

Abstract
The nuclear DNA transcriptional programming of the mitochondria proteome varies dramatically between tissues depending on its functional requirements. This programming generally regulates all of the proteins associated with a metabolic or biosynthetic pathway associated with a given function, essentially regulating the maximum rate of the pathway while keeping the enzymes at the same molar ratio. This may permit the same regulatory mechanisms to function at low and high flux capacity situations. This alteration in total protein content results in rather dramatic changes in the mitochondria proteome between tissues. A tissues mitochondria proteome also changes with disease state, in Type 1 diabetes the liver mitochondrial proteome shifts to support ATP production, urea synthesis and fatty acid oxidation. Acute flux regulation is modulated by numerous post-translational events that also are highly variable between tissues. The most studied post-translational modification is protein phosphorylation which is found all of the Complexes of oxidative phosphorylation and most of the major metabolic pathways. The functional significance of these modifications is currently a major area of research along with the kinase and phosphatase regulatory network. This near ubiquitous presence of protein phosphorylations, and other post-translational events, in the matrix suggest that not all post-translational events have functional significance. Screening methods are being introduced to detect the active or dynamic post-translational sites to focus attention on sites that might provide insight into regulatory mechanisms.
Keywords: Protein phosphorylation, Diabetes, Oxidative phosphorylation, Metabolic regulation
In recent years, the mitochondrial proteome has begun to be appreciated as a dynamic program generated by the nuclear (nDNA) and mitochondrial DNA (mtDNA) to adapt the mitochondrial functional capacity to meet the needs of the tissue or disease state. In addition to the amounts of protein deposited into the mitochondrion, it is also clear that post-translational modifications are tissue and disease specific and modify the function and localization of mitochondria proteins and enzymes. Thus, any interpretation of mitochondrial events in disease should include a careful examination of translational and post-translational modifications of the mitochondrial proteome.
The Differential Protein Content of Mitochondria
The first determinations of the mitochondrial proteome were conducted mostly on a protein by protein basis where the general distribution of proteins in different tissues was appreciated. That is, the liver mitochondrion was known to contain large amounts of urea cycle enzymes, brain contained GABA-metabolizing enzymes, while heart specialized in ATP production. Differences were also known in numerous metabolic pathways and even in ion transport, especially Ca2+, as been well demonstrated in the brain (Brown et al. 2006;Oliveira and Goncalves 2009). However, it was not until the proteomic screening tools became available that the magnitude of the differences in this protein program was fully realized. Taylor et al (Taylor et al. 2003) published one of the first screens of the mammalian mitochondrial proteome of the human heart. In the same year, Mootha et al (Mootha et al. 2003) qualitatively evaluated the mitochondrial proteome from different tissues of the mouse using mass spectroscopic approaches. Though these authors focused on the common proteins in this study, they proposed that many quantitative differences in the protein contents likely exist between tissues. This was followed by another more quantitative mass spectroscopic study from Mann’s laboratory investigating the differences in the mitochondrial proteome in rat tissues (Forner et al. 2006) that supported the notion that the programming of the mitochondria is clearly different depending on the tissue. In our lab, Johnson et al (Johnson et al. 2007a;Johnson et al. 2007b) evaluated the quantitative differences in rat tissues using both quantitative mass spectrometry and 2-Dimensional Differential Gel Electrophoresis (2D-DIGE) approaches that revealed a remarkable variation in the quantitative distribution of mitochondrial proteins in the kidney, brain, heart and skeletal muscle of the rat. The 2D-DIGE approach was used to provide better quantitative approaches not available in mass spectrometry at the time, though the quantitative methods have significantly improved recently. These advantages of 2D-DIGE include: visualization of the protein differences, improved quantitative dynamic range, resolution of iso-electric variants (IEV) or molecular weight shifts representing differences in post-translational modifications or protein breakdown that are difficult to quantitatively detect using mass spectrometry techniques. Indeed, we find the 2D-DIGE methodology extremely useful in evaluating the contamination content, proteolysis artifacts, IEV modifications and quick global view of the differences difficult to obtain from mass spectroscopic approaches. Thus, many of our initial screens involve a 2D-DIGE that helps characterize the sample. 2D-DIGE takes protein from two samples and labels them differentially with a red or green dye. The protein extracts are then combined and run together on a 2D gel so that any variances in this rather unreliable format are the same for both protein samples. An example of the mouse liver versus mouse heart is presented in Figure 1. As seen in this Figure, the quantitative differences between the heart and liver are striking to the point that it is difficult to believe that these are really the same organelle. Outside of a few proteins, such as MnSOD, most of the proteins in the gel are either red or green indicating that they are not equal in the two tissues. These data demonstrate that each tissue alters its mitochondrial protein program depending on the specific functional needs of the tissue. The major reason that the differences in liver and heart appear so profound that when a particular function is altered by the nDNA translation all of the proteins involved in a pathway changes, not just one or two. The functional analysis of these data reveal that the heart is designed to have a high potential for creating ATP while the liver is more adapted for biosynthetic processes such as urea synthesis and processing of amino acids. Differences in nDNA programming of mitochondria have been observed for different tissues as discussed above as well as through development. Studies on whole mammalian sperm also show developmental alterations in the mitochondrial proteome through development and differentiation of the sperm (see review (Aitken and Baker 2007)). Forner et al demonstrated a striking difference between white and brown fat cells with the white cells having a specific increase in xenobiotic metabolism capacity(Forner et al. 2009).
Figure 1.
