

Apoptosis is a process present in a variety of cardiovascular diseases, and oxidative stress is a major apoptotic stimulus in these diseases. Glutathione S-transferase (GST) plays a crucial role against oxidative injury; it also regulates glutathione homeostasis. Recently, new roles for GST have been discussed such as in gene expression, protein glutathionylation and nitric oxide metabolism. However, its role with regard to cardiomyocyte apoptosis and alteration of signalling cascades of cardiomyocytes has not been determined. This article evaluates the effect of GST inhibition on cardiomyocyte apoptosis and on the alteration of proteins and mitogen-activated protein kinase pathways in rat models.
Keywords: Glutathione S-transferase, MAP kinases, Oxidative stress, Signalling
Abstract
Oxidative stress and ischemia-reperfusion (I/R) injury are crucial in the pathogenesis of cardiovascular diseases. The antioxidant glutathione S-transferase (GST) is responsible for the high-capacity metabolic inactivation of electrophilic compounds and toxic substrates. The main objective of the present study was to examine the effect of GST inhibition (with the administration of ethacrynic acid [EA]) on the viability and apoptosis of cardiomyocytes when these cells are exposed to various stress components of I/R and mitogen-activated protein kinase (c-Jun N-terminal kinase, p38 and extracellular signal-regulated kinase [ERK]) inhibitors. The primary culture of neonatal rat cardiomyocytes was divided into six experimental groups: control group of cells (group 1), cells exposed to H2O2 (group 2), I/R (group 3), I/R and EA (group 4), H2O2 coupled with EA (group 5), and EA alone (group 6). The viability of cardiomyocytes was determined using a colorimetric MTT assay. The apoptosis ratio was evaluated via fluorescein isothiocyanate-labelled annexin V and propidium iodide staining. c-Jun N-terminal kinase, p38, Akt/protein kinase B and ERK/p42-p44 transcription factors were monitored with flow cytometry. c-Jun N-terminal kinase activation increased due to GST inhibition during I/R. EA administration led to a significant increase in p38 activation following both H2O2 treatment and I/R. ERK phosphorylation increased when GST was exposed to I/R. A pronounced decrease in Akt phosphorylation was observed when cells were cotreated with EA and H2O2. GST plays an important role as a regulator of mitogen-activated protein kinase pathways in I/R injury.
A large collection of experimental data support the presence of apoptosis in a variety of cardiovascular diseases. It has also been well investigated that oxidative stress is a major apoptotic stimulus in many cardiac diseases (1–3). Among numerous defence mechanisms against oxidative injury, glutathione S-transferase (GST) plays a crucial role. The GST family, which comprises a relatively high amount of total cytosolic protein, is responsible for the high-capacity metabolic inactivation of electrophilic compounds and toxic substrates (4–6). Thus, glutathione homeostasis is essentially regulated by GST activity, and the glutathione redox status is critical for various biological events. Recently, novel roles for glutathione homeostasis and GST in signal transduction, gene expression, apoptosis, protein glutathionylation, nitric oxide metabolism and inflammation have been discussed (5,7,8).
It is important to note that alterations of cellular-reduced glutathione (GSH) metabolism and activity of GST can influence several signalling pathways (5,9). Certain types of GST play a key role in regulating mitogen-activated protein (MAP) kinase pathways involved in the cellular response to stress, apoptosis and proliferation (10,11), thus altering activity of apoptotic signal-regulating kinase-1 (ASK1) and influencing the decision regarding cell fate.
At least five of the human GST genes display functional polymorphisms. These polymorphisms are likely to contribute to interindividual differences in response to xenobiotics and clearance of oxidative stress products and, therefore, may determine susceptibility to various inflammatory pathologies including cancer, and cardiovascular and respiratory diseases (12,13). In addition, some GST polymorphisms have also been associated with increased risk of lung adenocarcinoma (12,14). Recent studies highlight the potentially unique roles of GST enzymes as crucial determinants of the development of ischemia-reperfusion (I/R). An association was found between different donor GST genotypes and primary graft dysfunction in patients following heart and lung transplantation (15,16). Other studies described the damaging effect of GST inhibition on peripheral and central motor neurons, cerebral astrocytes, isolated hepatocytes and vascular smooth muscle cells (17–20). Although the effect of GSH depletion in cardiomyocytes has been well described to be a result of different pathological states (5,21,22), the exact role of GST activity on cardiomyocyte apoptosis and alteration of signalling cascades of cardiomyocytes has not been determined.
Therefore, the present study was conducted to identify the biological role of GST in cardiomyocytes under oxidative stress conditions. Principally, the aim of the study was to evaluate the effect of GST inhibition (using ethacrynic acid [EA]) on cardiomyocyte apoptosis and on the alteration of proteins and MAP kinase pathways.
