

Abstract
Both autophagy and pro-inflammatory cytokines are involved in the host defence against mycobacteria, but little is known regarding the effect of autophagy on Mycobacterium tuberculosis (MTB)-induced cytokine production. In the present study, we assessed the effect of autophagy on production of monocyte-derived and T-cell-derived cytokines, and examined whether two functional polymorphisms in autophagy genes led to altered cytokine production. Blocking autophagy inhibited tumour necrosis factor-α (TNF-α) production, while enhancing interleukin-1β (IL-1β) production in peripheral blood mononuclear cells stimulated with MTB. Induction of autophagy by starvation or interferon-γ (IFN-γ) had the opposite effect. The modulation of both TNF-α and IL-1β production by autophagy was induced at the level of gene transcription. Functional polymorphisms in the autophagy genes ATG16L1 and IRGM did not have a major impact on MTB-induced cytokine production in healthy volunteers, although a moderate effect was observed on IFN-γ production by the ATG16L1 T300A polymorphism. These data demonstrate the interplay between autophagy and inflammation during host defence against mycobacteria, and future studies to investigate the clinical implications of these effects for the susceptibility to tuberculosis are warranted.
Keywords: cytokines, innate immunity, mycobacteria/mycobacterium
Introduction
Tuberculosis (TB) is a major health problem with 10 million new cases diagnosed each year, causing nearly 2 million deaths.1 However, from the estimated 2 billion individuals that have been initially infected with Mycobacterium tuberculosis (MTB), most develop asymptomatic infection, also called ‘latent tuberculosis’. The pathogen is often not eliminated and may persist for years inside macrophages because of its capacity to evade the host immune response. This persistence allows progression to active tuberculosis either as the primary disease or, years later, as reactivation when cellular immunity fails.
The interaction between MTB and cells of both the innate and adaptive immune systems results in secretion of pro-inflammatory cytokines such as tumour necrosis factor-α (TNF-α), interleukin-1β (IL-1β), IL-18, IL-12 and interferon-γ (IFN-γ). The TNF-α, produced by mononuclear cells, is crucial for host defence because TNF-α-deficient mice succumb rapidly after MTB infection, with significantly higher mycobacterial outgrowth in different organs compared with wild-type animals.2 The TNF-α is important for formation of granuloma, a mechanism for containing and restricting the replication of the bacilli.2,3 The crucial role played by TNF-α in human TB is also demonstrated by the increased susceptibility to TB in patients treated with anti-TNF-α therapy.4
Interleukin-1β is an essential component of the host defence to mycobacteria.5–7 MTB-infected IL-1 receptor type 1 knockout mice exhibit lower production of IFN-γ, defective granuloma formation, and decreased survival.6 Interferon-γ is secreted by natural killer, CD4+ and CD8+ T cells upon release of endogenous IL-12 and IL-18 by macrophages and dendritic cells. The IFN-γ activates macrophages to kill and eliminate the mycobacteria. It also enhances their expression of MHC class II molecules, which results in improved antigen presentation to T cells. The crucial importance of IFN-γ for human anti-mycobacterial defence is demonstrated by the increased susceptibility to mycobacterial infections in patients with IFN-γ receptor or IL-12 receptor deficiencies.8–10 As cytokines are therefore crucial for host defence against MTB, it is essential to understand the molecular pathway of the MTB-induced cytokine production.
Autophagy is a pathway through which cytoplasmic components, including organelles and intracellular pathogens, are sequestered in a double-membrane-bound autophagosome and delivered to the lysosome for degradation.11,12 Elimination of dysfunctional cell components is one of the main roles of autophagy, besides the recycling of cytoplasmic material, allowing the cell to maintain macromolecular synthesis and energy homeostasis during starvation and other stressful conditions. In addition to its role in cell survival, autophagy is also involved in host defence, with important effects on both innate and adaptive immune systems.13,14 Autophagy has an essential role for anti-mycobacterial host defence by fusing the autophagosomes containing mycobacteria with lysosomes,15,16 leading to antigen presentation and T-cell activation.17,18 Recent experiments in mice suggest that autophagy also modulates cytokine production: macrophages from ATG16L1 knockout mice with no functional autophagy exhibit elevated IL-1β production, suggesting an inhibitory role of autophagy on the production of this cytokine.19 However, so far this has not been studied in humans: although both autophagy and pro-inflammatory cytokines have an important role in host defence against mycobacteria, there are no reported data regarding the effect of autophagy on MTB-induced cytokine production. Therefore, in the present study, we assessed the effect of autophagy on production of monocyte-derived and T-cell-derived cytokines induced by MTB, and examined if two functional polymorphisms lead to altered MTB-induced cytokine production.