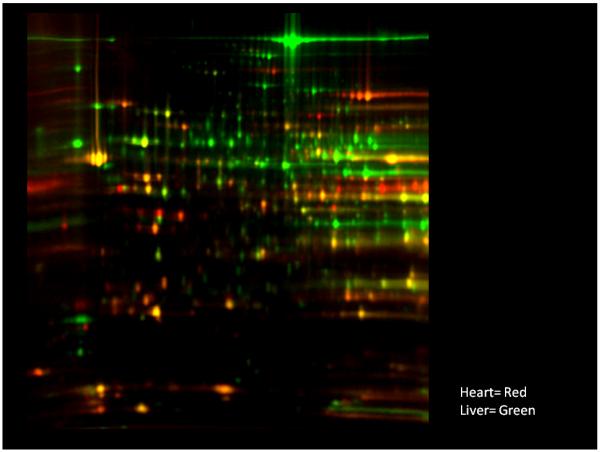
2D-DIGE of mouse heart and liver mitochondria using approaches described by Johnson et al (Johnson et al. 2008). Mitochondria were isolated from mouse liver and heart and subjected to identical protein extraction conditions with the exception of the liver mitochondrial protein being stained green while the heart protein was stained red.
Does the nDNA programming of the mitochondria change in disease states? It is quickly becoming clear that the mitochondria are reprogrammed in many different clinical conditions from diabetes (Johnson et al. 2008), obesity (Claessens et al. 2007), cancer (Krieg et al. 2004) and numerous genetic defects (Pagliarini et al. 2008) as well as viral infections (Diamond et al. 2007) and wound healing (Aden et al. 2008). Johnson et al (Johnson et al. 2008) conducted a comprehensive differential proteomic examination of a rat model of type 1 diabetes using both 2D-DIGE and mass spectroscopic techniques. Of the tissues studied, liver, heart and skeletal muscle, the liver revealed the largest number of protein level and IEV changes in diabetes. An example from a 2D-DIGE of the liver from control and diabetic animals is presented in Figure 2. A couple of the numerous IEV shifts are presented in the zoom regions at the right of the figure likely representing post-translational modifications in the diabetic liver. Protein assignments can be found in Johnson et al (Johnson et al. 2008). As clearly seen in this screen, a large number of proteins were modified in the liver. This study of mitochondrial protein variations within tissues during diabetes revealed general observations that were similar to that obtained in the tissue specific studies. That is, when alterations in the translational programming occur, they involve the entire pathway, not just one or two enzymes within a pathway. This global pathway up- or down-regulation is one of the reasons that the 2D-DIGE gel reveals such widespread change in protein contents since major changes in oxidative phosphorylation, glucose synthesis, methionine metabolism, urea cycle, and fatty acid oxidation were observed in this model. As shown by the tissue variation studies and disease state modifications, these proteomic studies are capable of nearly complete analysis of the metabolic flux potential of the tissue in one experiment. This provides an almost unparalleled examination of the major metabolic pathways flux potential in a control or diseased tissue (Johnson et al. 2007a;Johnson et al. 2008).
Figure 2.
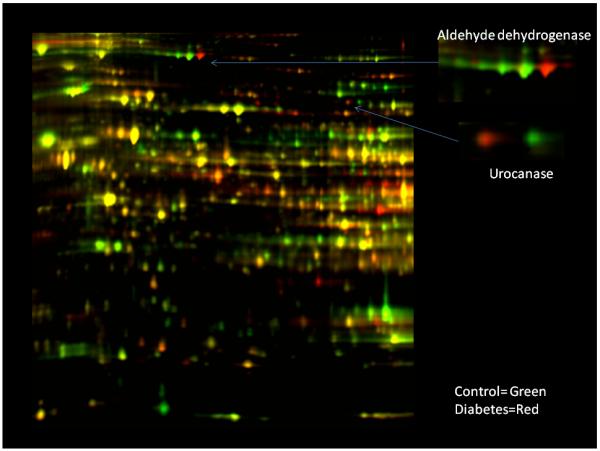
2D-DIGE of whole rat liver tissue extract from control and Type 1 Diabetic rats from Johnson et al (Johnson et al. 2008). The inserts are simply zoomed regions indicated by the arrows that represent regions where isoelectric shifts occurred. Experimental details along with protein identification are found in the original literature source.
Recently, an excellent relational database for the mitochondrial proteome was published from Mootha’s group (Pagliarini et al. 2008) that provides the relative protein content in many different tissues of the mouse as well as attempts to eliminate many cytosolic contaminates to the database using differential isolation techniques. Using this comprehensive database together with an analysis of the evolutionary aspects of electron transport chain formation, they were able to make predictions concerning genetic defects in Complex 1 function illustrating the power of analyzing genetic-based diseases using this approach.
The mitochondria proteome is also apparently altered in rapidly dividing cells where the mitochondria is enhanced in proteins involved in replication. This has been observed in cell culture lines (Schieke et al. 2006) as well as direct comparative studies in human tumors where mitochondrial chaperone proteins and elongation factors are up-regulated (Lin et al. 2007). Thus, the mitochondrial proteome may also be “tuned” to match the mitochondrial biosynthetic rate with the nuclear/cellular replicating rate in rapidly-dividing cells like cancer or embryological tissues. Differentiated mitochondrial function may be de-emphasized in favor of a program favoring mitochondria replication.