METHODS
Cell culture
The primary culture of neonatal rat cardiomyocytes was prepared as previously described (23,24). Briefly, cells were obtained from ventricular myocytes of two- to four-day-old Wistar rats (Charles-River Ltd, Hungary), using collagenase (Gibco Collagenase Type II, Invitrogen Corporation, USA). Isolated cells were plated on collagen I-coated plates (Coll Typ 1 cell coat, Greiner, Germany) at a density of 200,000/cm2. Cells were incubated in DMEM/F12 medium (Sigma-Aldrich, USA) and supplemented with 10% fetal bovine serum (Gibco, USA). The following day, when the cells firmly attached to the plate, the medium was replaced with a complete serum-free medium (CSFM) containing the following supplements: bovine serum albumin (2.5%; AlbuMax 1, Invitrogen Corporation), insulin (1 μM), transferrin (5.64 μg/mL), selenium (32 nM; insulin-transferrin-sodium-selenite media supplement, Sigma, Hungary), sodium pyruvate (2.8 mM, Sigma, Hungary), 3,3′,5′-triiodo-L-thyronine sodium salt (1 nM, Sigma), penicillin (100 IU/mL) and streptomycin (0.1 mg/mL) (PS solution, Sigma). Experiments began 24 h after incubation with CSFM, and the medium was changed every 24 h.
Cultured cardiomyocytes were randomly assigned to one of six experimental groups: control group of cells that were incubated in CSFM without treatment (group 1), cells treated with 150 μM of EA alone (group 2); cells exposed to 1 mM of H2O2 (group 3); cells exposed to I/R (group 4); cells treated with 1 mM of H2O2 together with 150 μM of EA (group 5); and cells exposed to I/R and 150 μM of EA (group 6).
In groups receiving I/R, cells were exposed to 1.5 h of ischemia using an ischemic buffer as described previously (25) followed by 2.5 h of reperfusion using normal CSFM. In group IV (cells were exposed to both I/R and EA), both the ischemic buffer and the reperfusion medium (CSFM) contained 150 μM of EA.
Based on the pilot experiments, a concentration of 150 μM and a treatment time of 4 h was chosen.
Cells were exposed to the above-mentioned concentration of chemicals for 4 h. MTT assay evaluation of cell survival was performed immediately after treatment termination. Assessment of apoptotic signalling markers also began after treatments until permeabilization, and samples were stored at –20°C until further processing according to the protocol supplied by the manufacturer. Experiments were repeated six times in duplicate wells.
Cell viability test
The viability of cardiomyocytes was determined using a colorimetric MTT assay (3-[4,5-dimethylthiazol-2-yl]-2,5-diphenyltetrazolium bromide, Sigma). The assay is based on the reduction of MTT into a blue formazan dye by the functional mitochondria of viable cells. At the end of the treatments, the medium was discarded from the plates, and the cells were subsequently washed twice with phosphate-buffered saline (PBS; Sigma). Cells were then incubated with PBS containing 0.5 mg/mL of MTT for 3 h at 37°C in an atmosphere of 5% CO2. The solution was carefully aspirated, and 1 mL of dimethylsulfoxide was added to dissolve the blue-coloured formazan particles. The samples from duplicate wells were transferred to a 96-well plate, and absorbance was measured by an ELISA reader (Sirio microplate reader, Seac Corporation, Italy) at 570 nm, and presented in arbitrary units. The results are expressed as percentages of control values.
Annexin V and propodium iodide staining of cells
The apoptosis ratio was evaluated after double staining with fluorescein isothiocyanate-labelled annexin V (BD Biosciences, Pharmingen, USA) and propidium iodide (PI; BD Biosciences) using flow cytometry, as previously described (26). First, the medium was discarded, and the wells were washed twice with an isotonic sodium chloride solution. Cells were removed from the plates using a mixture of 0.25% trypsin (Sigma, Hungary), 0.2% ethylene-diamine tetra-acetate (Serva, Hungary), 0.296% sodium citrate and 0.6% sodium chloride in distilled water. This medium was applied for 15 min at 37°C. The removed cells were washed in cold PBS, and were resuspended in binding buffer containing 10 mM Hepes NaOH, pH 7.4, 140 mM NaCl and 2.5 mM CaCl2. The cell count was determined in the Bürker’s chamber – 1 mL of solution contained 106 cells. One hundred microlitres of buffer (105 cells) was transferred into 5 mL round-bottom polystyrene tubes. Cells were incubated for 15 min with fluorescein-isothiocianate-conjugated annexin V molecules and PI, according to the manufacturer’s instructions. After this incubation period, 400 μL of annexin-binding buffer (BD Biosciences) was added to the tubes, as described by the manufacturer. The samples were immediately measured using a BD FacsCalibur flow cytometer (BD Biosciences), and analyzed using BD CellQuest software. Cells in each category were expressed as percentages of the total number of stained cells.
Statistics
All data were presented as mean ± SEM. Differences between groups were assessed with one-way ANOVA and Student’s t test. P<0.05 was considered to be statistically significant.
RESULTS
An MTT assay was performed to measure the absolute number of living cells in the groups. In the control group, the amount of living cells was increased to 100%. EA alone reduced the ratio of living cells to 43.41±11.15%, measured by MTT assay. Both I/R and H2O2 alone caused a marked reduction in the amount of living cells. The effect of cell death was significantly stronger on EA administration in groups treated with H2O2 or exposed to I/R (Figure 1).
Figure 1).
