

Scientific Abstract
Structural and sequence variation have been described in several members of the contactin (CNTN) and contactin associated protein (CNTNAP) gene families in association with neurodevelopmental disorders, including autism. Using array comparative genome hybridization (CGH), we identified a maternally inherited ~535 kb deletion at 3p26.3 encompassing the 5′ end of the contactin 4 gene (CNTN4) in a patient with autism. Based on this finding and previous reports implicating genomic rearrangements of CNTN4 in autism spectrum disorders (ASDs) and 3p− microdeletion syndrome, we undertook sequencing of the coding regions of the gene in a local ASD cohort in comparison with a set of controls. Unique missense variants were identified in 4/75 unrelated individuals with an ASD, as well as in 1/107 controls. All of the amino acid substitutions were nonsynonomous, occurred at evolutionarily conserved positions, and were, thus, felt likely to be deleterious. However, these data did not reach statistical significance, nor did the variants segregate with disease within all of the ASD families. Finally, there was no detectable difference in binding of two of the variants to the interacting protein PTPRG in vitro. Thusadditional, larger studies will be necessary to determine whether CNTN4 functions as an autism susceptibility locus in combination with other genetic and/or environmental factors.
Keywords: contactin 4, autism, autism spectrum disorder, 3p26 deletion, contactins, susceptibility locus
Introduction
Autism spectrum disorders (ASDs) are a heterogeneous and common group of developmental disorders. Affected infants and children demonstrate severe and persistent impairments in expressive and receptive language and social interaction skills that are evident before age 3, as well as a variety of stereotypic and repetitive behaviors. There is overwhelming scientific evidence demonstrating high heritability (≥90%) and a strong genetic component to the etiologies of autism (reviewed in [Abrahams & Geschwind, 2008]). However, identifiable causes, such as Fragile X syndrome and chromosome abnormalities, account for ≤10% of cases. It is estimated that 10 to >100 genes, in addition to environmental effects, may contribute to disease susceptibility. Recently, small de novo genomic rearrangements or copy number variations (CNVs), that are likely pathogenic, have been identified in an additional ~10% of ASD cases [Marshall et al., 2008; Sebat et al., 2007; Shen et al., 2010; Szatmari et al., 2007]. Selected inherited CNVs likely represent additional susceptibility loci [Marshall et al., 2008; Shen et al., 2010; Szatmari et al., 2007]. A unifying theme among many of the CNVs identified, as well as susceptibility loci ascertained by linkage and association studies and direct gene sequencing, is localization of the involved protein at neural synapses.
Contactin 4 (CNTN4), also called BIG-2, is a member of the immunoglobulin (Ig) superfamily of axon-associated adhesion molecules. The structure for the six human contactins consists of an N-terminal signal peptide followed by 6 Ig-like and 4 fibronectin type III-like domains, and a C-terminal glycosylphosphatidylinositol (GPI) binding site that anchors the protein to the external surface of the plasma membrane [Shimoda & Watanabe, 2009]. Cell surface contactins complex with one or more of 5 human contactin-associated proteins (CNTNAPs) that are members of the neurexin gene family (reviewed in [Burbach & van der Zwaag, 2009; Katidou, Vidaki, Strigini, & Karagogeos, 2008]). These complexes are believed to participate in neuronal migration, axon guidance, and neuronal and glial cell interactions, with exact functions dependent on the developmental stage and exact localization of the interacting proteins.
Recent research implicates contactins and contactin-associated proteins (CNTNAPs) in neurodevelopmental disorders, including autism. Linkage and association analyses [Alarcon et al., 2008; Arking et al., 2008], as well as deep resequencing [Bakkaloglu et al., 2008], identified CNTNAP2 as a susceptibility locus for autism. Homozygosity mapping identified a homozygous deletion in the 5′ UTR of CNTN3 (BIG-1) in an autistic individual from a consanguineous mating, further implicating this gene family [Morrow et al., 2008].
