

SUMMARY
Mutations in the imprinted CDKN1C gene are associated with the childhood developmental disorder Beckwith-Wiedemann syndrome (BWS). Multiple mouse models with deficiency of Cdkn1c recapitulate some aspects of BWS but do not exhibit overgrowth of the newborn, a cardinal feature of patients with BWS. In this study, we found that Cdkn1c mutants attained a 20% increase in weight during gestation but experienced a rapid reversal of this positive growth trajectory very late in gestation. We observed a marked effect on placental development concurrently with this loss of growth potential, with the appearance of large thrombotic lesions in the labyrinth zone. The trilaminar trophoblast layer that separates the maternal blood sinusoids from fetal capillaries was disordered with a loss of sinusoidal giant cells, suggesting a role for Cdkn1c in maintaining the integrity of the maternal-fetal interface. Furthermore, the overgrowth of mutant pups decreased in the face of increasing intrauterine competition, identifying a role for Cdkn1c in the allocation of the maternal resources via the placenta. This work explains one difficulty in precisely replicating BWS in this animal model: the differences in reproductive strategies between the multiparous mouse, in which intrauterine competition is high, and humans, in which singleton pregnancies are more common.
INTRODUCTION
Beckwith-Wiedemann syndrome (BWS; MIM 130650) is a complex congenital overgrowth disorder that occurs in approximately 1/13,700 live births. A diagnosis of BWS is usually based on the presence of two out of five major characteristics in the infant: macrosomia (birth weight >97th percentile), macroglossia, neonatal hypoglycaemia, ear creases or pits and/or abdominal wall defects. BWS can include other features such as hemihypertrophy, visceromegaly, hepatoblastoma, embryonal tumours, nevus flammeus, cleft palate, cardiac abnormalities, advanced bone age, enlarged placenta and abnormalities in placental vasculature (Weksberg et al., 2010). There is a high incidence of premature birth for BWS infants, sometimes in combination with polyhydramnios and gestational hypertension (Wangler et al., 2005), with some BWS mother’s suffering the more serious complication of HELLP and preeclampsia (Romanelli et al., 2009).
BWS patients display multiple genetic and epigenetic mutations that mainly disrupt the expression of a cluster of imprinted genes located at human chromosome 11p15 (Cooper et al., 2005; Weksberg et al., 2005). Nearly half of patients with familial BWS carry germline mutations in the coding sequence of the maternally expressed cyclin-dependent kinase inhibitor 1c (CDKN1C; p57KIP2) gene (Hatada et al., 1996; Hatada et al., 1997; Lee et al., 1997; O’Keefe et al., 1997; Engel et al., 2000). However, BWS is predominantly seen as a sporadic occurrence and, in this group of patients, direct DNA mutations within CDKN1C mutations are relatively infrequent (<5%) (Cooper et al., 2005). The most frequent alteration in BWS, reported in >50% of patients, is loss of DNA methylation at the promoter of a long, non-coding RNA, KCNQ1OT (also known as LIT1), which lies 220 kb distant to CDKN1C. Loss of methylation of this region is associated with downregulation of CDKN1C (Diaz-Meyer et al., 2003). Studies on the corresponding mouse imprinted domain on distal chromosome 7 demonstrate that this region, termed KvDMR1 or IC2 (imprinting centre 2), acts as the imprinting centre for Cdkn1c (Caspary et al., 1998; Feinberg, 2000; Fitzpatrick et al., 2002). All these data suggest that loss of CDKN1C is a factor in the majority of BWS cases. However, although three independent studies examining loss of Cdkn1c function in mice identified several developmental abnormalities consistent with BWS, including abdominal wall defects, cleft palate, placentomegaly, renal dysplasia, adrenal cytomegaly, maternal preeclampsia and prematurity, none reported the cardinal feature of BWS, that of somatic overgrowth at birth (Yan et al., 1997; Zhang et al., 1997; Takahashi et al., 2000a; Takahashi et al., 2000b; Kanayama et al., 2002).
In mice, Cdkn1c is expressed in derivatives of all three germ layers – the endoderm, mesoderm and ectoderm – and in all major organs of the body during embryonic development (Lee et al., 1995; Matsuoka et al., 1995; Westbury et al., 2001). Cdkn1c is primarily expressed in cells that are exiting cell cycle but are not terminally differentiated. In extraembryonic tissues, Cdkn1c is dynamically expressed during mid-to-late placental development in the giant trophoblast cells that abut the maternal decidua, the glycogen cells within the junctional zone, the fetal endothelium, the syncytiotrophoblast and some larger sinusoidal nuclei (Riley et al., 1998; Westbury et al., 2001; Georgiades et al., 2002; Coan et al., 2006). The spatial and temporal expression profile of Cdkn1c probably reflects the multiple functional roles that Cdkn1c plays during development. Cdkn1c, also known as p57Kip2, encodes a cyclin-dependant kinase inhibitor (CDKi) belonging to the same family as Cdkn1a (encoding p21) and Cdkn1b (encoding p27) (Hatada and Mukai, 1995; Lee et al., 1995; Matsuoka et al., 1995; Matsuoka et al., 1996). As with all CDKis, excess Cdkn1c induces cell cycle arrest (Lee et al., 1995; Matsuoka et al., 1995). In addition, Cdkn1c also directs differentiation in some cell types (Dyer and Cepko, 2000; Reynaud et al., 2000; Joseph et al., 2003; Joseph et al., 2009), influences cell migration (Sakai et al., 2004; Itoh et al., 2007) and modifies the actin cytoskeleton (Yokoo et al., 2003; Vlachos and Joseph, 2009). CDKN1C therefore functions in numerous processes to ensure correct development.
We have previously demonstrated that early embryonic growth is exquisitely sensitive to the precise dosage of Cdkn1c on the inbred mouse strain background 129S2/SvHsd (129) (Andrews et al., 2007). Mice that expressed Cdkn1c at just twofold the normal endogenous level were growth restricted from embryonic day 13.5 (E13.5), whereas mice deficient for Cdkn1c were heavier at E13.5. Several groups have reported that Cdkn1c deficiency does not result in an overgrowth phenotype at birth (Yan et al., 1997; Zhang et al., 1997; Takahashi et al., 2000a; Takahashi et al., 2000b; Kanayama et al., 2002). These studies were performed with the isogenic mouse strain C57BL/6 (BL6). Consequently, there are two possible explanations for the absence of overgrowth reported in Cdkn1c mutant mice. Either there are differences in strain background that account for a disparity in growth data between different studies, or Cdkn1c mutant mice experience a loss of growth potential late in development. The purpose of this study was to differentiate between these two possibilities by examining the growth potential of Cdkn1c mutants from E15.5 until birth on a pure (>99%) 129 strain background.