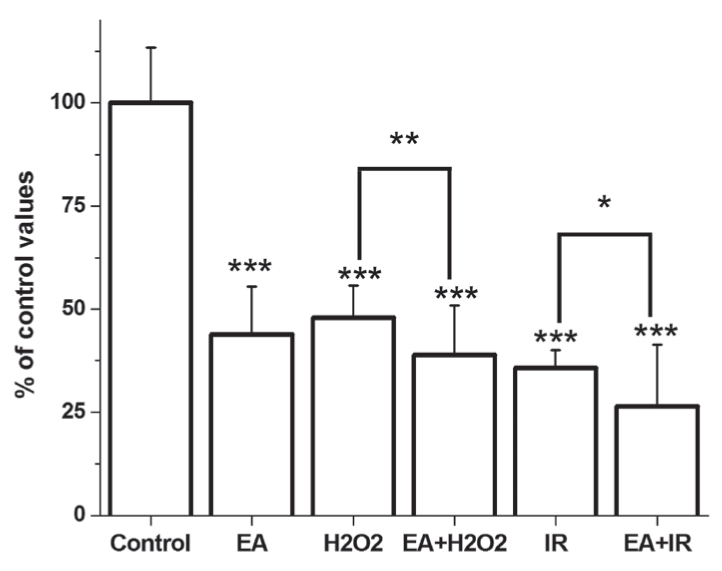
Viability of cardiomyocytes as measured by the MTT assay. *P<0.05; **P<0.001; ***P<0.05 compared with the control group. EA Ethacrynic acid; I/R Ischemia and reperfusion
The control group had 85.7±1.94% of intact, living cells (annexin V and PI negative) and 4.6±0.82% of cells in the early phase of apoptosis (annexin V positive and PI negative) (Figures 2 and 3). EA administration decreased the amount of living cells, and increased the percentage of apoptotic cells. A significant increase in the amount of apoptotic cells was observed in both the H2O2-treated and I/R groups, with a lower number of living cells (Figures 2 and 3). When EA was added in groups treated with H2O2 or I/R, the quantity of apoptotic cells was further increased and the amount of living cells was decreased. Interestingly, EA increased the amount of necrotic cells (annexin V negative and PI positive) during I/R and decreased the number of living cells.
Figure 2).
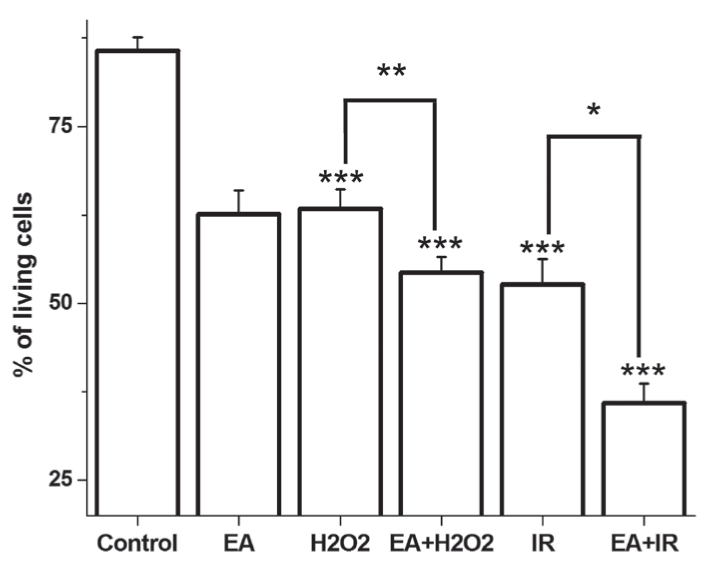
The mean percentage of living cells. Data expressed as mean percentage ± SEM. *P<0.05; **P<0.01; ***P<0.05 compared with the control group. EA Ethacrynic acid; I/R Ischemia and reperfusion
Figure 3).
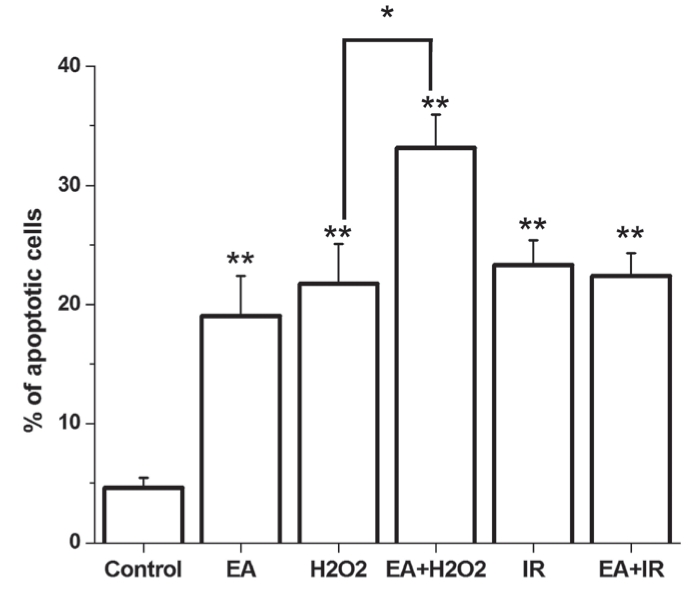
The mean percentage of apoptotic cells. Data are expressed as mean percentage ± SEM. *P<0.05; **P<0.05 compared with the control group. EA Ethacrynic acid; I/R Ischemia and reperfusion
c-Jun N-terminal kinase (JNK) activation increased markedly on EA administration to cardiomyocytes (Figure 4). H2O2 treatment increased the level of activated JNK; however, this difference was not significant. I/R caused a noticeable increase in JNK activation. On the other hand, EA was capable of augmenting the activation of JNK significantly when cells were cotreated with H2O2 or when cells were exposed to I/R (Figure 4).