Material and methods
Peripheral blood mononuclear cell stimulation assays
After obtaining informed consent, venous blood was drawn from the cubital vein of healthy volunteers into 10 ml EDTA tubes (BD, Franklin Lakes, NJ). The mononuclear cell fraction was isolated by density centrifugation of blood, diluted 1 : 1 in pyrogen-free saline over Ficoll-Paque (Pharmacia Biotech, PA). Cells were washed twice in saline and resuspended in culture medium (RPMI; Invitrogen, CA) supplemented with gentamicin 10 μg/ml, l-glutamine 10 mm and pyruvate 10 mm. Cells were counted in a Coulter counter (Coulter Electronics, Buckinghamshire, UK) and the number was adjusted to 5 × 106 cells/ml. A total of 5 × 105 mononuclear cells in a 100-μl volume was added to round-bottom 96-wells plates (Greiner, Frickenhausen, Germany) in duplicate. Cells were incubated with either RPMI or increasing doses of 3-methyl adenine (3MA; Sigma, St. Louis, MO) ranging from 1 to 10 mm for 24 hr, 48 hr or 7 days at 37° (incubation time was dependent on the cytokines measured). Cytokine concentrations were assessed in the supernatants using ELISA.
For autophagy inhibition and induction experiments, cells were pre-incubated for 1 hr at 37° in RPMI or in the presence of 3MA (10 mm) or IFN-γ (400 IE/ml; Immukine, Boehringer, Ingelheim, Germany), respectively. After pre-treatment, 100 μl RPMI (negative control) or sonicated MTB H37Rv (1 μg/ml end concentration) was added to the cells. Cells were incubated for 24 hr and cytokine production was measured in supernatant using ELISA.
Autophagy was also induced by incubation of peripheral blood mononuclear cells (PBMCs) in starvation medium (Earle's balanced salt solution, EBSS; Invitrogen, Carlsbad, CA). In some wells, PBMCs were pre-incubated for 1 hr at 37° in the presence of 3MA (10 mm), to block the induction of autophagy. Subsequently, cells were stimulated for 3 hr with sonicated MTB H37Rv (1 μg/ml). Thereafter, supernatants were discarded and TRIzol reagent was added to the cells, after which they were stored at −80° until analysed.
To investigate the effect of 3MA on the ATP-dependent IL-1β release, PBMCs were pre-treated for 1 hr with either RPMI or 3MA (10 mm). Subsequently, cells were stimulated with RPMI or MTB H37Rv (1 μg/ml) for 3 hr. After stimulation, supernatants were discarded and refreshed with RPMI containing 1 mm ATP, after which the cells were incubated for another 15 min. The ATP-dependent IL-1β secretion after the additional 15 min was assessed in the supernatant.
Inhibition of autophagy by small interfering RNA
As a complementary approach to inhibit the autophagy PBMCs were transfected with control (scrambled) or ATG7 small interfering RNA (siRNA) with a nucleofector kit using an Amaxa electroporation chamber (Lonza, Basel, Switzerland), After 24 hr of incubation at 37°, cells were stimulated with MTB H37Rv (1 μg/ml), and after an additional incubation of 24 hr the supernatants were collected and stored at −80° until analysis.
Cytokine measurements
Measurements of TNF-α, IL-1β, IL-6, IFN-γ, IL-10, IL-22 and IL-23 were performed in the supernatants using commercial ELISA kits (R&D Systems, Minneapolis, MN: TNF-α, IL-1β, IL-22; Sanquin, Amsterdam, the Netherlands: IL-6, IL-10, IFN-γ; or E-Bioscience, San Diego, CA: IL-23).
Real-time PCR for IL-1, TNF-α and ATG7
RNA purification was performed from TRIzol according to the manufacturer's instructions. Isolated RNA was transcribed into complementary DNA using iScript cDNA. Synthesis kit (Bio-Rad, Hercules, CA) followed by quantitative PCR using SYBR Green. (Applied Biosystems, Foster City, CA). The following primers were used: IL-1β, forward 5′-GCCCTAAACAGATGAAGTGCTC-3′ and reverse 5′-GAACCAGCATCTTCCTCAG-3′; TNF-α, forward 5′- TGGCCCAGGCAGTCAGA-3′ and reverse 5′-GGTTTGCTACAACATGGGCTACA-3′; and ATG7, forward 5′-CAGTTTGCCCCTTTTAGTAGTGC-3′and reverse 5′-CCTTAATGTCCTTGGGAGCTTCA-3′. Data were corrected for expression of the housekeeping gene β2-microglobulin, for which the primers forward 5′-ATGAGTATGCCTGCCGTGTG-3′ and reverse 5′-CCAAATGCGGCATCTTCAAAC-3′ were used.
Genotyping for ATG16L1 Thr300Ala and IRGM polymorphisms
DNA was isolated from whole blood by using the Puregene isolation kit (Gentra Sytems, Minneapolis, MN), according to the manufacturer's protocol. Genotyping for the presence of the ATG16L1 Thr300Ala (rs2241880),and IRGM promoter polymorphisms rs4958847 and rs13361189 polymorphisms was performed using TaqMan single nucleotide polymorphism (SNP) assays C_9095577_20, C_1398968_10 and C_31986315_10, respectively, on the 7300 ABI Real-Time PCR system (Applied Biosystems).