RESULTS
Characterising newborn Cdkn1c mutant (loss of maternal allele) phenotype on the 129 strain background
On a predominantly BL6 strain background, the majority of Cdkn1c mutant pups die around the time of birth (Yan et al., 1997; Zhang et al., 1997; Takahashi et al., 2000a). We found that the survival frequency of Cdkn1c mutant pups on the 129 background was similar to this, with four out of 60 survivors carrying the targeted allele (Table 1). To determine newborn weights, a total of 130 pups were recovered at birth from 22 litters. Similar to previous studies, newborn Cdkn1c mutant pups were not heavier than wild-type littermates (Fig. 1A). Typically, recovered wild-type pups had suckled (55/84), whereas only 4/46 (11.5%) Cdkn1c mutant pups showed evidence of milk in their stomach (Fig. 1B). Cdkn1c mutant pups displayed phenotypes that have previously been reported for the BL6 background, with various levels of limb shortening, cleft palate, abdominal muscle abnormalities and air inflated abdomens (data not shown). Cleft palate was not present in the four mutants that fed but the absence of overgrowth in Cdkn1c mutant pups could not be explained by a failure to suckle because, when fed animals were excluded from the analysis, mutants were still not significantly heavier than wild-type littermates (Fig. 1C). Thus, as with the BL6 studies, Cdkn1c mutant mice were not overgrown at birth on the 129 strain background.
Table 1.
Survival data for Cdkn1c mutants on a 129 strain background

Fig. 1.

Growth trajectory of Cdkn1c129 mutants. (A) Postnatal weights for wild-type (WT) and Cdkn1c mutant pups on a pure 129 background (WT, 1561±167 mg, n=84; Cdkn1c−m/+p, 1586±185 mg, n=46; 22 litters; P=0.44). Error bars show standard deviation. (B) Number of wild-type compared with mutant newborns that showed evidence of suckling. (C) Postnatal weights for wild-type and Cdkn1c mutant pups after excluding data from fed animals (WT, 1510±296 mg, n=29 versus Cdkn1c−m/+p 1567±287 mg, n=42; P=0.132). (D,E) Embryonic weights for wild type and Cdkn1c mutant at (D) E15.5 (WT, 407±84 mg, n=24; Cdkn1c−m/+p, 468±113 mg, n=21; P=6.27×10−6) and (E) E18.5 (WT, 1196±129 mg, n=63; Cdkn1c−m/+p, 1169±169, n=63; P=0.001).
In order to identify the time point after E13.5 at which Cdkn1c mutant embryonic growth was constrained, weights were examined at E15.5 and E18.5. In contrast to immediately after birth, there was no evidence for loss of viability at these earlier stages (Table 1), but Cdkn1c mutant embryos were 15% heavier than wild-type embryos at E15.5 (P=6.28×10−6) and 8% heavier at E18.5 (P=0.001) (Fig. 1D,E). This work identified a slowdown in embryonic growth very late in gestation.
Defects in the trilaminar trophoblast cell structure that separates the maternal blood sinusoids from the fetal capillaries
Loss of growth potential late in gestation is a classic indicator of placental insufficiency. The previously reported characteristics of the Cdkn1c mutant placenta include overgrowth with overproliferation of trophoblast cells, reduced vascularisation of the labyrinth zone and reduced diameter of mutant fetal capillaries, suggesting impaired function (Zhang et al., 1998; Takahashi et al., 2000a). Similarly, Cdkn1c mutant 129 placentas were substantially overgrown (Fig. 2A). Examination of the Cdkn1c mutant 129 placentas additionally exposed substantial areas of pooled maternal blood in the labyrinth at E15.5 and, more dramatically, at E18.5 in all samples examined (Fig. 2B), with extensive collagen deposition adjacent to these areas in mutants but not wild-type control placentas (Fig. 2C). The presence of these substantial thrombotic lesions precluded a meaningful quantification of the maternal sinusoids, but a general and irregular dilation was revealed by membrane-localised E-cadherin immunostaining (Fig. 2D). Furthermore, the large, polyploid nuclei that normally line the maternal blood spaces at E18.5 (Coan et al., 2005) were discernible in the labyrinth of both wild-type and mutant placenta at E18.5 but there were relatively fewer in the mutant labyrinth (Fig. 2F; Table 2).
Fig. 2.

Maternal blood space defect in Cdkn1c mutant placentas. (A) Wet weights for wild-type (WT) and Cdkn1c mutant placentas on a pure 129 background at the indicated gestational stages. Cdkn1c mutant placentas are 46% heavier at E13.5 (WT, 52±11 mg, n=8; Cdkn1c−m/+p, 76±9 mg, n=9; P=0.0001), 48% heavier at E15.5 (WT, 74±16 mg, n=24; Cdkn1c−m/+p, 110±25 mg, n=21; P=8.86×10−7) and 51% heavier at E18.5 (WT, 80±24 mg, n=24; Cdkn1c−m/+p, 117±24 mg, n=24; P=2.37×10−7). (B) Haematoxylin and eosin staining of Cdkn1c mutant and wild-type midline representative placental sections at E18.5, as indicated. Jz, junctional zone; Lab, labyrinth; mbs, maternal blood spaces. (C) Masson’s trichrome staining of Cdkn1c mutant and wild-type midline placental sections at E18.5. Collagen fibres stain blue (arrow). (D) E18.5 sections stained for E-cadherin followed by haematoxylin, revealing disordered maternal sinusoids in the labyrinth. (E) Higher power images of the labyrinth stained with DAPI. Large polyploid nuclei are readily apparent in wild-type but not mutant placenta.
Table 2.
Relative proportion of diploid to giant nuclei in the labyrinth at E18.5

Greatly enlarged maternal blood sinuses have previously been reported in mutant placenta lacking Cdkn1c in combination with excess expression of Igf2 (Caspary et al., 1999), and in placenta carrying a paternal duplication of distal chromosome 7 (Rentsendorj et al., 2010), but not in those with isolated loss of Cdkn1c. On the 129 strain, the maternal blood spaces defect arose from isolated loss of Cdkn1c. This was not explained by changes in Igf2 and Igf1r mRNA levels in mutant placenta or differences in Igf2 expression between BL6 and 129 placenta (data not shown) but probably reflects inherent differences in the structure of the placenta between the BL6 and 129 strains (Dackor et al., 2009).