Figure 4).
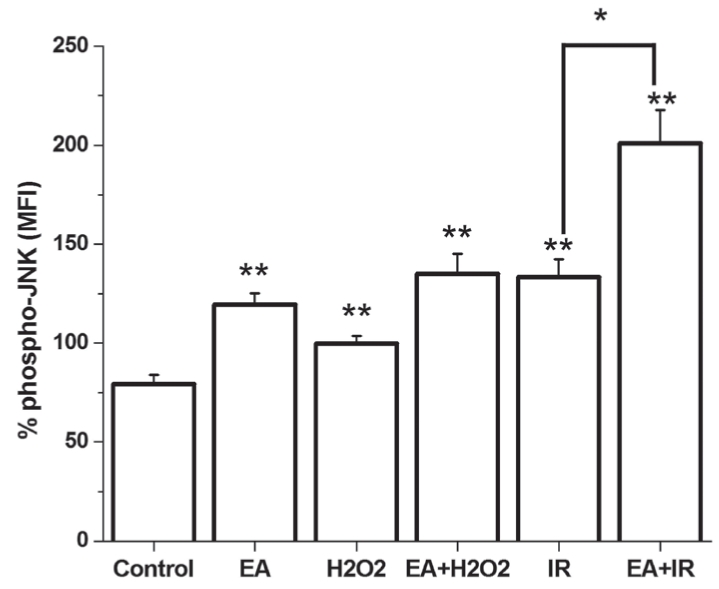
Phosphorylation of c-Jun N-terminal kinase (JNK) is demonstrated in cultured cardiomyocytes. *P<0.01; **P<0.05 compared with the control group. EA Ethacrynic acid; I/R Ischemia and reperfusion; MFI Mean fluorescence intensity
GST inhibition led to a significant increase in p38 activation related to nontreated cells. Both H2O2 incubation and I/R resulted in a significant increase in p38 MAP kinase activation. EA administration during I/R increased p38 activity to 357.57±5.39% of control values. Likewise, when cells were incubated with H2O2 together with EA, the level of phophorylated p38 markedly increased; however, this difference was not statistically significant compared with the group treated with H2O2 alone (Figure 5).
Figure 5).
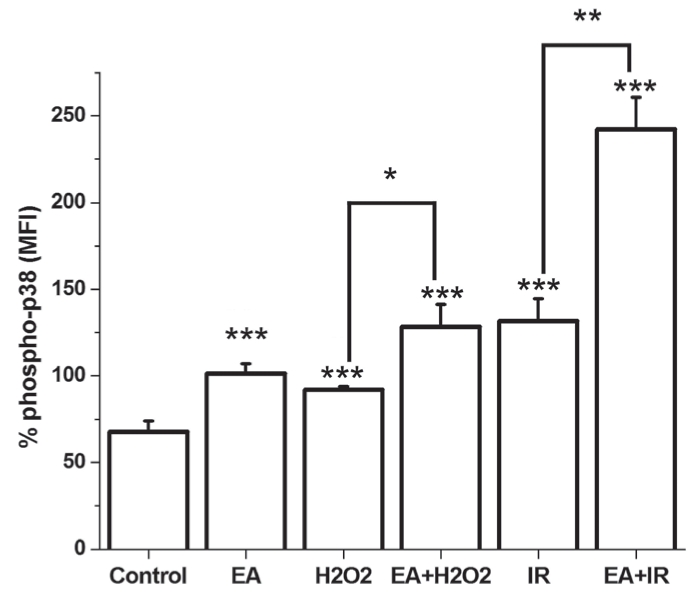
Phosphorylation of p38 (phospho-p38) mitogen-activated protein kinase is demonstrated in cultured cardiomyocytes. *P<0.05; **P<0.01; ***P<0.05 compared with the control group. EA Ethacrynic acid; I/R Ischemia and reperfusion; MFI Mean fluorescence intensity
Extracellular signal-regulated kinase (ERK) phosphorylation increased in GST-inhibited groups (incubated with EA) that were either treated with H2O2 or exposed to I/R, without any statistically significant difference. ANOVA failed to evaluate significant differences between groups. Moreover, the analysis of difference (using Student’s t test) between the group receiving I/R and the group incubated with EA during I/R revealed statistically significant divergence (Figure 6).
Figure 6).