CNTN4 maps to 3p26.3 and is deleted in individuals with 3p− syndrome, a rare microdeletion syndrome associated with developmental delay/mental retardation, microcephaly, and characteristic facies [Fernandez et al., 2008; Malmgren, Sahlen, Wide, Lundvall, & Blennow, 2007]. The CNTN4 gene has been considered a strong candidate for the developmental delay and microcephaly associated with 3p− syndrome, based on its postulated role in neurodevelopment and its disruption in patients with features of 3p− syndrome and a balanced 3;10 or unbalanced 3;13 chromosome translocation, respectively [Fernandez et al., 2004; Fernandez et al., 2008]. Several studies performing molecular characterization of patients with 3p− syndrome have concluded that CNTN4 lies within the minimal critical region [Dijkhuizen et al., 2006; Malmgren et al., 2007]. However, in a more recent investigation, CNTN4 appears to be excluded from the critical region based on a single patient with an interstitial 3p26 deletion [Shuib et al., 2009]. In addition, rare phenotypically normal individuals have been reported with visible deletions involving 3p26 that include CNTN4 ([Pohjola, de Leeuw, Penttinen, & Kaariainen, 2010; Shuib et al., 2009] and references therein).
More recently, an intragenic deletion within CNTN4 [Roohi et al., 2009] was reported in two siblings with an ASD and their clinically unaffected father. A third affected child did not carry the deletion. Similarly, inherited and de novo intragenic duplications disrupting CNTN4 in individuals with an ASD have also recently been reported [Glessner et al., 2009; Roohi et al., 2009].
We recently identified a maternally inherited deletion of 3p26.3 that includes the 5′ UTR of CNTN4 in a child with severe autism. Based on the increasing evidence implicating contactins in ASDs, as well as localization of CNTN4 within the 3p− syndrome critical region, we undertook sequencing of the coding regions of the gene in a local cohort of ASD patients.
Materials and Methods
Patient sample
Our patient cohort consisted of 87 children affected with an ASD from 75 unrelated families recruited to the Central Ohio Registry for Autism (CORA) at Nationwide Children’s Hospital (NCH). Ethnicities of affected probands included Caucasian (84%), African American/Black (5%) and mixed race/other [11%, that included 2 Caucasian/Hispanic, 2 African American/Caucasian, 1 Asian/European, 1 Egyptian, 1 Pakistani, and 1 Arab (not otherwise specified)]. Patients were recruited through the Autism Center or genetics clinics at NCH (72%) or through Developmental Pediatrics at Wright-Patterson Air Force Base Medical Center (28%). The voluntary, fully informed consent of the subjects used in this research was obtained as required by 32 CFR 219 and AFI 40-402, according to institutional guidelines. For inclusion in the registry, families provided demographic and medical information, including psychological testing, as well as blood samples for the preparation of DNA and lymphoblastoid cell lines. The diagnosis of an ASD was made by the evaluating clinician, usually a psychologist and/or developmental pediatrician, based on DSM-IV criteria. Confirmatory scores on The Autism Diagnostic Observation Scale (ADOS) were available for 66/87 (76%) and ADI-R scores on 44/87 (51%). A detailed pedigree was available in all cases. Thirty-nine percent of families reported a history of an ASD in a member of their nuclear or extended family.
There was a history of psychiatric disease (depression, bipolar disorder, anxiety disorder, OCD, or mental illness, not otherwise specified) in 47% (35/75) of the nuclear and 35% (26/75) of the extended families, respectively. A history of alcohol and/or drug abuse was noted in 3% (2/75) of nuclear and 17% (13/75) of extended families. Learning disabilities, including ADHD, mental retardation, or school problems, were reported in 17% (13/75) of nuclear and in 31% (23/75) of extended families. Some families reported psychiatric, learning, or substance abuse issues in both the nuclear and extended families; thus, the numbers and percentages are not additive. Twelve of 68 patients (18%) for whom a head circumference (HC) measurement was available were macrocephalic (HC ≥ 98%). Fifty-one of the probands (68%) had a genetics evaluation, including physical examination, as part of their clinical care. Five of these patients were felt to be dysmorphic.
High-resolution blood karyotype analysis and fragile X DNA testing were both performed on 88% of enrolled probands. Three karyotypic abnormalities were present: mos 45,X[6]/46,XX[14]; mos 47,XX,+r(8)(p12q11.2)[28] / 46,XX[4]; and 45,XY,der(14;15)(q10;q10). Only the ring 8 was felt to be causally related to the diagnosis of an ASD.
Controls
A set of 107 control DNAs consisted of Caucasian CEPH samples and included 52 males and 55 females not known to have autism in the family.