A study of key markers of placental development at E15.5, when the placental phenotype was well established, revealed significant reductions in the expression level of genes that are either required for, or primarily expressed in, the junctional zone of the placenta (Fig. 3A). This included markers specific to spongiotrophoblast cells (Prl8a8 and Prl3a1), markers expressed in both spongiotrophoblast and glycogen cells (Tpbpa), markers expressed in both spongiotrophoblast and giant cells (Hand1) and Flt1, which is expressed in spongiotrophoblast, fetal endothelial cells and maternal endothelial cells at this time point (Lescisin et al., 1988; Breier et al., 1995; Riley et al., 1998; Simmons et al., 2008a) (Fig. 3A). By contrast, and despite the obvious abnormalities, the majority of markers assayed for the labyrinth were expressed at levels similar to wild type (Fig. 3B). The exceptions were cathepsin Q (Ctsq), an exclusive marker of the sinusoidal trophoblast giant cells (Simmons et al., 2007), whose expression was reduced to 60% of the wild-type level (P<0.014), and Prl3b1, a marker of spongiotrophoblast cells, parietal giant trophoblast cells and sinusoidal giant trophoblast cells (Simmons et al., 2008a), whose expression was reduced to 48% of the wild-type level. In situ characterisation with Prl3b1 at E15.5 suggested a defect in the sinusoidal trophoblast giant cell population (Fig. 3C).
Fig. 3.

Relative expression levels of representative placental markers at E15.5. (A) Junctional zone markers. Achaete-scute complex homolog 2 (Ascl2) is required for spongiotrophoblast differentiation (Guillemot et al., 1994). Trophoblast specific protein-α (Tpbpa) is expressed in spongiotrophoblast and glycogen cells (Lescisin et al., 1988). Heart and neural crest derivatives expressed transcript 1 (Hand1) expressed in spongiotrophoblast cells and giant cells, including sinusoidal trophoblast giant cells (Riley et al., 1998; Simmons et al., 2008a). Prolactin family 8, subfamily a, member 8 (Prl8a8) and Prl3a1 are spongiotrophoblast-cell-specific markers (Simmons et al., 2008a). FMS-like tyrosine kinase 1 (Flt1) is expressed in spongiotrophoblast cells, and fetal and maternal endothelial cells (Breier et al., 1995). *P=0.014. (B) Labyrinth zone markers with schematic illustrating the organisation of the trilaminar trophoblast layer with gene expression markers. Dlx3 is expressed broadly in the labyrinth without indication of layer-specificity at E14.5 (Simmons et al., 2008a). Kinase insert domain protein receptor (Kdr; also known as Flk1) is expressed in fetal endothelial cells with a key role in vascular development (Shalaby et al., 1995). Glial cell missing 1 (Gcm1) encodes a transcription factor required for the initiation of fetal blood vessel branching (Anson-Cartwright et al., 2000) and is expressed specifically in syncytiotrophoblast II cell layer by E14.5 (Simmons et al., 2008a). Pleckstrin homology-like domain, family A, member 2 (Phlda2) is expressed in syncytiotrophoblast layers I and II (Qian et al., 1997; Frank et al., 1999; Dunwoodie and Beddington, 2002). Syna is expressed in syncytiotrophoblast layer I (Simmons et al., 2008a). Placental lactogen II (Prl3b1) is expressed in both the parietal giant cell layer and sinusoidal trophoblast cells of the labyrinth (Jackson et al., 1986). Cathepsin Q (Ctsq) is expressed exclusively in the sinusoidal trophoblast giant cells (Simmons et al., 2007). Solute carrier family 2, member A1 (Slc22a1) is expressed both in the junctional zone and the labyrinth zone, and Sc22a3 expression is restricted to the sinusoidal trophoblast cells (Shin et al., 1997). *P=0.014. Data expressed as relative fold change in comparison with wild type. n=4+4. Error bars show standard deviation. (C) In situ hybridisation with Prl3b1 riboprobe of wild-type and Cdkn1c mutant placental sections at E15.5. Lower magnification images (4×) illustrate diminished staining in regions containing spongiotrophoblast cells and parietal and sinusoidal giant trophoblast cells, all of which are cell types that express this marker. Higher magnification (10×) specifically illustrates Prl3b1 staining of sinusoidal giant trophoblast cells in wild-type and mutant labyrinth.
Depleted glycogen stores in mutant placenta
It has been proposed that Cdkn1c might be required for the progression of pre-glycogen cells to mature glycogen cells by inhibiting their proliferation and promoting differentiation (Coan et al., 2006). Loss of function of Cdkn1c might consequently result in either an expansion of this cell population (overproliferation), with increased glycogen storage, or a failure in differentiation, with diminished glycogen storage. Quantification at E15.5 of the mRNAs of two genes expressed exclusively in the glycogen cells of the mouse placenta [protocadherin 12 (Pcdh12) and gap junction protein, beta 3 (Gjb3; also known as Cx31)] (Bouillot et al., 2006; Zheng-Fischhofer et al., 2007) revealed no difference in the expression of Pcdh12 (Fig. 4A), an early marker of this lineage (Bouillot et al., 2006), suggesting that Cdkn1c was not absolutely required for proliferation. However, expression of Gjb3, an exclusive marker of mature glycogen cells (Coan et al., 2006), was diminished significantly, suggesting that the differentiation of this population was affected by loss of Cdkn1c. Using periodic acid Schiff (PAS), a glycogen-specific stain, we observed a lower intensity of glycogen staining in placentas from mutant mice compared with controls at E15.5 (Fig. 4B). The concentration of glycogen in mutant placentas was significantly lower than in controls at E15.5 (69%) and at E18.5 (62%) (Fig. 4C). Defective glycogen storage might therefore contribute to the loss of embryonic growth potential very late in gestation.
Fig. 4.