The co-ordination of the mtDNA and nDNA expression levels must be highly orchestrated since most of the Complexes assembled for oxidative phosphorylation have elements from mtDNA and nDNA that are generally represented in a fixed ratio in normal tissues. The mechanisms of the coordination of mtDNA and nDNA transcription are poorly understood, but several of the nuclear transcription factors have been identified (for example see (Martinez-Diez et al. 2006)). It is clear that if this coordination of mtDNA and nDNA was disrupted, adverse events could occur in the formation of the electron transport chain. It is of interest that the ratio of the mtDNA- and nDNA-encoded subunits in cytochrome oxidase (Krieg et al. 2004;Mazzanti and Giulivi 2006) and other subunits (Mazzanti and Giulivi 2006) seem to be mis-matched in some cancers. These results suggest that some disruption in the normal coordination of mitochondrial biosynthesis might occur in these rapidly-dividing cancer cells along with the previously discussed alterations associated with enhancing mitochondrial biosynthetic rates.
From this brief review, it is clear that there is no one simple mitochondrial proteome. The mitochondrial proteome is tuned to the specific needs of the tissue as well as the specific environment or disease state in which the cell is experiencing. This “tuning of function” involves setting the level of nearly all the proteins associated with a given metabolic pathway, not individual rate-limiting enzymes. Thus, regulation of flux via translation seems to regulate the total amount of enzyme in a given pathway, allowing post-translational modifications and other factors to control flux acutely. Keeping the ratio of the different enzymes within a given pathway constant, by increasing all enzymes simultaneously, may be important in the coordination of these acute regulatory mechanisms in the matrix. Thus, the translational control of metabolism is a regulation of Vmax by controlling the concentration of enzymes while the relative ratio of most of the enzymes is kept constant allowing one flux control network to work under low and high capacity conditions. An example of this global regulation mechanism is illustrated in Figure 3, where the relative level of the complexes of oxidative phosphorylation are compared between liver and heart and changes within the liver during the progression of type 1 Diabetes for oxidative phosphorylation. This analysis is basically extracted from the data presented in Figures 1 and 2. As seen in this example, most of the detected Complexes associated with oxidative phosphorylation are increased in the heart versus liver (as well as brain and kidney). I have excluded Complex II from this analysis since it involves the oxidation of FADH that is involved in synthetic reactions and not required for generation of ATP from NADH. A similar increase in these Complexes is observed within the liver alone when the ATP production is increased in Type 1 Diabetes. Thus, to increase the capacity for ATP production, most of the enzymes associated with oxidative phosphorylation are increased when comparing between tissues or during the adaptation within a given tissue. This is true for most of the metabolic pathways including fatty acid oxidation, amino acid processing, and urea cycle between tissues or adaptation within a given tissue such as the liver (Johnson et al. 2007a;Johnson et al. 2008). Having established several general principles of translational control of mitochondrial maximum flux capacity via protein content, how is acute flux modulated?
Figure 3.
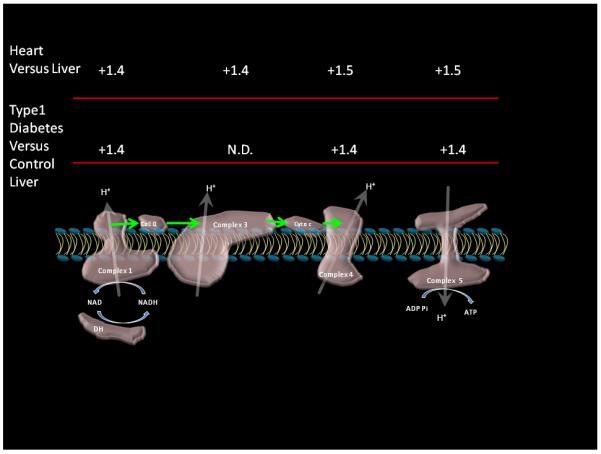
Relative changes in oxidative phosphorylation Complexes content between tissues and in the diabetic liver. Porcine liver and heart data as reported by Johnson et al (Johnson et al. 2007b). Diabetes data is from matched controls of rat liver under diabetic and control conditions (Johnson et al. 2008). Data were derived from the means of experimental data taking the mean difference of all of the subunits detected within a given complex. Complex 3 was not detected in the diabetes study that was on whole tissue decreasing the sensitivity to mitochondrial proteins.
Matrix Protein Post-Translational Modifications
The previous discussion focused on the levels of mitochondrial proteins in different tissues and disease states. This basically focuses on the nuclear transcriptional regulation of mitochondrial function by providing the required amount of protein to accomplish a given task. However, the acute regulation of flux must be controlled by post-translational events either in the form of direct modifications of the proteins, assembly of protein complexes, or via the concentrations of metabolites or allosteric regulatory factors. Recently, it has become evident that the pallet of post-translational modifications available to modify matrix enzyme function is extensive. These modifications include phosphorylation (Hopper et al. 2006;Kerbey et al. 1976;Schulenberg et al. 2003;Struglics et al. 2000), sumoylation (Braschi et al. 2009;Harder et al. 2004;Zunino et al. 2009), acylation (Kostiuk et al. 2008;Stucki et al. 1989), ADP-ribosylation (Scovassi 2004), glycosylation (Anello et al. 2004), S-nitrosylation (Foster and Stamler 2004;Moon et al. 2006;Sun et al. 2007), oxidation (Lin et al. 2002;Moon et al. 2006), sulferation (Ogata 1986) and many protein-protein interactions (Galante et al. 1981;Gledhill and Walker 2005;Green and Grover 2000).