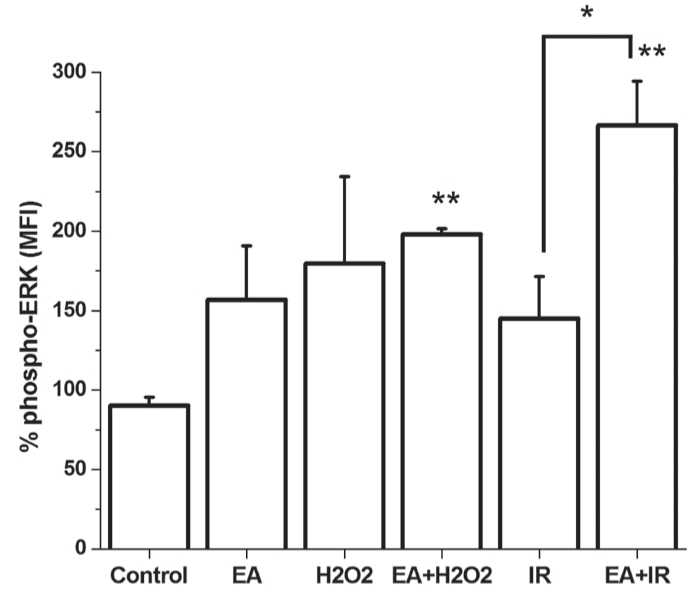
Phosphorylation of extracellular signal-regulated kinase (phospho-ERK) is demonstrated in cultured cardiomyocytes. *P<0.05; **P<0.05 compared with the control group. EA Ethacrynic acid; I/R Ischemia and reperfusion; MFI Mean fluorescence intensity
Both administration of EA, H2O2 and I/R caused nonsignificant reduction of Akt activity. On the other hand, H2O2 treatment resulted in a more pronounced decrease (40.49±5.68%) of Akt phosphorylation when GST was inhibited by EA (Figure 7). There was no significant difference among groups as evaluated by ANOVA.
Figure 7).
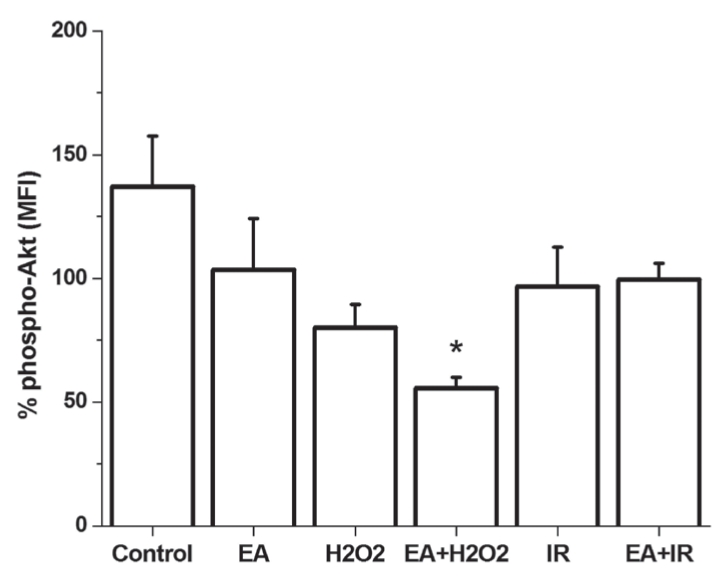
Phosphorylation of Akt (phospho-Akt) is demonstrated in cultured cardiomyocytes. *P<0.05 compared with the control group. EA Ethacrynic acid; I/R Ischemia and reperfusion; MFI Mean fluorescence intensity
DISCUSSION
The present study showed that pharmacological inhibition of GST could markedly exaggerate oxidative stress-induced apoptosis in cardiomyocytes. GST inhibition was associated with increased activation of MAP kinases under stress conditions.
A key determinant of the cellular response to oxidative stress relates to the level and molecular form of glutathione (27). A crucial factor that affects the level of glutathione is its utilization via GST (28). GSTs function by conjugating reduced GSH and catalyzing the attack on foreign compound or oxidative stress products, generally forming less-reactive materials that can be readily excreted. There are three distinct families of GSTs in mammals: cytosolic, mitochondrial and membrane-associated proteins. Cytosolic GSTs represent the largest family, which is subdivided into seven classes based on their amino acid sequence (mu, pi, theta, alpha, sigma, omega and zeta). Those within a particular class typically share more than 40% of their identity, but at least five of the human GST genes display functional polymorphisms. These polymorphisms are likely to contribute to interindividual differences in response to xenobiotics and clearance of oxidative stress products and, therefore, may determine susceptibility to various inflammatory pathologies including cancer, and cardiovascular and respiratory diseases (13). Several studies indicate that loss of mu, pi and theta GST genes increases susceptibility to inflammatory diseases such as asthma (29). In addition, some GST polymorphisms, such as GSTM1 null genotype, have also been associated with increased risk of lung adenocarcinoma, especially when found in combination with GSTP1 Val polymorphism (14). Recent studies highlight the potentially unique roles of GST enzymes as crucial determinants of the development of I/R. In the area of heart and lung transplantation, investigators found an association between donor GST mu null genotypes and primary graft dysfunctions in patients. Particularly, the donor GST mu null and GST pi_313 AA genotypes were individually associated with increased development of graft dysfunction; however, the combined phenotype was associated with 70% occurrence of graft failure (15). Further investigation validated the clinical significance of these findings because results showed that the donor GST mu genotype was independently associated with graft failure, which significantly improved the ability to predict it (16).