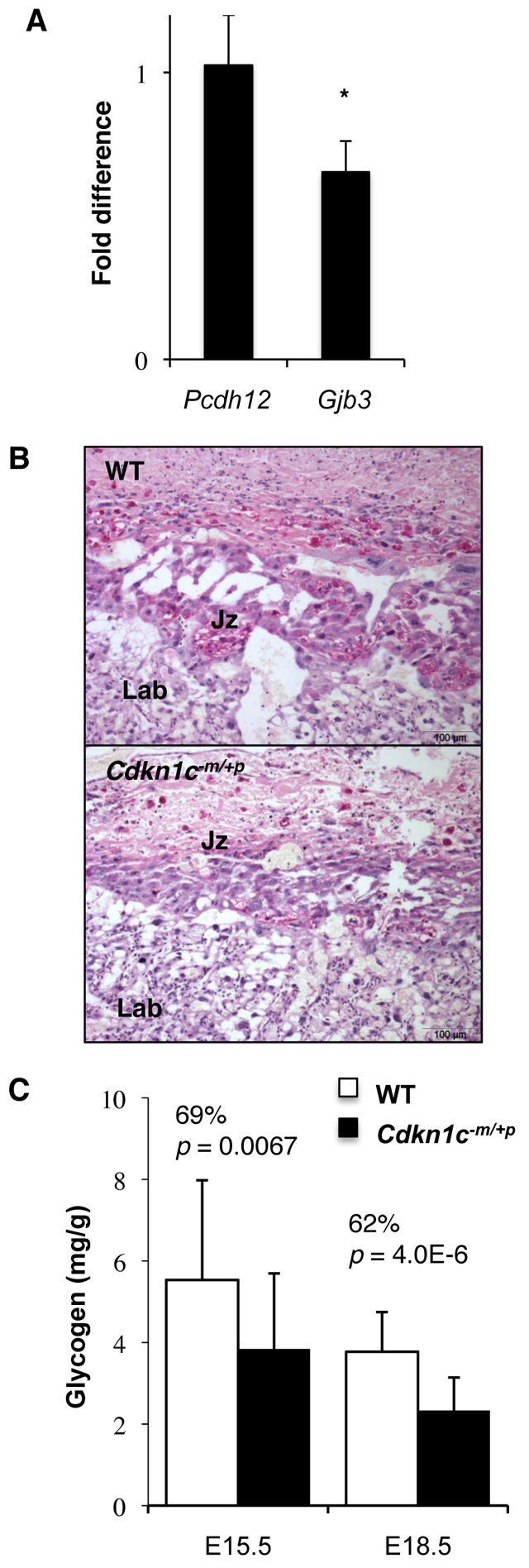
Glycogen levels in Cdkn1c mutants. (A) Relative expression levels of glycogen-cell-specific marker genes at E15.5. *P=0.014. Data expressed as relative fold change in comparison with wild type. n=4+4. Error bars show standard deviation. (B) Periodic acid Schiff staining of Cdkn1c mutant and wild-type midline placental sections at E15.5 and E18.5 reveals reduced glycogen staining in the transgenic junctional zone in mutants. (C) Direct biochemical determination of stored glycogen at E15.5 and E18.5 expressed per gram placental weight.
Overgrowth of mutants in small litters
There is a long established inverse correlation between litter size and the weight of individual pups within a litter, which suggests intrauterine competition for a limited supply of maternal resources (McLaren, 1965). Because a functionally defective placenta might be expected to diminish the ability of an embryo to solicit nutrients from the mother, we investigated whether there was evidence for this by examining fetal weights in litters with few pups (three to five), in which intrauterine competition would be lower, versus larger litters (>five pups), in which intrauterine competition would be greater (Fig. 5). Cdkn1c mutant embryos were more overgrown, by 19%, in smaller litters (Fig. 5). Furthermore, we did not find the anticipated inverse correlation between litter size and the weight in wild-type embryos, which were a similar average weight irrespective of litter size. This suggested that the mutants bore the brunt of increased intrauterine competition and thus might explain why Cdkn1c mutant mice, unlike singleton BWS babies with loss of function of CDKN1C, are not overgrown at birth.
Fig. 5.

Cdkn1c mutant embryos are able to reach their intrinsic growth potential when intrauterine competition is low. Embryonic weights for pups in litters with three to five pups versus litters of six or more pups. Mutant pups from litters with three to five pups were 119% the weight of wild type (1314±166 mg versus 1102±151 mg; six litters; P=0.002 by Student’s t-test), whereas the mutants from litters of six or more pups were 107% the weight of wild type (1095±105 mg versus 1156±158 mg; 15 litters; P=0.016). The weights of mutants in smaller litters were significantly different to the weights of mutants in larger litters (P<0.05 by ANOVA, Tukey-Kramer post hoc test), whereas wild-type weights did not differ between litter sizes (P>0.05).
DISCUSSION
The key finding of this study is that embryos deficient for Cdkn1c are nearly 20% heavier than their wild-type counterparts. Thus, loss of function of CDKN1C can result in somatic overgrowth in both humans and mice. However, in mice, overgrowth was attenuated by intrauterine competition. We also observed several striking defects of the placenta, suggesting that, despite its increased size, the function of this organ was compromised, which is consistent with the loss of growth potential of mutant embryos very late in gestation.
Cdkn1c mutant embryos were overgrown at E15.5 and at E18.5, just prior to birth, but, as with earlier studies, we observed no difference in the weight of mutant and wild-type pups after birth. Although this loss of growth potential might reflect developmental abnormalities of the mutant embryo, placental dysfunction provides an explanation both for the loss of growth potential late in gestation and the differences in growth potential of mutants in small litters to those in larger litters, in which there is more intrauterine competition via the placenta for maternal resources. Consistent with this interpretation, severe placental abnormalities preceded the attenuated overgrowth of Cdkn1c mutant mice. In particular, maternal blood seemed to be haemorrhaging into the mutant placenta, indicating a breakdown in the integrity of the trilaminar trophoblast layer that separates this maternal blood from the fetal circulation. This layer consists of three differentiated trophoblast cell types that are thought to arise from distinct precursors (Simmons et al., 2008b). Expression of Ctsq and Prl3b1, markers of the sinusoidal trophoblast giant cells that line the maternal sinusoids, was significantly reduced, suggesting a specific defect in this component of the trilaminar trophoblast layer. Large nuclei have been observed in these cells very late in gestation and are thought to result from endoreduplication (Coan et al., 2005). Cdkn1c has a known role in triggering endoreduplication in trophoblast stem (TS) cells through inhibition of Cdk1 (Ullah et al., 2008). There were fewer giant cells in the labyrinth of the mutant placenta, providing the first in vivo evidence that the process of endoreduplication in the sinusoidal giant trophoblast cells is regulated by Cdkn1c, being delayed or diminished in mutants lacking this protein.
