. Science. 2006;312(5779):1533–1547. doi: 10.1126/science.1124669. [DOI] [PubMed] [Google Scholar]
- Gilles FH, Averill DR, Kerr CS. Neonatal endotoxin encephalopathy. Annals Neurol. 1977;2:49–56. doi: 10.1002/ana.410020108. [DOI] [PubMed] [Google Scholar]
- Graziano Alessandro, Liu X, Murray KD, Jones EG. Vesicular glutamate transporters define two sets of glutamatergic afferents to the somatosensory thalamus and two thalamocortical projections in the mouse. J Comp Neurol. 2008;507:1258–1276. doi: 10.1002/cne.21592. [DOI] [PubMed] [Google Scholar]
- Hagberg H, Peebles D, Mallard C. Models of white matter injury: comparison of infectious, hypoxic-ischemic, and excitotoxic insults. Ment Retard Dev Disabil Res Rev. 2002;8:30–38. doi: 10.1002/mrdd.10007. [DOI] [PubMed] [Google Scholar]
- Kinney HC, Back SA. Human oligodendroglial development: relationship to periventricular leukomalacia. Semin Pediatr Neurol. 1998;5:180–189. doi: 10.1016/s1071-9091(98)80033-8. [DOI] [PubMed] [Google Scholar]
- Kopp EB, Medzhitov R. The Toll-receptor family and control of innate immunity. Curr Opin Immunol. 1999;11:13–18. doi: 10.1016/s0952-7915(99)80003-x. [DOI] [PubMed] [Google Scholar]
- Kukley M, Capetillo-Zarate E, Dietrich D. Vesicular glutamate release from axons in white matter. Nat Neurosci. 2007;10:311–320. doi: 10.1038/nn1850. [DOI] [PubMed] [Google Scholar]
- Lechpammer M, Manning SM, Samonte F, Nelligan J, Sabo E, Talos DM, Volpe JJ, Jensen FE. Minocycline treatment following hypoxic/ischaemic injury attenuates white matter injury in a rodent model of periventricular leucomalacia. Neuropathol Appl Neurobiol. 2008;34:379–393. doi: 10.1111/j.1365-2990.2007.00925.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S, Lachance C, Patrizi S, Lefebvre S, Follett PL, Jensen FE, Rosenberg PA, Volpe JJ, Vartanian T. The toll-like receptor TLR4 is necessary for lipopolysaccharide-induced oligodendrocyte injury in the CNS. J Neurosci. 2002;22:2478–2486. doi: 10.1523/JNEUROSCI.22-07-02478.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehnardt S, Henneke P, Lien E, Kasper DL, Volpe JJ, Bechmann I, Nitsch R, Weber JR, Golenbock DT, Vartanian T. A mechanism for neurodegeneration induced by group B streptococci through activation of the TLR2/MyD88 pathway in microglia. J Immunol. 2006;177:583–492. doi: 10.4049/jimmunol.177.1.583. [DOI] [PubMed] [Google Scholar]
- Leviton A, Paneth N, Reuss ML, Susser M, Allred EN, Dammann O, Kuban K, Van Marter LJ, Pagano M, et al. Maternal infection, fetal inflammatory response, and brain damage in very low birth weight infants. Pediatr Res. 1999;46:566–575. doi: 10.1203/00006450-199911000-00013. [DOI] [PubMed] [Google Scholar]
- Liu B, Liao M, Mielke JG, Ning K, Chen Y, Li L, El-Hayek YH, Gomez E, Zukin RS, Fehlings MG, Wan Q. Ischemic insults direct glutamate receptor subunit 2-lacking AMPA receptors to synaptic sites. J Neurosci. 2006;26:5309–5319. doi: 10.1523/JNEUROSCI.0567-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marret S, Mukendi R, Gadisseux JF, Gressens P, Evrard P. Effect of ibotenate on brain development: an excitotoxic mouse model of microgyria and posthypoxic-like lesions. J Neuropathol Exp Neurol. 1995;54:358–370. doi: 10.1097/00005072-199505000-00009. [DOI] [PubMed] [Google Scholar]
- Martin JA, Kung HC, Mathews TJ, Hoyert DL, Strobino DM, Guyer B, Sutton SR. Annual summary of vital statistics: 2006. Pediatrics. 2008;121:788–801. doi: 10.1542/peds.2007-3753. [DOI] [PubMed] [Google Scholar]
- McDonald JW, Althomsons SP, Hyrc KL, Choi DW, Goldberg MP. Oligodendrocytes are highly vulnerable to AMPA/kainate receptor-mediated excitotoxicity. Nat Med. 1998;4:291–297. doi: 10.1038/nm0398-291. [DOI] [PubMed] [Google Scholar]
- Muse ED, Jurevics H, Toews AD, Matsushima GK, Morell P. Parameters related to lipid metabolism as markers of myelination in mouse brain. J Neurochem. 2001;76:77–86. doi: 10.1046/j.1471-4159.2001.00015.x. [DOI] [PubMed] [Google Scholar]
- Nijboer CH, Heijnen CJ, Willemen HL, Groenendaal F, Dorn GW, 2nd, van Bel F, Kavelaars A. Cell-specific roles of GRK2 in onset and severity of hypoxic-ischemic brain damage in neonatal mice. Brain Behav Immun. 2010;24:420–426. doi: 10.1016/j.bbi.2009.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Shea TM, Klinepeter KL, Mels PJ, Dillard RG. Intrauterine infection and the risk of cerebral palsy in very low birth weight infants. Pediatr Perinatal Epidemiol. 1998;12:72–83. [PubMed] [Google Scholar]