These genotyping assays were performed in a 25 μl total reaction volume, containing 2 μl genomic DNA as well as primers, two specific probes (with either VIC or FAM label) and Universal PCR 2 × Master mix (Applied Biosystems). Cycling conditions were 2 min at 50° and 10 min at 95° followed by 40 cycles of 95° for 15 seconds and 1 min at 60°. Fluorescence intensities were corrected using a post-read/pre-read method for 1 min at 60° before and after the amplification. The software automatically plotted genotypes based on a two-parameter plot with an overall success rate of > 95%. Intermediate samples were excluded from the analysis.
Two cohorts of healthy volunteers without known TB contact have been assessed (n = 73 and n = 104), consisting of foresters from the ‘Geldersch Landschap’ and ‘Kroondomein’ departments in the Netherlands. The individuals were between 23 and 73 years old, and 77% were men. All volunteers gave written informed consent, and the study was approved by the Ethical Committee of the Radboud University Nijmegen Medical Centre, Nijmegen, the Netherlands. The cytokine production induced by sonicated MTB H37Rv in cells isolated from volunteers bearing various ATG16L1 and IRGM genetic variants was compared.
Statistical analysis
Differences were analysed using the Wilcoxon signed rank test or Friedman test for paired samples and Mann–Whitney or Kruskal–Wallis test for unrelated samples. P < 0·05 was considered statistically significant. Data are shown as cumulative results of level obtained in all volunteers (means + SEM).
Results
Inhibition of autophagy enhances MTB-induced production of IL-1β and IL-6, but decreases production of TNF-α
Inhibition of autophagy with 3MA (a blocker of the Beclin1 complex that is crucial for the initiation of autophagy) decreased TNF-α production induced by MTB H37Rv (Friedman test P = 0·003). In contrast, autophagy inhibition increased the production of IL-1β and IL-6 (Friedman test P = 0·017 and P = 0·035, respectively) in a dose-dependent manner (Fig. 1). No changes were observed in the induction of IL-10 (range 46–50 pg/ml) or IL-18 (range 2–4 pg/ml) production when autophagy was inhibited (data not shown). The T-cell-derived cytokines IFN-γ, IL-17, IL-22 and IL-23 levels were not detectable when cells were stimulated with MTB H37Rv, most likely as a result of the naive TB status of the individuals tested.
Figure 1.
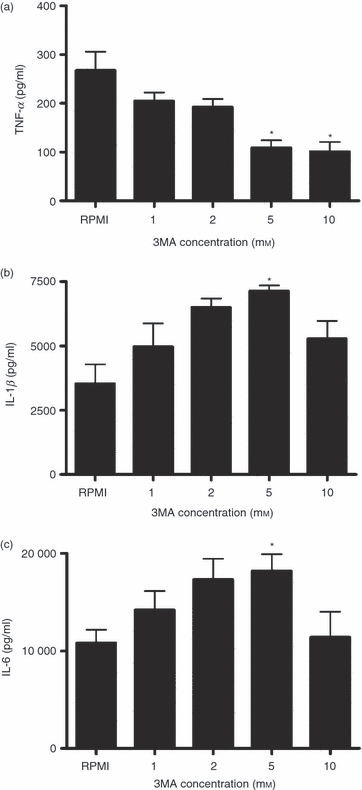
The effect of the autophagy inhibitor 3-methyl adenine (3MA) on Mycobacterium tuberculosis (MTB) -induced cytokine production. Freshly isolated human peripheral blood mononuclear cells (PBMCs) were pre-incubated with increasing doses of 3MA (1, 2, 5, or 10 mm) for 1 hr at 37° in culture medium and subsequently stimulated with sonicated MTB H37Rv (1 μg/ml). After 24 hr of incubation, tumour necrosis factor-α (TNF-α) (a), interleukin-1β (IL-1β) (b) and IL-6 (c) were measured in the supernatant using specific ELISA. Data are presented as means + SEM, n = 4 *P < 0·05 compared with RPMI.
Induction of autophagy enhances TNF-α production
In line with the experiments showing that inhibition of autophagy leads to decreased production of TNF-α, induction of autophagy with IFN-γ enhanced TNF-α production in MTB-stimulated PBMCs. This effect was abolished when autophagy was inhibited by 3MA (P < 0·01), suggesting that normal autophagy is necessary for the effects of IFN-γ on TNF-α production (Fig. 2a). In contrast, treatment of PBMCs with IFN-γ did not significantly modify IL-1β production (Fig. 2b).
Figure 2.
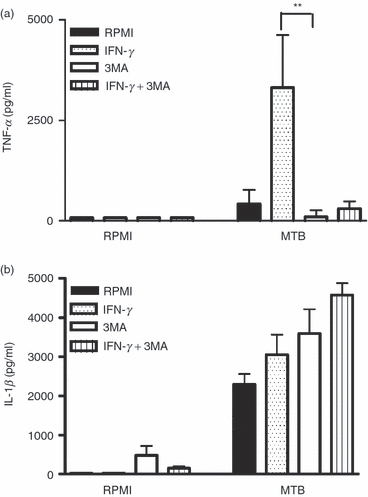
Modulation of inflammatory cytokine production by inhibition and induction of autophagy. Peripheral blood mononuclear cells (PBMCs) were pre-incubated for 1 hr with RPMI (negative control), 3-methyl adenine (3MA) 10 mm (inhibition of autophagy), interferon-γ (IFN-γ) (400 IE/ml; induction of autophagy) or both 3MA and IFN-γ at 37°. Thereafter, cells were stimulated for 24 hr at 37° with either RPMI or sonicated Mycobacterium tuberculosis (MTB; 1 μg/ml). Tumour necrosis factor-α (TNF-α) (a) and interleukin-1β (IL-1β) (b) concentrations were measured in the supernatant using specific ELISA. Data are presented as means + SEM, n = 6, **P < 0·01.