Since protein phosphorylation is currently the major post-translational modification under investigation, I will focus this review on the current status of understanding this process. Though the protein phosphorylation regulation of pyruvate dehydrogenase was one of the first demonstrations and characterizations of protein phosphorylation regulating a metabolic enzyme (Denton et al. 1975), it was not believed to be widespread within the matrix. However, recent studies with dyes (Hopper et al. 2006;Schulenberg et al. 2003), 32P labeling (Aponte et al. 2009a;Aponte et al. 2009b;Bender and Kadenbach 2000;Papa et al. 1996;Struglics et al. 2000), mass spectrometry (Boja et al. 2009;Fang et al. 2007;Reinders et al. 2007); (Villen et al. 2007), antibodies (Lee et al. 2005) and IEV analysis of mitochondria proteins (Aponte et al. 2009b;Schieke et al. 2006) have suggested that protein phosphorylation is much more extensive than previously believed. Indeed, every Complex within oxidative phosphorylation has been found to have at least one, but more commonly many, phosphorylation sites detected by most of the methods available. Screening of protein phosphorylations using phosphopeptide-enriching schemes has resulted in a vast number of protein phosphorylation sites. The almost ubiquitous nature of the protein phosphorylations in the mitochondria raises the question as whether they are all significant or not. Clearly, the consensus sequences of many kinases are rather non-specific, resulting in the statistical structure of the web-based programs to predict phosphorylation sites. Thus, many sites might be venerable to a given kinase. Secondly, as long as the phosphorylation does not affect the protein function and does not turn over rapidly (thereby consuming significant amounts of ATP), it is not unreasonable that some of these phosphorylations detected in screens might actually just be “decorations” and not lead to significant functional events. Some of these sites could be used in the protein folding confirmation stages of the extensive processing that mitochondrial proteins undergo on entry into the mitochondria and are “locked” in place as structural elements deep within the protein. Finally, since most of these proteins traverse the cytosol, it is also possible that they are phosphorylated in the cytosol before ever entering the mitochondria. Phosphorylation has actually been proposed as involved in the signaling process targeting proteins to the mitochondria. With these concerns and the discovery of all of these phosphorylation sites with these different approaches, which protein phosphorylations should be further examined for the difficult task of establishing the functional significance of these modifications? There are several approaches that might prove useful, first to just study those proteins that play critical roles in the important pathways. However, as discussed above, all of the proteins seem to be important in most of the reaction pathways as illustrated by the up-regulation of most of the enzymes in the network, and regrettably most of these enzymes have phosphorylation sites. It would be useful if we knew the structure and the implications of a given phosphorylation site on protein function so we could predict the impact of a phosphorylation, but this is still well out the realm of protein bioinformatics for most proteins at the current time. Another approach is to use newly-emerging quantitative peptide phosphorylation methods to determine whether a mass spectrometry detected protein phosphorylaton, or any post-translational modification, is labile to physiological perturbations. We have accomplished this task using i-TraQ combined with an Orbit-trap mass spectrometer to monitor the changes in some protein phosphorylations with perturbations in intact mitochondria (Boja et al. 2009). However, the statistical requirements and preparation requirements, potential differential enhancement of phosphorylated peptides, impedes the detection limits of this approach. Possibly the next generation of detection schemes will overcome these limitations and permit the detection of phosphorylation lability as a criterion for further investigation.
The approach we have focused on that overcomes many of the limitations of simply detecting protein phosphorylation is the use of 32P incorporation from ATP synthesized by the mitochondria from added inorganic 32P. This essentially uses the mitochondria to create the γ-32P ATP from 32P at high activity within the mitochondrial matrix and then observe what turns over with regard to protein phosphorylation. This assures that: 1) The phosphorylation site is turning over and not locked in some structural requirement and 2) That the phosphorylation occurs in the matrix and 3) Can determine if the phosphorylation is labile to perturbations by following the labeling procedure with physiological perturbations. Using this approach, we have been successful demonstrating a wide range of protein phosphorylations in the mitochondrial matrix. The phosphorylation patterns in the heart and liver mitochondria are presented in Figure 4 as adapted from Aponte et al(Aponte et al. 2009b). In these experiments, 32P inorganic phosphate is added to fully energized mitochondria and permitted to incubate for 20 minutes. The protein is then extracted using standard methods and autoradiograms of the 2-dimensional gels run to detect 32P incorporation. The number of proteins reflecting a degree of phosphorylation turnover in both the heart and liver is impressive with over 100 spots being detected. Most could not be identified since they represented proteins that were of too low abundance to collect mass spectrometry identifications. In addition, we find that the pattern of 32P labeling does simply reflect the relative protein content differences as discussed above, but reveals a different degree of 32P incorporation for many of the proteins. This implies that the post-translational modifications are quantitatively different, implying that these events might reflect differences in the acute differential modification of the enzymatic activity. We have evaluated the time courses and performed cold and hot chase experiments to determine that freshly-isolated mitochondria are not at steady state with regard to protein phosphorylation. It seems that the mitochondria are generally dephosphorylated after isolation, likely due to the depletion of ATP. Upon warming and energization, the ATP pools rebuild as well as the protein phosphorylation pools. Thus, some of the differences between the liver and the heart could be due to the differences in the depletion at isolation rather than differences in the control network. This screen provides a large list of potential protein phosphorylations that dynamically turnover the matrix that may warrant further investigation. We have selected two enzymes at this stage to investigate further, succinyl-CoA synthetase (SCS), that is apparently highly phosphorylated in heart but not liver, and Complex V of mitochondrial oxidative phosphorylation, since it revealed several apparent phosphorylation sites.
Figure 4.