In addition to the obvious defect in the maternal-fetal interface, we identified a reduction in the amount of glycogen stored in the placenta at E15.5 and E18.5. Although the glycogen cell lineage was present in both wild-type and mutant placenta, diminished expression of Gjb3, which marks mature glycogen cells, in the mutants suggested a role for Cdkn1c in the maturation of these cells, which might account for their lower glycogen content. Placental glycogen stored in the late gestational placenta is thought to provide a rapidly mobilisable energy depot for the final stages of gestation, when the fetus is placing the highest demands on maternal resources for growth (Coan et al., 2006). Our data indicate that loss of function of Cdkn1c alone does not boost glycogen stores to match placental overgrowth and the intrinsic growth potential of the embryo. This mismatch might contribute to the failure of Cdkn1c mutants to attain their maximum intrinsic growth potential. We have previously shown that an adjacent imprinted gene, Phlda2, regulates the storage of glycogen in the placenta, and we suggested that imprinting Cdkn1c and Phlda2 would have a synergistic outcome for in utero growth with the reduced dosage of Cdkn1c acting intrinsically to increase the fetal size and the reduced dosage of Phlda2 providing the necessary placental modifications to support this enhanced growth (Tunster et al., 2010). In this respect, it will be interesting to determine the consequences of combined loss of expression of Cdkn1c and Phlda2 on late fetal growth.
The placental defects that we have observed and the unusual growth dynamics of Cdkn1c mutant embryos suggest an explanation for why it has been difficult to precisely replicate BWS in this animal model. First, there are comparative differences between the human and mouse placenta: the human placenta has a hemomonochorial structure lacking a cell type equivalent to the sinusoidal giant trophoblast cell in mice (Georgiades et al., 2002; Carter and Enders, 2004), so the consequence of loss of function of CDKN1C in the human placenta might differ from that in the mouse. More importantly, in mice there is competition between individual foetuses for maternal resources. In the mouse model, mutants were able to overgrow by 19% in small litters but were only able to overgrow by 7% in the larger litters, whereas there was no difference in the growth of the wild-type embryos with respect to intrauterine competition, indicating that mutants were less able to compete than their wild-type littermates. Human pregnancies are generally singleton; therefore, the consequences of placental dysfunction might be less apparent in some pregnancies. In twin pregnancies, there is competition between fetuses and, in a study on pregnancies in which twins were discordant for BWS, in 7/11 cases the BWS twin was not heavier than the unaffected twin (Bliek et al., 2009), suggesting that the BWS fetus might experience a loss of growth potential when in competition with an unaffected fetus in utero.
Conclusions
BWS is traditionally regarded as an overgrowth disorder, with babies classified primarily on the basis of birth weight over the 97th percentile. Our data provides further support for the role of CDKN1C in this disorder but also suggests that, in some circumstances, BWS infants with inactivating mutations at CDKN1C might not display overt overgrowth at birth. Reassessing the birth weights in existing BWS patients with known inactivating mutations in CDKN1C and those that have an epigenetic mutation at KvDMR (balanced loss of expression of CDKN1C and PHLDA2) will be an important step forward in our understanding of the aetiology of BWS. This work will be important because some BWS infants might not be identified by clinicians as classic BWS cases owing to the lack of somatic overgrowth. Moreover, it might be that loss of function of CDKN1C is a more common mutation than currently realised, for example, in cases of cleft palate or abdominal wall defects in the absence of fetal overgrowth and in cases in which the mother develops preeclampsia.
METHODS
Mice
The mutant mice used in this study (Cdkn1ctm1Sje), which were generated by targeted removal of exons 1 and 2 of the Cdkn1c gene, have been described previously (Zhang et al., 1997). To avoid ambiguity, we indicate the parental origin of the alleles in heterozygotes by a superscript m for maternal or p for paternal. Male Cdkn1c+m/−p heterozygotes (absence of paternal allele; regarded as phenotypically wild type) were crossed to wild-type 129S2/SvHsd (129) females for >12 generations and the offspring genotyped as described previously (Andrews et al., 2007). To generate offspring without expression from the maternal allele (Cdkn1c−m/+p; termed Cdkn1c mutants), female Cdkn1c+m/−p heterozygotes were mated with wild-type 129 males.
Growth analysis
Embryonic and placental wet weights were taken at the stated time points after a discernible plug. Embryos and placentas were dissected free from extraembryonic membranes, immersed in cold fixative, briefly mopped dry and weighed. In order to obtain postnatal weights, pregnant females were monitored from E17.5. Litters were collected as found.
Histological analyses
Placentas were fixed overnight in phosphate-buffered 4% paraformaldehyde (PFA), paraffin-embedded and 10 μm sections taken. Haematoxylin and eosin (H&E) staining, in situ hybridisation and periodic acid Schiff staining for glycogen were performed as described (Tunster et al., 2010). Masson’s trichrome staining was performed using a standard protocol with the substitution of light green for aniline blue, staining collagen green. E-cadherin immunohistochemistry was performed on 5 μm midline placental sections. Sections were dewaxed in xylene and rehydrated through a series of ethanol washes. Antigen retrieval was performed by boiling slides in citrate buffer (Dako) for 20 minutes. Slides were blocked in Envision+ peroxidase block (Dako) and then 10% normal rabbit serum. Slides were incubated overnight at 4°C with a 1:150 dilution of purified mouse anti E-cadherin primary antibody (BD Transduction Laboratories). Sections were incubated in anti-mouse HRP-polymer secondary antibody (Dako), and positivity observed by incubating with DAB chromogen (Dako). Slides were counterstained with Mayer’s haematoxylin (Sigma), dehydrated and mounted in DPX. Slides were similarly processed without the primary antibody as controls for non-specific staining. Counting of placental cells was performed on morphologically equivalent areas of the labyrinth at E18.5. Owing to the irregularities of the maternal blood spaces, it was not feasible to determine the cell number per placenta. Instead, a count was made of the giant trophoblast cells as a proportion of the diploid trophoblast cells within a fixed field of view.
mRNA analysis
Quantitative PCR of reverse transcribed RNA was performed as described (Tunster et al., 2010). n=4 wild type + 4 Cdkn1c mutant, obtained from two litters (2+2 for each litter).
Biochemical determination of placental glycogen concentration
Glycogen was extracted from whole placenta according to the method of Lo et al. (Lo et al., 1970) and resuspended in 1 ml of H2O. Glycogen extract was diluted 1:60 in H2O and glycogen concentration determined using a glycogen assay kit (BioAssay Systems) as described (Tunster et al., 2010).