Inhibition of autophagy using siRNA against ATG7 influences both TNF-α and IL-1β production
Figure 3 shows inhibition of autophagy with siRNA against ATG7. In line with experiments using a pharmacological agent (3MA) to inhibit autophagy, inhibition of autophagy by ATG7 siRNA showed the same trends of diminished production of TNF-α (Fig. 3a) and increased production of IL-1β (Fig. 3b).
Figure 3.
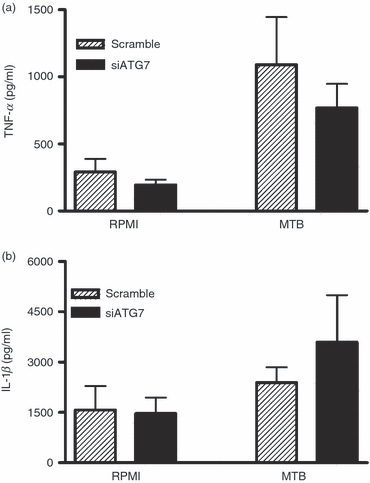
Modulation of inflammatory cytokine production by inhibition of autophagy with small interfering RNA (siRNA) against ATG7. Peripheral blood mononuclear cells (PBMCs) were transfected with siRNA ATG7 or scrambled siRNA after 24 hr stimulated with sonicated Mycobacterium tuberculosis (MTB) (1 μg/ml). After another 24 hr incubation tumour necrosis factor-α (TNF-α) (a) and interleukin-1β (IL-1β) (b) concentrations were measured in the supernatant using specific ELISA. Data are presented as means + SEM, n = 6.
Modulation of IL-1β and TNF-α production is regulated at the transcriptional level
In the following set of experiments we assessed the level at which autophagy regulates cytokine production. Both induction (with starvation medium EBSS) and inhibition (with 3MA) of autophagy modulated the transcription of pro-inflammatory cytokines. The TNF-α mRNA levels significantly increased when incubating stimulated PBMCs in starvation medium (EBSS) (Fig. 4a), whereas induction of autophagy seemed to decrease IL-1β mRNA (Fig. 4b). The opposite effect was exerted by autophagy inhibition through 3MA: TNF mRNA was decreased (Fig. 4c), wheeras a trend towards an increase in IL-1β mRNA was observed (Fig. 4d). In contrast, inhibition of autophagy had no effect on IL-1β processing and release induced by the inflammasome activation with ATP 20 (Fig. 4e).
Figure 4.
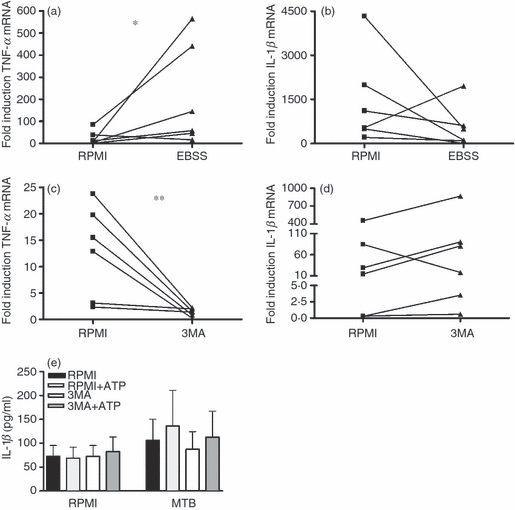
The modulatory effect of autophagy on cytokine production is exerted at a transcriptional level. Cells pre-treated for 1 hr with RPMI or starvation medium, in the presence or absence of 3-methyl adenine (3MA; 10 mm), were stimulated for 3 hr with sonicated Mycobacterium tuberculosis (MTB; 1 μg/ml). Reverse transcription-PCR was performed and relative levels of tumour necrosis factor-α (TNF-α) (a, c) and interleukin-1β (IL-1β) (b, d) mRNA were determined. Data are presented as fold change, n = 6, *P < 0·05, **P < 0·01. Separately, peripheral blood mononuclear cells (PBMCs) were pre-incubated for 1 hr in the presence or absence of 3MA (10 mm) and were stimulated for 3 hr with MTB (1 μg/ml) at 37°. After the stimulation, supernatants were discarded and refreshed with RPMI or RPMI containing 1 mm ATP, and cell were incubated for an additional 15 min. ATP-dependent IL-1β production was measured with ELISA (e). Data are presented as means + SEM, n = 6.