The present study used EA to pharmacologically inhibit GST. EA has been shown to be a substrate of many GST isoezymes; furthermore, nonenzymatic GSH conjugation of EA also exists. Moreover, it was shown that the EA glutathione conjugate is a GST inhibitor due to its greater affinity for the enzymes, whereas EA itself inhibits GST through reversible covalent interactions (6).
Administration of EA resulted in a marked increase of apoptotic cells, principally when cells were cotreated with H2O2. The amounts of necrotic cells were elevated following EA treatment and in the group receiving I/R and EA simultaneously. The increased level of reactive oxygen species and a more unfavourable glutathione state may exaggerate the intensity of insult and may explain the increased amount of necrotic cells in GST-inhibited groups during I/R.
On the other hand, GST acts as regulator of MAP kinase pathways. For example, GST has been shown to be an endogenous inhibitor of JNK via protein-protein interaction; thus influencing cellular stress response and apoptosis (27). JNK has been implicated in apoptotic signalling and mediates cytotoxicity in various conditions including I/R, oxidative and nitrosative stress (10,30). In normal cells, JNK activity is maintained at a low level through interaction with GST (4,8,31). Under oxidative or nitrosative stress, GST and JNK dissociate, thus activating JNK and oligomerizing GST. Meanwhile, the liberated JNK regains its activity by phophorylation during oxidative or nitrosative stress, and further phosphorylates c-Jun. Similar processes occur with c-Jun, the downstream effector of JNK (32). Moreover, several phosphatases interact with GST and are subject to glutathionylation, thus regulating the phophorylation state of signalling pathways (4,8). We have found that pharmacological inhibition of GST augments JNK activity itself. This could be explained by elimination of JNK sequestration within a protein complex with GST, and inhibition of S-glutathionylation. On the other hand, effective inhibition of GST may cause oxidative injury, even on otherwise unstressed cells due to hindered elimination of trivially developing oxidant and toxic materials. This may occur as a result of JNK phosphorylation. It has already been described that GST knockout mice have high basal JNK activity; furthermore, treatment of cells with a potent GST inhibitor causes activation of JNK (4,33).
The same paradigm seems to hold true for other kinases such as thioredoxin or ASK1, influencing cellular stress response and cell fate decision (4,33,34). Similar to JNK, ASK1 activity is also reduced by protein-protein sequestration under unstressed conditions (11). However, oxidative stress triggers dissociation of the thioredoxin-ASK1 complex and further catalyzes the dissociation of GST-ASK1, leading to ASK1 activation. Liberation of thioredoxin causes p38 activation (11). On the other hand, ASK triggers the apoptogenic kinase cascade, leading to phophorylation of JNK and p38 MAP kinase. It also regulates the dynamic balance between apoptotic (JNK and p38) and survival (AKT and ERK) pathways (35,36). Recently, a novel role of GST has been described, since it has been identified that GST also heterodimerizes with tumour necrosis factor receptor-associated factor 2, thus reducing activation of both JNK and p38 (4,11).
The signalling pathway through p38 MAPK is activated by oxidative stress and is associated with cellular damage, mediation stress response and cytokine production. We found that oxidative injury and I/R cause noticeable induction of p38 activity in cardiomyocytes, which is further increased by EA administration. Our results regarding p38 activation on GST inhibition can be explained by the above-described processes.
According to our results, ERK is activated on GST inhibition in the presence of H2O2 administration or during reperfusion. The level of phosphorylated ERK of GST-inhibited cells receiving I/R exceeded the ERK phosphorylation level of cells that have undergone I/R alone. These findings may represent the association between ERK and GST. It has already been described that an immortalized fibroblast isolated from a GST mu genotype expressed significantly elevated ERK activity. Moreover, treatment with a potent GST inhibitor resulted in activation of ERK (33), and vice versa, the transcriptional induction of the GST gene is orchestrated by signalling pathways, such as ERK, which might deteriorate due to GST inhibition (37).
Although similar relationships between the synthesis of GST and Akt have been well investigated, the effect of GST inhibition on Akt-mediated cellular survival has not been fully described (37,38). Akt activity reduced significantly in groups receiving the GST inhibitor (EA) when compared with control values. Our results failed to show any further association between GST inhibition and Akt activity. The hindered antioxidant, antitoxic defense of cells treated with EA and the activation of ASK1 may explicate the results described by us.
SUMMARY
The present study showed that inhibition of GST by EA augments apoptosis as a result of oxidative injury and I/R. EA administration also induces the activation of the JNK, p38 and ERK pathways. These findings highlight the importance of GST and its polymorphisms in cardiac diseases. Therefore, further studies are needed to investigate the in vivo effect of GST inhibition and the role of GST polymorphism on myocardial damage under different pathological conditions in humans.
Acknowledgments
This work was supported by the Grants OTKA 78434, and the Hungarian Academy of Sciences.