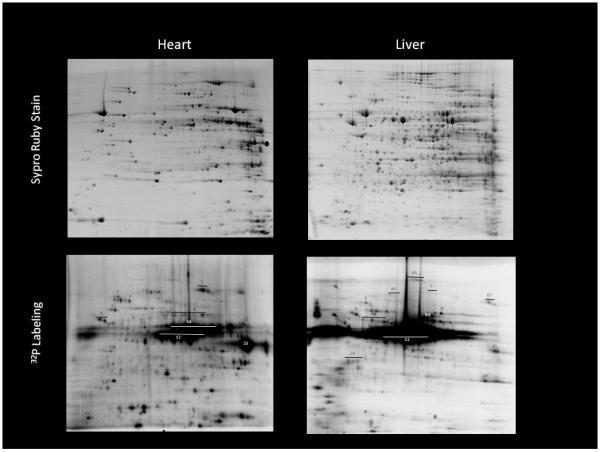
Protein content (upper panels) and autoradiograph (lower panels) of isolated heart and liver mitochondria. Mitochondria were exposed to 20 minutes of 32P labeling and then extracted identically. Protein identification and method details found in Aponte et al (Aponte et al. 2009b). Some key identifications: 12: PDH, 25:SCS, 9: β subunit Complex V, 1: aconitase
Succinyl-CoA synthetase is an element in the Krebs cycle that is the only source of substrate-level phosphorylation of ADP to ATP without using the mitochondria membrane potential. This protein was heavily labeled with 32P in our initial studies, but in running several controls looking for metabolite binding, we found that inorganic phosphate would bind to SCS tenaciously through the SDS denaturing process. Thus, the apparent phosphorylation was actually direct binding of phosphate to SCS that survived the SDS and electrophoresis process. This was a very surprising result on its own; however, further studies revealed that phosphate was acting as a V-Type allosteric activator of SCS, increasing the Vmax of the reaction by likely stabilizing its dimer formation (Phillips et al. 2009). This was an interesting finding, but reveals that great care must be taken with numerous controls to interpret 32P labeling. It should be stressed, however, that SCS is the only mitochondrial protein we have found that will carry hot 32P through the SDS-2D-Gel electrophoresis system at this time.
Complex V plays a major role in ATP synthesis as the enzyme that actually converts the potential energy in the membrane potential to the potential energy in ATP. We noted several previously unreported 32P associations with Complex V that were further investigated. To confirm our assignments in the complicated 2D gel of all mitochondrial proteins, we purified Complex V by immuno-capture from the protein homogenates of mitochondria exposed to 32P. This approach permitted the detection of even more phosphorylation sites by eliminating all of the overlapping spots (see Figure 5 adapted from Aponte et al(Aponte et al. 2009b). This includes the gamma and d chain phosphorylations which were not detected in the whole mitochondria protein gel. This simple observation likely indicates that the actual number of 32P incorporation sites has been grossly underestimated in the full protein 2D gels. The apparent phosphorylation of the beta subunit is highlighted in Figure 5 since the position of this 32P label is well correlated with the majority of beta subunit protein observed in the Ruby-stained gel. This implies that only a small fraction of the beta subunit is retaining 32P and whether this is a phosphorylation or another metabolite binding phenomenon is still under investigation. In any event, all of the other sites detected seem conventional and await further investigation to establish the functional significance of these events.
Figure 5.

The 32P labeling of Complex V purified after 32P labeling in the intact mitochondria. Energized porcine heart mitochondria were incubated for 20 minutes with 32P. Mitochondrial proteins were extracted and Complex V purified using immuno-capture procedures. The insert is a zoom of the beta subunit autoradiogram of the beta subunit demonstrating the lack of correlation with the majority of the beta subunit protein spots. Experimental details in Aponte et al (Aponte et al. 2009b)
As illustrated in these examples, 32P incorporation has several experimental limitations. Its sensitivity is remarkably high in detecting proteins that cannot be seen with the best protein stains. However, this suggests that tiny fractions of the phosphorylated protein would result in a positive hit, which might not reflect a biochemically significant event. In addition, we have shown that in some cases even the binding of phosphate metabolites, not phosphorylation, can generate a false positive with these assays. Thus, careful controls are advised, as outlined by Aponte et al (Aponte et al. 2009a), before interpreting any results.
The finding of these numerous protein phosphorylation events occurring in the matrix brings up another important area of research that has just begun to be evaluated. What is the protein kinase/phosphatase system in the mitochondrial matrix? Beyond the isoforms of PDH kinases and phosphatases, very few of these signaling molecules have been unequivocally uniquely located to the matrix space. The isoforms of PDH kinase might be suspicious as not just operating on PDH since the sequence homology of these enzymes is quite low approaching only 50% for PDK-4(Harris et al. 1995). Thus, these kinases may do more than suspected. Another possibility is that cytosolic kinases are translocated into the matrix space under different conditions through undefined mechanisms and could contribute to the kinase pool. It remains unclear how these kinases enter then matrix to phosphorylate proteins and then exit the matrix when their chore is complete. Very little information on the matrix phosphatases are available, leaving this an unsatisfactory gap in our knowledge. Thus, in addition to establishing the functional significance, if any, of the detected protein phosphorylations in the matrix, the issue of the enzymes responsible for these phosphorylation events remain.