Autophagy gene polymorphisms and cytokine production induced by MTB
Polymorphisms in two genes involved in the process of autophagy, ATG16L1 and IRGM, have been associated with auto-inflammatory disorders,21–23 and IRGM polymorphisms also with susceptibility to TB.24–26 Considering the possible role of autophagy in modulating cytokine production induced by MTB, we examined if ATG16L1 and IRGM polymorphisms lead to altered cytokine production. However, no statistically significant differences in MTB-induced cytokine production were observed when PBMCs were used from healthy volunteers with different ATG16L1 genotypes, although PBMCs from individuals bearing the ATG16L1 300G variant tended to produce less IFN-γ (Fig. 5). In addition, no significant differences in cytokine production were observed between individuals bearing the different IRGM alleles (Fig. 6). Although the cohorts were relatively large (57–104 subjects), only one (in the case of TNF-α and IFN-γ) or two (in the case of IL-1β) individuals were homozygous for the IRGM rs4958847 AA and rs13361189 CC allele, precluding a definitive conclusion regarding the effect of these two IRGM SNPs on cytokine production.
Figure 5.
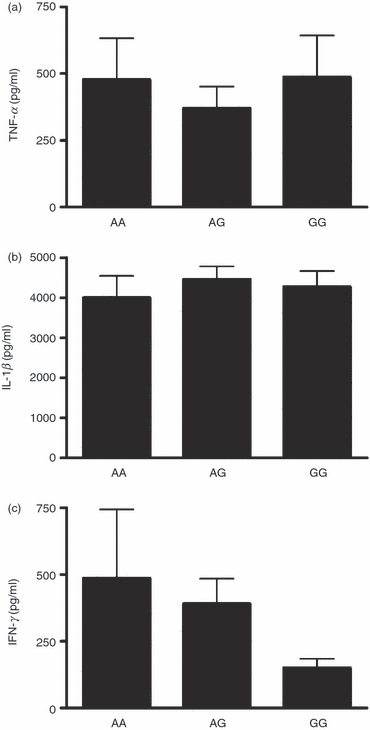
ATG16L1 polymorphisms and Mycobacterium tuberculosis (MTB) -induced cytokine production. Cytokine production capacity of tumour necrosis factor-α (TNF-α) (a), interleukin-1β (IL-1β) (b) and interferon-γ (IFN-γ) (c) by PBMCs obtained from healthy volunteers after stimulation for 24 hr (TNF-α and IL-1β) or 48 hr (IFN-γ) with sonicated MTB stratified for ATG16L1 Thr300Ala genotype (the A allele equals 300Thr allele, G allele equals 300Ala allele). Data are presented as means + SEM, n = 73 (13 AA, 37 AG, 23 GG).
Figure 6.
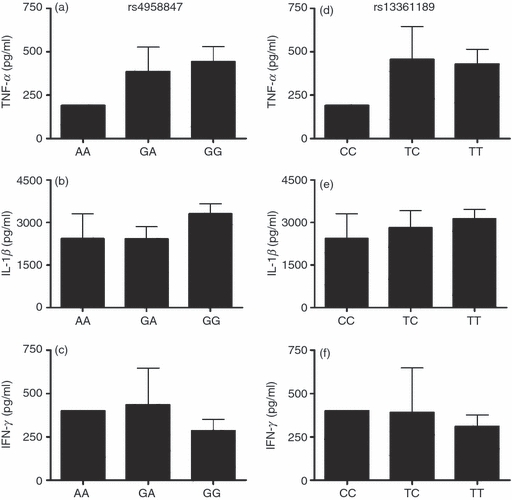
IRGM1 polymorphisms and Mycobacterium tuberculosis (MTB) -induced cytokine production. Cytokine production capacity of tumour necrosis factor-α (TNF-α) (a, d), interleukin-1β (IL-1β) (b, e) and interferon-γ (IFN-γ) (c, f) by peripheral blood mononuclear cells (PBMCs) obtained from healthy volunteers after stimulation for 24 hr (TNF-α and IL-1β) or 48 hr (IFN-γ) with sonicated MTB stratified for IRGM genotypes. Data are presented as means + SEM. n = 104 (2 AA, 25 GA, 77 GG for rs4958847, and 2 CC, 17 TC, 83 TT for rs13361189).
Discussion
Autophagy is essential for host defence against MTB.15,16,24 In the present study, we show that autophagy modulates pro-inflammatory cytokine production after stimulation with MTB. Autophagy showed differential effects on MTB-induced cytokine production; autophagy stimulated TNF-α production and inhibited IL-1β and IL-6 production. These effects of autophagy on MTB-induced pro-inflammatory cytokines were exerted at the transcriptional level.