Statistical analyses
Fold change in expression was calculated, using ddCt values, for each gene compared with two housekeeping genes for 4+4 samples. Mann-Whitney tests were used to determine whether these fold changes were significantly different from 1. Within litters, statistical significance [probability (P) values] was determined using the Student’s t-test (two-tailed distribution and two sample unequal variance). To compare weight data between litters, Anderson-Darling was used as a basic test for normality (P=0.239) and Bartlett’s test as a test for unequal variance (P=0.350). The Tukey-Kramer method was used in conjunction with an ANOVA to find which means were significantly different from one another. The difference between mutants and wild type in small litters and also between mutants in small litters versus mutants in large litters were both significant (P<0.05), whereas the difference between mutants and wild type within the larger litters was not significant (although this was significant by a standard t-test), nor was the difference between wild type in small litters and those in large litters significant. The χ2 test was performed to determine the significance in the difference between observed over the expected appearance of a particular genotype.
TRANSLATIONAL IMPACT.
Clinical issue
Beckwith-Wiedemann syndrome (BWS), which affects 1/13,700 live births, is a congenital disorder causing large body size, large organs and a number of other symptoms. However, mouse models based on the most common mutation reported in BWS patients, loss of function of the cell cycle inhibitor CDKN1C, do not display overgrowth at birth, calling into question the validity of the mouse as an appropriate model organism for studying this disorder.
Results
In this paper, the authors re-examine the CDKN1C-deficient mouse model. They find that mutants do in fact display significant overgrowth early on in gestation, but that embryonic growth slows down shortly before birth. The extent of overgrowth noted at birth is inversely proportional to the size of the litter, owing to CDKN1C-specific placental defects that constrain overgrowth in large litters.
Implications and future directions
These findings confirm that loss of CDKN1C function causes somatic overgrowth in mice, modelling this key aspect of BWS. This has previously been masked by placental defects that constrain overgrowth when there are multiple foetuses, as is normal for mice, but not as common in humans. This finding might be of great importance for human BWS, because human BWS patients also present with placental defects. It is possible that, in some instances, such as in twin pregnancies, overgrowth of human CDKN1C mutant foetuses might be restrained, similar to what is observed in the mouse. Consequently, BWS and CDKN1C loss of function might be substantially underdiagnosed because the main diagnostic indicator is lost.
Acknowledgments
We thank Derek Scarborough for assistance with Masson’s trichrome staining and Zoe Basset for help with the biochemical determination of placental glycogen. We are grateful to Steve Elledge and Pumin Zhang for originally supplying us with their Cdkn1c knockout mouse model in 1997. We particularly thank Graham Burton and Louis Lefebvre for their valuable and insightful comments. S.J.T. was supported by BBSRC grant BB/G015465 and M.V.d.P. holds a BBSRC PhD studentship.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
R.M.J. conceived the study, performed the growth analysis and drafted the manuscript. S.J.T. performed the histological analyses, quantitative PCR and glycogen measurements. M.V.d.P. assisted with data accumulation and statistical analysis.
REFERENCES
- Andrews S. C., Wood M. D., Tunster S. J., Barton S. C., Surani M. A., John R. M. (2007). Cdkn1c (p57Kip2) is the major regulator of embryonic growth within its imprinted domain on mouse distal chromosome 7. BMC Dev. Biol. 7, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anson-Cartwright L., Dawson K., Holmyard D., Fisher S. J., Lazzarini R. A., Cross J. C. (2000). The glial cells missing-1 protein is essential for branching morphogenesis in the chorioallantoic placenta. Nat. Genet. 25, 311–314 [DOI] [PubMed] [Google Scholar]
- Bliek J., Alders M., Maas S. M., Oostra R. J., Mackay D. M., van der Lip K., Callaway J. L., Brooks A., van ‘t Padje S., Westerveld A., et al. (2009). Lessons from BWS twins: complex maternal and paternal hypomethylation and a common source of haematopoietic stem cells. Eur. J. Hum. Genet. 17, 1625–1634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouillot S., Rampon C., Tillet E., Huber P. (2006). Tracing the glycogen cells with protocadherin 12 during mouse placenta development. Placenta 27, 882–888 [DOI] [PubMed] [Google Scholar]
- Breier G., Clauss M., Risau W. (1995). Coordinate expression of vascular endothelial growth factor receptor-1 (flt-1) and its ligand suggests a paracrine regulation of murine vascular development. Dev. Dyn. 204, 228–239 [DOI] [PubMed] [Google Scholar]
- Carter A. M., Enders A. C. (2004). Comparative aspects of trophoblast development and placentation. Reprod. Biol. Endocrinol. 2, 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspary T., Cleary M. A., Baker C. C., Guan X. J., Tilghman S. M. (1998). Multiple mechanisms regulate imprinting of the mouse distal chromosome 7 gene cluster. Mol. Cell. Biol. 18, 3466–3474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caspary T., Cleary M. A., Perlman E. J., Zhang P., Elledge S. J., Tilghman S. M. (1999). Oppositely imprinted genes p57(Kip2) and igf2 interact in a mouse model for Beckwith-Wiedemann syndrome. Genes Dev. 13, 3115–3124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coan P. M., Ferguson-Smith A. C., Burton G. J. (2005). Ultrastructural changes in the interhaemal membrane and junctional zone of the murine chorioallantoic placenta across gestation. J. Anat. 207, 783–796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coan P. M., Conroy N., Burton G. J., Ferguson-Smith A. C. (2006). Origin and characteristics of glycogen cells in the developing murine placenta. Dev. Dyn. 235, 3280–3294 [DOI] [PubMed] [Google Scholar]