REFERENCES
- 1.Chen QM, Tu VC. Apoptosis and heart failure: Mechanisms and therapeutic implications. Am J Cardiovasc Drugs. 2002;2:43–57. doi: 10.2165/00129784-200202010-00006. [DOI] [PubMed] [Google Scholar]
- 2.Kumar D, Jugdutt BI. Apoptosis and oxidants in the heart. J Lab Clin Med. 2003;142:288–97. doi: 10.1016/S0022-2143(03)00148-3. [DOI] [PubMed] [Google Scholar]
- 3.Roth E, Hejjel L. Oxygen free radicals in heart disease. In: Pugsley MK, editor. Cardiac Drug Development Guide. Humana Press; 2003. pp. 47–66. [Google Scholar]
- 4.Townsend DM, Findlay VL, Tew KD. Glutathione S-transferases as regulators of kinase pathways and anticancer drug targets. Methods Enzymol. 2005;401:287–307. doi: 10.1016/S0076-6879(05)01019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–92. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- 6.Tirona RG, Pang KS. Bimolecular glutathione conjugation kinetics of ethacrynic acid in rat liver: In vitro and perfusion studies. J Pharmacol Exp Ther. 1999;290:1230–41. [PubMed] [Google Scholar]
- 7.Jones DP. Redox potential of GSH/GSSG couple: Assay and biological significance. Methods Enzymol. 2002;348:93–112. doi: 10.1016/s0076-6879(02)48630-2. [DOI] [PubMed] [Google Scholar]
- 8.Townsend DM, Tew KD, Tapiero H. The importance of glutathione in human disease. Biomed Pharmacother. 2003;57:145–55. doi: 10.1016/s0753-3322(03)00043-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sen CK. Cellular thiols and redox-regulated signal transduction. Curr Top Cell Regul. 2000;36:1–30. doi: 10.1016/s0070-2137(01)80001-7. [DOI] [PubMed] [Google Scholar]
- 10.Adler V, Yin Z, Fuchs SY, et al. Regulation of JNK signaling by GSTp. EMBO J. 1999;18:1321–34. doi: 10.1093/emboj/18.5.1321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Townsend DM, Tew KD. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene. 2003;22:7369–75. doi: 10.1038/sj.onc.1206940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.McIlwain CC, Townsend DM, Tew KD. Glutathione S-transferase polymorphisms: Cancer incidence and therapy. Oncogene. 2006;25:1639–48. doi: 10.1038/sj.onc.1209373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayes JD, Flanagan JU, Jowsey IR. Glutathione transferases. Annu Rev Pharmacol Toxicol. 2005;45:51–88. doi: 10.1146/annurev.pharmtox.45.120403.095857. [DOI] [PubMed] [Google Scholar]
- 14.Wang J, Deng Y, Cheng J, Ding J, Tokudome S. GST genetic polymorphisms and lung adenocarcinoma susceptibility in a Chinese population. Cancer Lett. 2003;201:185–93. doi: 10.1016/s0304-3835(03)00480-4. [DOI] [PubMed] [Google Scholar]
- 15.Christie JD, Aplenc R, DeAndrade J, Lung Transplant Outcome Group Donor glutathione S-transferase genotype is associated with primary graft dysfunction following lung transplantation. J Heart Lung Transplant. 2005;24:S80. [Google Scholar]
- 16.Hadjiliadis D, Lingaraju R, Mendez J, et al. Donor glutathione S-transferase (GST) mu null genotype in lung transplant recipients is associated with increased incidence of bronchiolitis obliterans (BOS) independent of acute rejection. J Heart Lung Transplant. 2007;26:S108. [Google Scholar]
- 17.Rizzardini M, Lupi M, Bernasconi S, Mangolini A, Cantoni L. Mitochondrial dysfunction and death in motor neurons exposed to the glutathione-depleting agent ethacrynic acid. J Neurol Sci. 2003;207:51–8. doi: 10.1016/s0022-510x(02)00357-x. [DOI] [PubMed] [Google Scholar]
- 18.Yamamoto K, Masubuchi Y, Narimatsu S, Kobayashi S, Horie T. Rat liver microsomal lipid peroxidation produced during the oxidative metabolism of ethacrynic acid. Pharmacol Toxicol. 2001;88:176–80. doi: 10.1034/j.1600-0773.2001.d01-100.x. [DOI] [PubMed] [Google Scholar]
- 19.Huang J, Philbert MA. Cellular responses of cultured cerebellar astrocytes to ethacrynic acid-induced perturbation of subcellular glutathione homeostasis. Brain Res. 1996;711:184–92. doi: 10.1016/0006-8993(95)01376-8. [DOI] [PubMed] [Google Scholar]
- 20.Cao Z, Hardej D, Trombetta LD, Li Y. The role of chemically induced glutathione and glutathione S-transferase in protecting against 4-hydroxy-2-nonenal-mediated cytotoxicity in vascular smooth muscle cells. Cardiovasc Toxicol. 2003;3:165–77. doi: 10.1385/ct:3:2:165. [DOI] [PubMed] [Google Scholar]