Summary
Proteomic approaches can now rapidly access the net translational impact on the potential capacity of the mitochondria to perform different tasks. The functional program of the mitochondria is varied primarily by the translation of nDNA and is different for every tissue and even within a given tissue under different environmental or clinical conditions. The translational program is apparently quite crude in that it regulates the levels of nearly the entire enzyme complement of a given pathway, suggesting that it is truly modulating Vmax of the pathway while not altering the ratio of enzymes. This method of regulation potentially leaves network control mechanisms intact since the system remains balanced. The acute regulation of flux at the post-translational level is just now beginning to be fully appreciated. The literature is now exploding with new post-translational modifications within the mitochondria with phosphorylation only being one of many. The ubiquitous nature of the protein phosphorylations, and other post-translational modifications, almost requires that not all of these sites are regulatory and new approaches to establish which sites warrant the expense and time required to investigate. The regulation of the post-translational signaling network within the mitochondria is also poorly understood with the possible exception of the PDH reaction. This area of post-translational modification of mitochondrial function likely represents one of the key regulatory elements that we will need to explore before we will understand the function of the mitochondria in health and disease.
Acknowledgements
I would like to thank most of the members of my lab in this effort. Rachel Hopper and Stephanie Carroll initiated the proteomics program in the laboratory. Thor Johnson provided the insight for generating the relational quantitative data sets as well as initiating the 32P labeling experiments. Ksenia Blinova developed critical aspects of the Blue Native and Ghost native projects. Darci Phillips perfected many of the assays and protein separations as well as the Complex isolation methods and radioisotopic labeling. Angel Aponte, (G.B.) from the NHLBI Proteomic core worked tirelessly on the development of the 2D gel autoradiograms as well as the protein isolation procedures, while Emily Boja taught us the mass spectrometry methods for quantitative determination of protein phosphorylation and protein content. I also thank David Chess and Darci Phillips for his editorial help.
Reference List
- Aden N, Shiwen X, Aden D, Black C, Nuttall A, Denton CP, et al. Proteomic analysis of scleroderma lesional skin reveals activated wound healing phenotype of epidermal cell layer. Rheumatology. 2008;47:1754–1760. doi: 10.1093/rheumatology/ken370. [DOI] [PubMed] [Google Scholar]
- Aitken RJ, Baker MA. The role of proteomics in understanding sperm cell biology. Int J Andrology. 2007;31:295–302. doi: 10.1111/j.1365-2605.2007.00851.x. [DOI] [PubMed] [Google Scholar]
- Anello M, Spampinato D, Piro S, Purrello F, Rabuazzo AM. Glucosamine-induced alterations of mitochondrial function in pancreatic beta-cells: possible role of protein glycosylation. Am J Physiol Endocrinol Metab. 2004;287:E602–E608. doi: 10.1152/ajpendo.00320.2003. [DOI] [PubMed] [Google Scholar]
- Aponte AM, Phillips D, Harris RA, Blinova K, French S, Johnson DT, et al. 32P labeling of protein phosphorylation and metabolite association in the mitochondria matrix. Methods Enzymol. 2009a;457:63–80. doi: 10.1016/S0076-6879(09)05004-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aponte AM, Phillips D, Hopper RK, Johnson DT, Harris RA, Blinova K, et al. Use of (32)P to study dynamics of the mitochondrial phosphoproteome. J Proteome Res. 2009b;8:2679–2695. doi: 10.1021/pr800913j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bender E, Kadenbach B. The allosteric ATP-inhibition of cytochrome c oxidase activity is reversibly switched on by cAMP-dependent phosphorylation. FEBS Lett. 2000;466:130–134. doi: 10.1016/s0014-5793(99)01773-1. [DOI] [PubMed] [Google Scholar]
- Boja ES, Phillips D, French SA, Harris RA, Balaban RS. Quantitative Mitochondrial Phosphoproteomics Using iTRAQ on an LTQ-Orbitrap with High Energy Collision Dissociation. J Proteome Res. 2009 doi: 10.1021/pr900387b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braschi E, Zunino R, McBride HM. MAPL is a new mitochondrial SUMO E3 ligase that regulates mitochondrial fission. EMBO Rep. 2009;10:748–754. doi: 10.1038/embor.2009.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown MR, Sullivan PG, Geddes JW. Synaptic mitochondria are more susceptible to Ca2+overload than nonsynaptic mitochondria. J Biol Chem. 2006;281:11658–11668. doi: 10.1074/jbc.M510303200. [DOI] [PubMed] [Google Scholar]
- Claessens M, Saris WHM, Bouwman FG, Evelo CTA, Hul GBJ, Blaak EE, et al. Differential valine metabolism in adipose tissue of low and high fat-oxidizing obese subjects. Proteomics Clin Appl. 2007;1:1306–1315. doi: 10.1002/prca.200700049. [DOI] [PubMed] [Google Scholar]