Activation of autophagy resulted in opposite effects on TNF-α and IL-1β, exerting an increase of TNF-α production and a decrease in IL-1β production. This is an important, yet puzzling, effect considering the role of both TNF-α and IL-1β in host defence against MTB.11,12,14–16,27 On the one hand, the increase in the production of TNF-α is likely to increase anti-mycobacterial innate immunity. On the other hand, one may speculate that the decrease in the production of IL-1β may result in decreased T helper type 17 (Th17) responses,28 and so indirectly shifting the response towards protective Th1 cellular immunity, as Th1 and Th17 are known to negatively regulate each other.29 We were not able to study directly the effect of autophagy on Th17 responses for two reasons. Inhibition of autophagy through 3MA inhibited T-cell proliferation, whereas prolonged incubation of cells, necessary for optimal Th17 responses, led to cell death when cells were kept for long periods of time in the EBSS (starvation) medium. Another mechanism that might influence cytokine levels in these experiments is IFN-γ-induced Toll-like receptor 2 (TLR2) and nucleotide-binding oligomerization domain containing 2 (NOD2) expression,30,31 which in turn can lead to more cytokine production. However, induction of autophagy with starvation medium instead of IFN-γ led to similar results, but the effects of IFN-γ on cytokine production were abolished when 3MA was added. Therefore, the effects of IFN-γ on cytokine production are at least partly mediated through autophagy.
In a separate set of experiments, we investigated the level at which the effects of autophagy on MTB-induced cytokines are exerted. Autophagy clearly influenced transcription of both TNF-α and IL-1β but no post-transcriptional effect was observed for modulation of IL-1β synthesis. However, a different effect was seen on TNF-α versus IL-1β transcription. There are several pathways that induce pro-inflammatory cytokines after MTB stimulation. The most important receptors involved in MTB recognition and subsequent induction of cytokine responses are TLRs (TLR2 in combination with TLR1 or TLR6, TLR4 and TLR9) and NOD2. We have previously shown that NOD2, but not TLR, responses are modulated by autophagy.32 Transcription of TNF-α and IL-1β is differently regulated, with IL-1β transcription being mediated by extracellular signal-regulated kinase (ERK) and p38, but not Jun N-terminal kinase (JNK) kinase. In contrast, TNF-α transcription is mediated through ERK-dependent and JNK-dependent pathways, with little effect through p38 kinase.33 Our data therefore suggest that differential modulation of the MAP kinase transcriptional pathways by autophagy may be responsible for the differences in TNF-α and IL-1β production, although this remains to be confirmed in future studies.
Interleukin-1β is processed and secreted after activation of a protein complex called the inflammasome, leading to activation of caspase-1 and processing of inactive pro-IL-1β into the active cytokine. ATP acting on the P2X7 receptor is a known activator of the inflammasome. No effect of autophagy on the IL-1β processing and secretion induced by ATP was observed, providing an additional argument that autophagy modulates cytokine production in humans at the transcriptional, rather than the post-transcriptional level.32 This is in contrast to studies in murine cells, in which inhibition of the inflammasome by autophagy seems to play an important role.19,34 This argues that important differences exist between the effects of autophagy in mice and humans.
Single nucleotide polymorphisms in the autophagy genes ATG16L1 and IRGM have been associated with Crohn's disease,21–23 and an association of IRGM SNPs and TB has also been reported.24–26 ATG16L1 is a component of a large protein complex formed together with ATG5 and ATG12 that is essential for autophagosome formation. IRGM is a downstream effector of IFN-γ and has been implicated in the process of autophagy induction in macrophages.16 Subsequently, reductions in IRGM1 levels are associated with decreased autophagy.24 However, these polymorphisms did not influence the induction of TNF-α or IL-1β production by cells isolated from healthy volunteers and stimulated with MTB. Whereas the T300A ATG16L1 polymorphism has been shown to modulate NOD2-induced IL-1β production, it does not affect TLR-induced cytokine synthesis.32 Although we should be careful interpreting these data, given the small number of subjects carrying the functional SNPs, this suggests that modulation of cytokine production by NOD2 ligands in MTB may be compensated by the unaffected TLR-dependent induction. Interestingly, a trend towards lower IFN-γ production was observed in cells isolated from individuals homozygous for the 300A allele of ATG16L1, and this may affect anti-mycobacterial host defence in the patients. Whether the T300A ATG16L1 polymorphism also modulates susceptibility to TB remains unclear. The effects of this ATG16L1 SNP may be exerted not only though IFN-γ production, but also through modulation of antigen presentation at the level of MHC class II, a process known to be influenced by autophagy.17,18
Our findings might have been strengthened by examination of the M. tuberculosis genotype in relation to host genotype. Another SNP in the IRGM gene, –261T, has been shown to be associated with protection from certain MTB genotypes but not with protection against Mycobacterium africanum.25 This supports the concept of co-evolution of M. tuberculosis and the human immune system described in a previous study.35 In this study we looked at the relation between host genotype and autophagy-mediated modulation of cytokine production, but in future patient studies we hope to investigate whether there is a relation between certain autophagy host genotypes and M. tuberculosis genotypes
In conclusion, the process of autophagy influences cytokine production induced in human PBMCs by MTB. These effects are different on TNF-α compared with IL-1β induction, and are induced at the transcriptional level. The next step will be to relate these effects on cytokine production to the direct anti-mycobacterial defence mechanisms in macrophages. Finally, an assessment of the clinical implications of this autophagy–inflammation interaction is needed, with the important question whether (partial) defects in autophagy may increase susceptibility to TB.