- Cooper W. N., Luharia A., Evans G. A., Raza H., Haire A. C., Grundy R., Bowdin S. C., Riccio A., Sebastio G., Bliek J., et al. (2005). Molecular subtypes and phenotypic expression of Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 13, 1025–1032 [DOI] [PubMed] [Google Scholar]
- Dackor J., Caron K. M., Threadgill D. W. (2009). Placental and embryonic growth restriction in mice with reduced function epidermal growth factor receptor alleles. Genetics 183, 207–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz-Meyer N., Day C. D., Khatod K., Maher E. R., Cooper W., Reik W., Junien C., Graham G., Algar E., Der Kaloustian V. M., et al. (2003). Silencing of CDKN1C (p57KIP2) is associated with hypomethylation at KvDMR1 in Beckwith-Wiedemann syndrome. J. Med. Genet. 40, 797–801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunwoodie S. L., Beddington R. S. (2002). The expression of the imprinted gene Ipl is restricted to extra-embryonic tissues and embryonic lateral mesoderm during early mouse development. Int. J. Dev. Biol. 46, 459–466 [PubMed] [Google Scholar]
- Dyer M. A., Cepko C. L. (2000). p57(Kip2) regulates progenitor cell proliferation and amacrine interneuron development in the mouse retina. Development 127, 3593–3605 [DOI] [PubMed] [Google Scholar]
- Engel J. R., Smallwood A., Harper A., Higgins M. J., Oshimura M., Reik W., Schofield P. N., Maher E. R. (2000). Epigenotype-phenotype correlations in Beckwith-Wiedemann syndrome. J. Med. Genet. 37, 921–926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg A. P. (2000). The two-domain hypothesis in Beckwith-Wiedemann syndrome. J. Clin. Invest. 106, 739–740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzpatrick G. V., Soloway P. D., Higgins M. J. (2002). Regional loss of imprinting and growth deficiency in mice with a targeted deletion of KvDMR1. Nat. Genet. 32, 426–431 [DOI] [PubMed] [Google Scholar]
- Frank D., Mendelsohn C. L., Ciccone E., Svensson K., Ohlsson R., Tycko B. (1999). A novel pleckstrin homology-related gene family defined by Ipl/Tssc3, TDAG51, and Tih1: tissue-specific expression, chromosomal location, and parental imprinting. Mamm. Genome 10, 1150–1159 [DOI] [PubMed] [Google Scholar]
- Georgiades P., Ferguson-Smith A. C., Burton G. J. (2002). Comparative developmental anatomy of the murine and human definitive placentae. Placenta 23, 3–19 [DOI] [PubMed] [Google Scholar]
- Guillemot F., Nagy A., Auerbach A., Rossant J., Joyner A. L. (1994). Essential role of Mash-2 in extraembryonic development. Nature 371, 333–336 [DOI] [PubMed] [Google Scholar]
- Hatada I., Mukai T. (1995). Genomic imprinting of p57KIP2, a cyclin-dependent kinase inhibitor, in mouse. Nat. Genet. 11, 204–206 [DOI] [PubMed] [Google Scholar]
- Hatada I., Ohashi H., Fukushima Y., Kaneko Y., Inoue M., Komoto Y., Okada A., Ohishi S., Nabetani A., Morisaki H., et al. (1996). An imprinted gene p57(KIP2) is mutated in Beckwith-Wiedemann syndrome. Nat. Genet. 14, 171–173 [DOI] [PubMed] [Google Scholar]
- Hatada I., Nabetani A., Morisaki H., Xin Z., Ohishi S., Tonoki H., Niikawa N., Inoue M., Komoto Y., Okada A., et al. (1997). New p57KIP2 mutations in Beckwith-Wiedemann syndrome. Hum. Genet. 100, 681–683 [DOI] [PubMed] [Google Scholar]
- Itoh Y., Masuyama N., Nakayama K., Nakayama K. I., Gotoh Y. (2007). The cyclin-dependent kinase inhibitors p57 and p27 regulate neuronal migration in the developing mouse neocortex. J. Biol. Chem. 282, 390–396 [DOI] [PubMed] [Google Scholar]
- Jackson L. L., Colosi P., Talamantes F., Linzer D. I. (1986). Molecular cloning of mouse placental lactogen cDNA. Proc. Natl. Acad. Sci. USA 83, 8496–8500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph B., Wallen-Mackenzie A., Benoit G., Murata T., Joodmardi E., Okret S., Perlmann T. (2003). p57(Kip2) cooperates with Nurr1 in developing dopamine cells. Proc. Natl. Acad. Sci. USA 100, 15619–15624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph B., Andersson E. R., Vlachos P., Sodersten E., Liu L., Teixeira A. I., Hermanson O. (2009). p57Kip2 is a repressor of Mash1 activity and neuronal differentiation in neural stem cells. Cell Death Differ. 16, 1256–1265 [DOI] [PubMed] [Google Scholar]
- Kanayama N., Takahashi K., Matsuura T., Sugimura M., Kobayashi T., Moniwa N., Tomita M., Nakayama K. (2002). Deficiency in p57Kip2 expression induces preeclampsia-like symptoms in mice. Mol. Hum. Reprod. 8, 1129–1135 [DOI] [PubMed] [Google Scholar]
- Lee M. H., Reynisdottir I., Massague J. (1995). Cloning of p57KIP2, a cyclin-dependent kinase inhibitor with unique domain structure and tissue distribution. Genes Dev. 9, 639–649 [DOI] [PubMed] [Google Scholar]
- Lee M. P., DeBaun M., Randhawa G., Reichard B. A., Elledge S. J., Feinberg A. P. (1997). Low frequency of p57KIP2 mutation in Beckwith-Wiedemann syndrome. Am. J. Hum. Genet. 61, 304–309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lescisin K. R., Varmuza S., Rossant J. (1988). Isolation and characterization of a novel trophoblast-specific cDNA in the mouse. Genes Dev. 2, 1639–1646 [DOI] [PubMed] [Google Scholar]
- Lo S., Russell J. C., Taylor A. W. (1970). Determination of glycogen in small tissue samples. J. Appl. Physiol. 28, 234–236 [DOI] [PubMed] [Google Scholar]
- Matsuoka S., Edwards M. C., Bai C., Parker S., Zhang P., Baldini A., Harper J. W., Elledge S. J. (1995). p57KIP2, a structurally distinct member of the p21CIP1 Cdk inhibitor family, is a candidate tumor suppressor gene. Genes Dev. 9, 650–662 [DOI] [PubMed] [Google Scholar]
- Matsuoka S., Thompson J. S., Edwards M. C., Bartletta J. M., Grundy P., Kalikin L. M., Harper J. W., Elledge S. J., Feinberg A. P. (1996). Imprinting of the gene encoding a human cyclin-dependent kinase inhibitor, p57KIP2, on chromosome 11p15. Proc. Natl. Acad. Sci. USA 93, 3026–3030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren A. (1965). Genetic and environmental effects on foetal and placental growth in mice. J. Reprod. Fertil. 9, 79–98 [DOI] [PubMed] [Google Scholar]