- 21.Das DK, Maulik N. Antioxidant effectiveness in ischaemia-reperfusion tissue injury. Methods Enzymol. 1994;233:601–10. doi: 10.1016/s0076-6879(94)33063-8. [DOI] [PubMed] [Google Scholar]
- 22.Sies H. Oxidative stress: Introduction. In: Sies H, editor. Oxidative stress, Oxidants and antioxidants. London: London Academic Press; 1991. [Google Scholar]
- 23.Tokola H, Salo K, Vuolteenaho O, Ruskoaho H. Basal and acidic fibroblast growth factor-induced atrial natriuretic peptide gene expression and secretion is inhibited by staurosporine. Eur J Pharm. 1994;267:195–206. doi: 10.1016/0922-4106(94)90171-6. [DOI] [PubMed] [Google Scholar]
- 24.Luodonpa M, Vuolteenaho O, Eskelinen S, Ruskoaho H. Effects of adrenomedullin on hypertrophic responses induced by angiotensin II, endothelin-1 and phenylephrine. Peptides. 2001;22:1859–66. doi: 10.1016/s0196-9781(01)00505-8. [DOI] [PubMed] [Google Scholar]
- 25.Gordon JM, Dusting GJ, Woodman OL, Ritchie RH. Cardioprotective action of CRF peptide urocortin against simulated ischemia in adult rat cardiomyocytes. Am J Physiol Heart Circ Physiol. 2003;284:330–6. doi: 10.1152/ajpheart.01121.2001. [DOI] [PubMed] [Google Scholar]
- 26.Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phospatidylserine expression on early apoptotic cells using fluorescein labelled annexin V. J Immunol Methods. 1995;184:39–51. doi: 10.1016/0022-1759(95)00072-i. [DOI] [PubMed] [Google Scholar]
- 27.Yin Z, Ivanov VN, Habelhah H, Tew K, Ronai Z. Glutathione S-transferase p elicits protection against H2O2-induced cell death via coordinated regulation of stress kinases. Cancer Res. 2000;60:4053–7. [PubMed] [Google Scholar]
- 28.Hayes JD, McLellan LI. Glutathione and glutathione-dependent enzymes represent a co-ordinately regulated defence against oxidative stress. Free Radical Res. 1999;31:273–300. doi: 10.1080/10715769900300851. [DOI] [PubMed] [Google Scholar]
- 29.Romieu I, Sienra-Monge JJ, Ramírez-Aguilar M, et al. Genetic polymorphism of GSTM1 and antioxidant supplementation influence lung function in relation to ozone exposure in asthmatic children in Mexico City. Thorax. 2004;59:8–10. [PMC free article] [PubMed] [Google Scholar]
- 30.Weston CR, Davis RJ. The JNK signal transduction pathway. Curr Opin Genet Dev. 2002;12:14–21. doi: 10.1016/s0959-437x(01)00258-1. [DOI] [PubMed] [Google Scholar]
- 31.Wang T, Arifoglu P, Ronai Z, Tew KD. Glutathione S-transferase P1-1 (GSTP1-1) inhibits c-Jun N-terminal kinase (JNK1) signaling through interaction with the C terminus. J Biol Chem. 2001;276:20999–1003. doi: 10.1074/jbc.M101355200. [DOI] [PubMed] [Google Scholar]
- 32.Townsend DM. S-glutathionylation: Indicator of cell stress and regulator of the unfolded protein response. Mol Interv. 2007;7:313–24. doi: 10.1124/mi.7.6.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ruscoe JE, Rosario LA, Wang T, et al. Pharmacologic or genetic manipulation of glutathione S-transferase P1-1 (GSTpi) influences cell proliferation pathways. J Pharmacol Exp Ther. 2001;298:339–45. [PubMed] [Google Scholar]
- 34.Davis W, Jr, Ronai Z, Tew KD. Cellular thiols and reactive oxygen species in drug-induced apoptosis. J Pharmacol Exp Ther. 2001;296:1–6. [PubMed] [Google Scholar]
- 35.Takeda K, Matsuzawa A, Nishutoh H, Ichido H. Roles of MAPKKK ASK1 in stress induced cell death. Cell Struct Funct. 2003;28:23–9. doi: 10.1247/csf.28.23. [DOI] [PubMed] [Google Scholar]
- 36.Matsuzawa A, Nishitoh H, Tobiume K, et al. Physiological roles of ASK1-mediated signal transduction in oxidative stress- and endoplasmic reticulum stress-induced apoptosis: Advanced findings from ASK1 knockout mice. Antioxid Redox Signal. 2002;3:415–25. doi: 10.1089/15230860260196218. [DOI] [PubMed] [Google Scholar]
- 37.Kim SG, Lee SJ. PI3K, RSK, and mTOR signal networks for the GST gene regulation. Toxicol Sci. 2007;96:206–13. doi: 10.1093/toxsci/kfl175. [DOI] [PubMed] [Google Scholar]
- 38.Kim SK, Novak RF. The role of intracellular signaling in insulin-mediated regulation of drug metabolizing enzyme gene and protein expression. Pharmacol Ther. 2007;113:88–120. doi: 10.1016/j.pharmthera.2006.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]