- Otomo E, Tohgi H, Kogure K, Hirai S, Terashi A, Gotoh F, Tazaki Y, Ito I, Swada T, Yamaguchi T, Kikuchi H, Kobayashi S, Fujishima H, Nakashima M. Effect of a novel free radical scavenger, edaravone (MCI-186), on acute brain infarction - Randomized, placebo-controlled, double-blind study at multicenters. Cerebrovasc Dis. 2003;15:222–229. doi: 10.1159/000069318. [DOI] [PubMed] [Google Scholar]
- Reynolds R, Dawson M, Papadopoulos D, Polito A, Cenci di Bello I, Pham-Dinh D, Levine J. The response of NG2-expressing oligodendrocyte progenitors to demyelination in MOG-EAE and MS. J Neurocytol. 2002;31:523–536. doi: 10.1023/a:1025747832215. [DOI] [PubMed] [Google Scholar]
- Rice JE, Vannucci RC, Brierley JB. The influence of immaturity on hypoxic-ischemic brain damage in the rat. Annals Neurol. 1981;9:131–141. doi: 10.1002/ana.410090206. [DOI] [PubMed] [Google Scholar]
- Rivkin MJ, Flax J, Mozel R, Osthanondh R, Volpe JJ, Villa-Komaroff L. Oligodendroglial development in human fetal cerebrum. Annals Neurol. 1995;38:92–101. doi: 10.1002/ana.410380116. [DOI] [PubMed] [Google Scholar]
- Rosenberg PA, Dai W, Gan X, Ali SB, Fu JM, Back SA, Sanchez RM, Segal MM, Follett PL, Jensen FE, Volpe JJ. Mature myelin basic protein-expressing oligodendrocytes are insensitive to kainate toxicity. J Neurosci Res. 2003;71:237–245. doi: 10.1002/jnr.10472. [DOI] [PubMed] [Google Scholar]
- Shen Y, Plane J, Deng W. Mouse models of periventricular leukomalacia. J Vis Exp. 2010;39 doi: 10.3791/1951. pii: 1951. http://www.jove.com/index/Details.stp?ID=1951. [DOI] [PMC free article] [PubMed]
- Talos DM, Fishman RE, Park H, Folkerth RD, Follett PL, Volpe JJ, Jensen FE. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. I. Rodent cerebral white matter and cortex. J Comp Neurol. 2006a;497:42–60. doi: 10.1002/cne.20972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talos DM, Follett PL, Folkerth RD, Fishman RE, Trachtenberg FL, Volpe JJ, Jensen FE. Developmental regulation of alpha-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid receptor subunit expression in forebrain and relationship to regional susceptibility to hypoxic/ischemic injury. II. Human cerebral white matter and cortex. J Comp Neurol. 2006b;497:61–77. doi: 10.1002/cne.20978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verney C, Monier A, Fallet-Bianco C, Gressens P. Early microglial colonization of the human forebrain and possible involvement in periventricular white-matter injury of preterm infants. J Anat. 2010;217:436–448. doi: 10.1111/j.1469-7580.2010.01245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Volpe JJ. Neurobiology of periventricular leukomalacia in the premature infant. Pediatric Res. 2001;50:553–562. doi: 10.1203/00006450-200111000-00003. [DOI] [PubMed] [Google Scholar]
- Wang X, Rousset CI, Hagberg H, Mallard C. Lipopolysaccharide-induced inflammation and perinatal brain injury. Semin Fetal Neonatal Med. 2006;11:343–353. doi: 10.1016/j.siny.2006.04.002. [DOI] [PubMed] [Google Scholar]
- Wang X, Stridh L, Li W, Dean J, Elmgren A, Gan L, Eriksson K, Hagberg H, Mallard C. Lipopolysaccharide sensitizes neonatal hypoxic-ischemic brain injury in a MyD88-dependent manner. J Immunol. 2009;183:7471–7477. doi: 10.4049/jimmunol.0900762. [DOI] [PubMed] [Google Scholar]
- Young RS, Yagel SK, Towfighi J. Systemic and neuropathologic effects of E. Coli endotoxin in neonatal dogs. Pediatr Res. 1983;17:349–353. doi: 10.1203/00006450-198305000-00008. [DOI] [PubMed] [Google Scholar]
- Young RS, Hernandez MJ, Yagel SK. Selective reduction of blood flow to white matter during hypotension in newborn dogs: a possible mechanism of periventricular leukomalacia. Annals Neurol. 1982;12:455–448. doi: 10.1002/ana.410120506. [DOI] [PubMed] [Google Scholar]
- Zhang N, Komine-Kobayashi M, Tanaka R, Liu MZ, Mizuno Y, Urabe T. Edaravone reduces early accumulation of oxidative products and sequential inflammatory responses after transient focal ischemia in mice brain. Stroke. 2005;36:2220–2225. doi: 10.1161/01.STR.0000182241.07096.06. [DOI] [PubMed] [Google Scholar]
- Zhang K, Sejnowski TJ. A universal scaling law between gray matter and white matter of cerebral cortex. Proc Natl Acad Sci U S A. 2000;97:5621–5626. doi: 10.1073/pnas.090504197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziskin JL, Nishiyama A, Fukaya M, Rubio M, Bergles D. Vesicular release of glutamate from unmyelinated axons in white matter. Nat Neurosci. 2007;10:321–330. doi: 10.1038/nn1854. [DOI] [PMC free article] [PubMed] [Google Scholar]