Acknowledgments
This study was part of the TI-Pharma project D-1-101. RvC was supported by a Vidi grant of the Netherlands Organization for Scientific Research. MGN was supported by a Vici grant of the Netherlands Organization for Scientific Research.
Disclosures
There are no conflicts of interest nor financial interest.
References
- 1.WHO. factsheet tuberculosis 2011. http://www.who.int/mediacentre/factsheets/fs104/en/
- 2.Flynn JL, Goldstein MM, Chan J, et al. Tumor necrosis factor-α is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity. 1995;2:561–72. doi: 10.1016/1074-7613(95)90001-2. [DOI] [PubMed] [Google Scholar]
- 3.Kindler V, Sappino AP, Grau GE, Piguet PF, Vassalli P. The inducing role of tumor necrosis factor in the development of bactericidal granulomas during BCG infection. Cell. 1989;56:731–40. doi: 10.1016/0092-8674(89)90676-4. [DOI] [PubMed] [Google Scholar]
- 4.Keane J, Gershon S, Wise RP, et al. Tuberculosis associated with infliximab, a tumor necrosis factor α-neutralizing agent. N Engl J Med. 2001;345:1098–104. doi: 10.1056/NEJMoa011110. [DOI] [PubMed] [Google Scholar]
- 5.Fremond CM, Togbe D, Doz E, et al. IL-1 receptor-mediated signal is an essential component of MyD88-dependent innate response to Mycobacterium tuberculosis infection. J Immunol. 2007;179:1178–89. doi: 10.4049/jimmunol.179.2.1178. [DOI] [PubMed] [Google Scholar]
- 6.Juffermans NP, Florquin S, Camoglio L, et al. Interleukin-1 signaling is essential for host defense during murine pulmonary tuberculosis. J Infect Dis. 2000;182:902–8. doi: 10.1086/315771. [DOI] [PubMed] [Google Scholar]
- 7.Yamada H, Mizumo S, Horai R, Iwakura Y, Sugawara I. Protective role of interleukin-1 in mycobacterial infection in IL-1 α/β double-knockout mice. Lab Invest. 2000;80:759–67. doi: 10.1038/labinvest.3780079. [DOI] [PubMed] [Google Scholar]
- 8.De Jong R, Altare F, Haagen IA, et al. Severe mycobacterial and Salmonella infections in interleukin-12 receptor-deficient patients. Science. 1998;280:1435–8. doi: 10.1126/science.280.5368.1435. [DOI] [PubMed] [Google Scholar]
- 9.Ottenhoff TH, Verreck FA, Lichtenauer-Kaligis EG, Hoeve MA, Sanal O, Van Dissel JT. Genetics, cytokines and human infectious disease: lessons from weakly pathogenic mycobacteria and salmonellae. Nat Genet. 2002;32:97–105. doi: 10.1038/ng0902-97. [DOI] [PubMed] [Google Scholar]
- 10.Altare F, Durandy A, Lammas D, et al. Impairment of mycobacterial immunity in human interleukin-12 receptor deficiency. Science. 1998;280:1432–5. doi: 10.1126/science.280.5368.1432. [DOI] [PubMed] [Google Scholar]
- 11.Todde V, Veenhuis M, Van der Kleij IJ. Autophagy: principles and significance in health and disease. Biochim Biophys Acta. 2009;1792:3–13. doi: 10.1016/j.bbadis.2008.10.016. [DOI] [PubMed] [Google Scholar]
- 12.Kundu M, Thompson CB. Autophagy: basic principles and relevance to disease. Annu Rev Pathol. 2008;3:427–55. doi: 10.1146/annurev.pathmechdis.2.010506.091842. [DOI] [PubMed] [Google Scholar]
- 13.Deretic V, Levine B. Autophagy, immunity and microbial adaptations. Cell Host Microbe. 2009;5:527–49. doi: 10.1016/j.chom.2009.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Münz C. Enhancing immunity through autophagy. Annu Rev Immunol. 2009;27:423–49. doi: 10.1146/annurev.immunol.021908.132537. [DOI] [PubMed] [Google Scholar]
- 15.Jagannath C, Lindsey DR, Dhandayuthapani S, Xu Y, Hunter RL, Jr, Eissa NT. Autophagy enhances the efficacy of BCG vaccine by increasing peptide presentation in mouse dendritic cells. Nat Med. 2009;15:267–76. doi: 10.1038/nm.1928. [DOI] [PubMed] [Google Scholar]
- 16.Gutierrez MG, Master SS, Singh SB, Taylor GA, Colombo MI, Deretic V. Autophagy is a defense mechanism inhibiting BCG and Mycobacterium tuberculosis survival in infected macrophages. Cell. 2004;119:753–66. doi: 10.1016/j.cell.2004.11.038. [DOI] [PubMed] [Google Scholar]
- 17.Paludan C, Schmid D, Landthaler M, et al. Endogenous MHC class II processing of a viral nuclear antigen after autophagy. Science. 2005;307:593–6. doi: 10.1126/science.1104904. [DOI] [PubMed] [Google Scholar]