- Denton RM, Randle PJ, Bridges BJ, Cooper RH, Kerbey AL, Pask HT, et al. Regulation of mammalian pyruvate dehydrogenase. Mol Cell Biochem. 1975;9:27–53. doi: 10.1007/BF01731731. [DOI] [PubMed] [Google Scholar]
- Diamond DI, Jacobs JM, Paeper B, Proll SC, Gritsenko MA, Carithers RI, et al. Proteomic Profiling of Human Liver Biopsies: Hepatitis C Virus-Induced Fibrosis and Mitochondrial Dysfunction. Hepatology. 2007;46:649–657. doi: 10.1002/hep.21751. [DOI] [PubMed] [Google Scholar]
- Fang JK, Prabu SK, Sepuri NB, Raza H, Anandatheerthavarada HK, Galati D, et al. Site specific phosphorylation of cytochrome c oxidase subunits I, IVi1 and Vb in rabbit hearts subjected to ischemia/reperfusion. FEBS Lett. 2007;581:1302–1310. doi: 10.1016/j.febslet.2007.02.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forner F, Foster LJ, Campanaro S, Valle G, Mann M. Quantitative proteomic comparison of rat mitochondria from muscle, heart, and liver. Mol Cell Proteomics. 2006;5:608–619. doi: 10.1074/mcp.M500298-MCP200. [DOI] [PubMed] [Google Scholar]
- Forner F, Kumar C, Luber CA, Fromme T, Klingenspor M, Mann M. Proteome differences between brown and white fat mitochondria reveal specialized metabolic functions. Cell Metab. 2009;10:324–335. doi: 10.1016/j.cmet.2009.08.014. [DOI] [PubMed] [Google Scholar]
- Foster MW, Stamler JS. New insights into protein S-nitrosylation. Mitochondria as a model system. J Biol Chem. 2004;279:25891–25897. doi: 10.1074/jbc.M313853200. [DOI] [PubMed] [Google Scholar]
- Galante YM, Wong SY, Hatefi Y. Mitochondrial adenosinetriphosphatase inhibitor protein: reversible interaction with complex V (ATP synthetase complex) Biochemistry. 1981;20:2671–2678. doi: 10.1021/bi00512a048. [DOI] [PubMed] [Google Scholar]
- Gledhill JR, Walker JE. Inhibition sites in F1-ATPase from bovine heart mitochondria. Biochem J. 2005;386:591–598. doi: 10.1042/BJ20041513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green DW, Grover GJ. The IF1 inhibitor protein of the mitochondrial F1F0-ATPase. Biochimica et Biophysica Acta-Bioenergetics. 2000;1458:343–355. doi: 10.1016/s0005-2728(00)00085-2. [DOI] [PubMed] [Google Scholar]
- Harder Z, Zunino R, McBride H. Sumo1 conjugates mitochondrial substrates and participates in mitochondrial fission. Curr Biol. 2004;14:340–345. doi: 10.1016/j.cub.2004.02.004. [DOI] [PubMed] [Google Scholar]
- Harris RA, Popov KM, Zhao Y, Kedishvili NY, Shimomura Y, Crabb DW. A new family of protein kinases--the mitochondrial protein kinases. Adv Enzyme Regul. 1995;35:147–162. doi: 10.1016/0065-2571(94)00020-4. [DOI] [PubMed] [Google Scholar]
- Hopper RK, Carroll S, Aponte AM, Johnson DT, French S, Shen RF, et al. Mitochondrial matrix phosphoproteome: effect of extra mitochondrial calcium. Biochemistry. 2006;45:2524–2536. doi: 10.1021/bi052475e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DT, Harris RA, Blair PV, Balaban RS. Functional consequences of mitochondrial proteome heterogeneity. Am J Physiol Cell Physiol. 2007a;292:C698–C707. doi: 10.1152/ajpcell.00109.2006. [DOI] [PubMed] [Google Scholar]
- Johnson DT, Harris RA, French S, Aponte A, Balaban RS. Proteomic Changes associated with Diabetes in the BB-DP rat. Am J Physiol Endocrinol Metab. 2008 doi: 10.1152/ajpendo.90352.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DT, Harris RA, French S, Blair PV, You J, Bemis KG, et al. Tissue heterogeneity of the mammalian mitochondrial proteome. Am J Physiol Cell Physiol. 2007b;292:C689–C697. doi: 10.1152/ajpcell.00108.2006. [DOI] [PubMed] [Google Scholar]
- Kerbey AL, Randle PJ, Cooper RH, Whitehouse S, Pask HT, Denton RM. Regulation of pyruvate dehydrogenase in rat heart. Mechanism of regulation of proportions of dephosphorylated and phosphorylated enzyme by oxidation of fatty acids and ketone bodies and of effects of diabetes: role of coenzyme A, acetyl-coenzyme A and reduced and oxidized nicotinamide-adenine dinucleotide. Biochem J. 1976;154:327–348. doi: 10.1042/bj1540327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kostiuk MA, Corvi MM, Keller BO, Plummer G, Prescher JA, Hangauer MJ, et al. Identification of palmitoylated mitochondrial proteins using a bio-orthogonal azido-palmitate analogue. FASEB J. 2008;22:721–732. doi: 10.1096/fj.07-9199com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krieg RC, Knuechel R, Schiffmann E, Liotta LA, Petricoin EF, herrmann PC. Mitochondrial proteome: Cancer-altered metabolism associated with cytochrome c oxidase subunit level variation. Proteomics. 2004;4:2789–2795. doi: 10.1002/pmic.200300796. [DOI] [PubMed] [Google Scholar]
- Lee I, Salomon AR, Ficarro S, Mathes I, Lottspeich F, Grossman LI, et al. cAMP-dependent tyrosine phosphorylation of subunit I inhibits cytochrome c oxidase activity. J Biol Chem. 2005;280:6094–6100. doi: 10.1074/jbc.M411335200. [DOI] [PubMed] [Google Scholar]