- O’Keefe D., Dao D., Zhao L., Sanderson R., Warburton D., Weiss L., Anyane-Yeboa K., Tycko B. (1997). Coding mutations in p57KIP2 are present in some cases of Beckwith-Wiedemann syndrome but are rare or absent in Wilms tumors. Am. J. Hum. Genet. 61, 295–303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian N., Frank D., O’Keefe D., Dao D., Zhao L., Yuan L., Wang Q., Keating M., Walsh C., Tycko B. (1997). The IPL gene on chromosome 11p15.5 is imprinted in humans and mice and is similar to TDAG51, implicated in Fas expression and apoptosis. Hum. Mol. Genet. 6, 2021–2029 [DOI] [PubMed] [Google Scholar]
- Rentsendorj A., Mohan S., Szabo P. E., Mann J. R. (2010). A genomic imprinting defect in mice traced to a single gene. Genetics 186, 686–699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynaud E. G., Guillier M., Leibovitch M. P., Leibovitch S. A. (2000). Dimerization of the amino terminal domain of p57Kip2 inhibits cyclin D1- cdk4 kinase activity. Oncogene 19, 1147–1152 [DOI] [PubMed] [Google Scholar]
- Riley P., Anson-Cartwright L., Cross J. C. (1998). The Hand1 bHLH transcription factor is essential for placentation and cardiac morphogenesis. Nat. Genet. 18, 271–275 [DOI] [PubMed] [Google Scholar]
- Romanelli V., Belinchon A., Campos-Barros A., Heath K. E., Garcia-Minaur S., Martinez-Glez V., Palomo R., Mercado G., Gracia R., Lapunzina P. (2009). CDKN1C mutations in HELLP/preeclamptic mothers of Beckwith-Wiedemann Syndrome (BWS) patients. Placenta 30, 551–554 [DOI] [PubMed] [Google Scholar]
- Sakai K., Peraud A., Mainprize T., Nakayama J., Tsugu A., Hongo K., Kobayashi S., Rutka J. T. (2004). Inducible expression of p57KIP2 inhibits glioma cell motility and invasion. J. Neurooncol. 68, 217–223 [DOI] [PubMed] [Google Scholar]
- Shalaby F., Rossant J., Yamaguchi T. P., Gertsenstein M., Wu X. F., Breitman M. L., Schuh A. C. (1995). Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376, 62–66 [DOI] [PubMed] [Google Scholar]
- Shin B. C., Fujikura K., Suzuki T., Tanaka S., Takata K. (1997). Glucose transporter GLUT3 in the rat placental barrier: a possible machinery for the transplacental transfer of glucose. Endocrinology 138, 3997–4004 [DOI] [PubMed] [Google Scholar]
- Simmons D. G., Fortier A. L., Cross J. C. (2007). Diverse subtypes and developmental origins of trophoblast giant cells in the mouse placenta. Dev. Biol. 304, 567–578 [DOI] [PubMed] [Google Scholar]
- Simmons D. G., Rawn S., Davies A., Hughes M., Cross J. C. (2008a). Spatial and temporal expression of the 23 murine Prolactin/Placental Lactogen-related genes is not associated with their position in the locus. BMC Genomics 9, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons D. G., Natale D. R., Begay V., Hughes M., Leutz A., Cross J. C. (2008b). Early patterning of the chorion leads to the trilaminar trophoblast cell structure in the placental labyrinth. Development 135, 2083–2091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi K., Kobayashi T., Kanayama N. (2000a). p57(Kip2) regulates the proper development of labyrinthine and spongiotrophoblasts. Mol. Hum. Reprod. 6, 1019–1025 [DOI] [PubMed] [Google Scholar]
- Takahashi K., Nakayama K., Nakayama K. (2000b). Mice lacking a CDK inhibitor, p57Kip2, exhibit skeletal abnormalities and growth retardation. J. Biochem. 127, 73–83 [DOI] [PubMed] [Google Scholar]
- Tunster S. J., Tycko B., John R. M. (2010). The imprinted Phlda2 gene regulates extraembryonic energy stores. Mol. Cell. Biol. 30, 295–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullah Z., Kohn M. J., Yagi R., Vassilev L. T., DePamphilis M. L. (2008). Differentiation of trophoblast stem cells into giant cells is triggered by p57/Kip2 inhibition of CDK1 activity. Genes Dev. 22, 3024–3036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlachos P., Joseph B. (2009). The Cdk inhibitor p57(Kip2) controls LIM-kinase 1 activity and regulates actin cytoskeleton dynamics. Oncogene 28, 4175–4188 [DOI] [PubMed] [Google Scholar]
- Wangler M. F., Chang A. S., Moley K. H., Feinberg A. P., Debaun M. R. (2005). Factors associated with preterm delivery in mothers of children with Beckwith-Wiedemann syndrome: a case cohort study from the BWS registry. Am. J. Med. Genet. 134A, 187–191 [DOI] [PubMed] [Google Scholar]
- Weksberg R., Shuman C., Smith A. C. (2005). Beckwith-Wiedemann syndrome. Am. J. Med. Genet. 137C, 12–23 [DOI] [PubMed] [Google Scholar]
- Weksberg R., Shuman C., Beckwith J. B. (2010). Beckwith-Wiedemann syndrome. Eur. J. Hum. Genet. 18, 8–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westbury J., Watkins M., Ferguson-Smith A. C., Smith J. (2001). Dynamic temporal and spatial regulation of the cdk inhibitor p57(kip2) during embryo morphogenesis. Mech. Dev. 109, 83–89 [DOI] [PubMed] [Google Scholar]
- Yan Y., Frisen J., Lee M. H., Massague J., Barbacid M. (1997). Ablation of the CDK inhibitor p57Kip2 results in increased apoptosis and delayed differentiation during mouse development. Genes Dev. 11, 973–983 [DOI] [PubMed] [Google Scholar]
- Yokoo T., Toyoshima H., Miura M., Wang Y., Iida K. T., Suzuki H., Sone H., Shimano H., Gotoda T., Nishimori S., et al. (2003). p57Kip2 regulates actin dynamics by binding and translocating LIM-kinase 1 to the nucleus. J. Biol. Chem. 278, 52919–52923 [DOI] [PubMed] [Google Scholar]
- Zhang P., Liegeois N. J., Wong C., Finegold M., Hou H., Thompson J. C., Silverman A., Harper J. W., DePinho R. A., Elledge S. J. (1997). Altered cell differentiation and proliferation in mice lacking p57KIP2 indicates a role in Beckwith-Wiedemann syndrome. Nature 387, 151–158 [DOI] [PubMed] [Google Scholar]
- Zhang P., Wong C., DePinho R. A., Harper J. W., Elledge S. J. (1998). Cooperation between the Cdk inhibitors p27(KIP1) and p57(KIP2) in the control of tissue growth and development. Genes Dev. 12, 3162–3167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng-Fischhofer Q., Kibschull M., Schnichels M., Kretz M., Petrasch-Parwez E., Strotmann J., Reucher H., Lynn B. D., Nagy J. I., Lye S. J., et al. (2007). Characterization of connexin31.1-deficient mice reveals impaired placental development. Dev. Biol. 312, 258–271 [DOI] [PubMed] [Google Scholar]