- 18.Schmid D, Pypaert M, Munz C. Antigen-loading compartments for major histocompatibility complex class II molecules continuously receive input from autophagosomes. Immunity. 2007;26:79–92. doi: 10.1016/j.immuni.2006.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saitoh T, Fujita N, Jang MH, et al. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature. 2008;456:264–8. doi: 10.1038/nature07383. [DOI] [PubMed] [Google Scholar]
- 20.Laliberte RE, Perregaux DG, McNiff P, Gabel CA. Human monocyte ATP-induced IL-1 β posttranslational processing is a dynamic process dependent on in vitro growth conditions. J Leukoc Biol. 1997;62:227–39. doi: 10.1002/jlb.62.2.227. [DOI] [PubMed] [Google Scholar]
- 21.Massey DC, Parkes M. Genome-wide association scanning highlights two autophagy genes, ATG16L1 and IRGM, as being significantly associated with Crohn's disease. Autophagy. 2007;3:649–51. doi: 10.4161/auto.5075. [DOI] [PubMed] [Google Scholar]
- 22.McCarroll SA, Huett A, Kuballa P, et al. Deletion polymorphism upstream of IRGM associated with altered IRGM expression and Crohn's disease. Nat Genet. 2008;40:1107–12. doi: 10.1038/ng.215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hampe J, Franke A, Rosenstiel P, et al. A genome-wide association scan of nonsynonymous SNPs identifies a susceptibility variant for Crohn disease in ATG16L1. Nat Genet. 2007;39:207–11. doi: 10.1038/ng1954. [DOI] [PubMed] [Google Scholar]
- 24.Singh SB, Davis AS, Taylor GA, Deretic V. Human IRGM induces autophagy to eliminate intracellular mycobacteria. Science. 2006;313:1438–41. doi: 10.1126/science.1129577. [DOI] [PubMed] [Google Scholar]
- 25.Intemann CD, Thye T, Niemann S, et al. Autophagy gene variant IRGM -261T contributes to protection from tuberculosis caused by Mycobacterium tuberculosis but not by M. africanum strains. PLoS Pathog. 2009;5:e1000577. doi: 10.1371/journal.ppat.1000577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Che N, Li S, Gao T, et al. Identification of a novel IRGM promoter single nucleotide polymorphism associated with tuberculosis. Clin Chim Acta. 2010;411:1645–9. doi: 10.1016/j.cca.2010.06.009. [DOI] [PubMed] [Google Scholar]
- 27.Deretic V, Delgado M, Vergne I, et al. Autophagy in immunity against Mycobacterium tuberculosis: a model system to dissect immunological roles of autophagy. Curr Top Microbiol Immunol. 2009;335:169–88. doi: 10.1007/978-3-642-00302-8_8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chung Y, Chang SH, Martinez GJ, et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity. 2009;30:576–87. doi: 10.1016/j.immuni.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stockinger B, Veldhoen M. Differentiation and function of Th17 T cells. Curr Opin Immunol. 2007;19:281–6. doi: 10.1016/j.coi.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 30.Rosenstiel P, Fantini M, Brautigam K, et al. TNF-α and IFN-γ regulate the expression of the NOD2 (CARD15) gene in human intestinal epithelial cells. Gastroenterology. 2003;124:1001–9. doi: 10.1053/gast.2003.50157. [DOI] [PubMed] [Google Scholar]
- 31.Wolfs TG, Buurman WA, van SA, et al. In vivo expression of Toll-like receptor 2 and 4 by renal epithelial cells: IFN-γ and TNF-α mediated up-regulation during inflammation. J Immunol. 2002;168:1286–93. doi: 10.4049/jimmunol.168.3.1286. [DOI] [PubMed] [Google Scholar]
- 32.Plantinga TS, Crisan TO, Oosting M, et al. Crohn′s disease associated ATG16L1 polymorphism modulates proinflammatory cytokine responses selectively upon activation of NOD2. Gut. 2011;60:1229–35. doi: 10.1136/gut.2010.228908. [DOI] [PubMed] [Google Scholar]
- 33.Kleinnijenhuis J, Joosten LA, Van d V, et al. Transcriptional and inflammasome-mediated pathways for the induction of IL-1β production by Mycobacterium tuberculosis. Eur J Immunol. 2009;39:1914–22. doi: 10.1002/eji.200839115. [DOI] [PubMed] [Google Scholar]
- 34.Petrilli V, Dostert C, Muruve DA, Tschopp J. The inflammasome: a danger sensing complex triggering innate immunity. Curr Opin Immunol. 2007;19:615–22. doi: 10.1016/j.coi.2007.09.002. [DOI] [PubMed] [Google Scholar]
- 35.van Crevel R, Parwati I, Sahiratmadja E, et al. Infection with Mycobacterium tuberculosis Beijing genotype strains is associated with polymorphisms in SLC11A1/NRAMP1 in Indonesian patients with tuberculosis. J Infect Dis. 2009;200:1671–4. doi: 10.1086/648477. [DOI] [PubMed] [Google Scholar]