- Lin J, Xu J, Tian H, Gao X, Chen Q, Gu Q, et al. Identification of candidate prostate cancer biomarkers in prostate needle biopsy specimens using proteomic analysis. Int J Cancer. 2007;121:2596–2605. doi: 10.1002/ijc.23016. [DOI] [PubMed] [Google Scholar]
- Lin TK, Hughes G, Muratovska A, Blaikie FH, Brookes PS, rley-Usmar V, et al. Specific modification of mitochondrial protein thiols in response to oxidative stress: a proteomics approach. J Biol Chem. 2002;277:17048–17056. doi: 10.1074/jbc.M110797200. [DOI] [PubMed] [Google Scholar]
- Martinez-Diez M, Santamaria G, Ortega AD, Cuezva JM. Biogenesis and dynamics of mitochondria during the cell cycle: significance of 3′UTRs. PLoS One. 2006;1:e107. doi: 10.1371/journal.pone.0000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzanti R, Giulivi C. Coordination of nuclear- and mitochondrial-DNA encoded proteins in cancer and normal colon tissues. Biochem Biophys Acta. 2006;1757:618–623. doi: 10.1016/j.bbabio.2006.04.005. [DOI] [PubMed] [Google Scholar]
- Moon KH, Hood BL, Kim BJ, Hardwick JP, Conrads TP, Veenstra TD, et al. Inactivation of oxidized and S-nitrosylated mitochondrial proteins in alcoholic fatty liver of rats. Hepatology. 2006;44:1218–1230. doi: 10.1002/hep.21372. [DOI] [PubMed] [Google Scholar]
- Mootha VK, Bunkenborg J, Olsen JV, Hjerrild M, Wisniewski JR, Stahl E, et al. Integrated analysis of protein composition, tissue diversity, and gene regulation in mouse mitochondria. Cell. 2003;115:629–640. doi: 10.1016/s0092-8674(03)00926-7. [DOI] [PubMed] [Google Scholar]
- Ogata KVM. Comparative Properties of Bovine Heart and Liver Rhodaneses and the Regulatory Role of the Rhodaneses in Energy Metabolism. J Prot Chem. 1986;5:239–246. [Google Scholar]
- Oliveira JM, Goncalves J. In situ mitochondrial Ca2+ buffering differences of intact neurons and astrocytes from cortex and striatum. J Biol Chem. 2009;284:5010–5020. doi: 10.1074/jbc.M807459200. [DOI] [PubMed] [Google Scholar]
- Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, Ong SE, et al. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papa S, Sardanelli AM, Cocco T, Speranza F, Scacco SC, Technikova-Dobrova Z. The nuclear-encoded 18 kDa (IP) AQDQ subunit of bovine heart complex I is phosphorylated by the mitochondrial cAMP-dependent protein kinase. FEBS Lett. 1996;379:299–301. doi: 10.1016/0014-5793(95)01532-9. [DOI] [PubMed] [Google Scholar]
- Phillips D, Aponte AM, French SA, Chess DJ, Balaban RS. Succinyl-CoA Synthetase Is a Phosphate Target for the Activation of Mitochondrial Metabolism. Biochemistry. 2009 doi: 10.1021/bi900725c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinders J, Wagner K, Zahedi RP, Stojanovski D, Eyrich B, van der LM, et al. Profiling phosphoproteins of yeast mitochondria reveals a role of phosphorylation in assembly of the ATP synthase. Mol Cell Proteomics. 2007;6:1896–1906. doi: 10.1074/mcp.M700098-MCP200. [DOI] [PubMed] [Google Scholar]
- Schieke SM, Phillips D, McCoy JP, Jr., Aponte AM, Shen RF, Balaban RS, et al. The mammalian target of rapamycin (mTOR) pathway regulates mitochondrial oxygen consumption and oxidative capacity. J Biol Chem. 2006;281:27643–27652. doi: 10.1074/jbc.M603536200. [DOI] [PubMed] [Google Scholar]
- Schulenberg B, Aggeler R, Beechem JM, Capaldi RA, Patton WF. Analysis of steady-state protein phosphorylation in mitochondria using a novel fluorescent phosphosensor dye. J Biol Chem. 2003;278:27251–27255. doi: 10.1074/jbc.C300189200. [DOI] [PubMed] [Google Scholar]
- Scovassi AI. Mitochondrial poly(ADP-ribosylation): from old data to new perspectives. FASEB J. 2004;18:1487–1488. doi: 10.1096/fj.04-1841rev. [DOI] [PubMed] [Google Scholar]
- Struglics A, Fredlund KM, Konstantinov YM, Allen JF, Moller IM. Protein phosphorylation/dephosphorylation in the inner membrane of potato tuber mitochondria. FEBS Letters. 2000;475:213–217. doi: 10.1016/s0014-5793(00)01680-x. [DOI] [PubMed] [Google Scholar]
- Stucki JW, Lehmann LH, Siegel E. Acylation of proteins by myristic acid in isolated mitochondria. J Biol Chem. 1989;264:6376–6380. [PubMed] [Google Scholar]
- Sun J, Morgan M, Shen RF, Steenbergen C, Murphy E. Preconditioning results in S-nitrosylation of proteins involved in regulation of mitochondrial energetics and calcium transport. Circ Res. 2007;101:1155–1163. doi: 10.1161/CIRCRESAHA.107.155879. [DOI] [PubMed] [Google Scholar]
- Taylor SW, Fahy E, Zhang B, Glenn GM, Warnock DE, Wiley S, et al. Characterization of the human heart mitochondrial proteome. Nature Biotechnology. 2003;21:281–286. doi: 10.1038/nbt793. [DOI] [PubMed] [Google Scholar]
- Villen J, Beausoleil SA, Gerber SA, Gygi SP. Large-scale phosphorylation analysis of mouse liver. Proc Natl Acad Sci U S A. 2007;104:1488–1493. doi: 10.1073/pnas.0609836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zunino R, Braschi E, Xu L, McBride HM. Translocation of SenP5 from the nucleoli to the mitochondria modulates DRP1-dependent fission during mitosis. J Biol Chem. 2009;284:17783–17795. doi: 10.1074/jbc.M901902200. [DOI] [PMC free article] [PubMed] [Google Scholar]