Array CGH
Array CGH was performed for clinical indications on 46 affected individuals (53%). A variety of platforms were employed due to rapidly evolving technology, including the Baylor Chromosomal Microarray V.6.1 (N=5), Spectral Genomics’ Constitutional Chip V3.0 (N=4), SignatureSelect V1.0 (N=10), SignatureSelect V2.0 (N=9), SignatureChip V3.0 (N=6), SignatureChip V4.0 (N=10), and NimbleGen CGX-3 (N=2). As part of this research, microarrays were performed using the Nimblegen CGX-3 oligonucleotide array on an additional 23 subjects (26% or a total 79% of affected subjects in the cohort with a microarray performed). All of the microarrays were performed and analyzed in the Clinical Molecular Genetics laboratory at NCH, with the exception of the Baylor, SignatureChip V3.0, and 5 of the SignatureChip V4.0 microarrays that were performed in the clinical laboratories of the respective institutions. The 3p26.3 deletion detected by array CGH was confirmed and visualized by metaphase fluorescence in situ hybridization (FISH) using BAC clone RP11-96G4 using previously published methods [Shaffer et al, 1994]..
DNA sequencing
The coding and flanking intronic sequence (22 exons) of CNTN4 was amplified by PCR using genomic DNA prepared from WBC or lymphoblastoid cell lines. Primers were designed with Primer Express (Applied Biosystems, Inc.) using genomic sequence for the largest transcript, CNTN4-001 (24 exons, with exons 1 and 2 non-coding) based on human genome build 18 sequence in the Ensembl database. Primer sequences and PCR conditions are provided in Supplemental Table S1. Genomic cycle sequencing was performed using Big Dye Terminator chemistry v3.1 (Applied Biosystems), and reaction products were separated by capillary gel electrophoresis on an ABI 3730 genetic analyzer. Sequence electropherograms were analyzed for the presence of rare variants or SNPs using Sequencher software v4.9 (GeneCodes), followed by manual scanning. Sequence variants were confirmed following bidirectional sequencing of a second PCR reaction. When a coding variant was observed, sequencing of the involved exon in other available family members was performed to determine if the variant was inherited or occurred de novo and if it segregated with other affected individuals in the family.
Upon detection, the effects of amino acid substitution were predicted using evolutionary conservation of amino acid sequence. Most CNTN family members were taken from TreeFam (accession number TF351103, www.treefam.org, Wellcome Trust Sanger Institute). Additional CNTN4 proteins were identified by Blastp analysis of the GenBank protein database [Altschul, Gish, Miller, Myers, & Lipman, 1990]. Evolutionary conservation was examined using multiple sequence alignment of all identified CNTN family members using Clustal W [Larkin, 2007].. This analysis was compared to that of SIFT [Ng & Henikoff, 2001] and Polyphen [Ramensky, Bork, & Sunyaev, 2002], programs for identifying potentially functional variants using protein homology along with amino acid or 3D-structural properties, respectively. SIFT was run in SIFT Sequence mode, where the program identifies the related proteins to be used in the analysis.
Variant Analysis
Common variants identified through sequencing were analyzed in Haploview 4.1 by single SNP and haplotype case-control association analyses. Haplotypes were constructed from the data using the four gamete rule. Rare variants in CNTN4 were collapsed into groups and compared by Fisher’s exact test using the method of Li and Leal [Li & Leal, 2008].
Site-directed mutagenesis and in vitro binding studies
Mutagenesis of the mouse CNTN4Ig1-4 cDNA cloned into the eukaryotic expression vector pSGHP1 to introduce the N178D and G221D changes was carried out by overlap extension. Wild-type and mutant forms of CNTN4 were transiently expressed in HEK293 cells as fusion proteins with human growth hormone (hGH). Affinity-isolation assays were carried out as described previously [Bouyain & Watkins, 2010]. Briefly, the purified carbonic anhydrase-like (CA-like) domain of mouse PTPRG was coupled to CNBr-activated sepharose at a density of 1 mg of protein per ml of resin. Conditioned media (250 μl) was mixed with an equal volume of 50 mM Tris-HCl pH 7.4, 200 mM NaCl and 0.2% (v/v) Tween-20. Samples were incubated with 30 μl of a 1:1 slurry of quenched CNBr-activated sepharose to deplete non-specific binding material. After centrifugation, 30 μl of a 1:1 slurry of PTPRG affinity resin was added to each sample followed by a 2h incubation at room temperature. The beads were washed with 25 mM Tris-HCl pH 7.4, 100 mM NaCl and 0.1% (v/v) Tween-20 and incubated in Laemmli loading buffer for 3 minutes at 100°C. Bound proteins were resolved by SDS-PAGE and detected by immunoblotting against hGH.
Results
Case report
The proband in this family (A154) is a 4 year old male referred to NCH genetics clinic for evaluation and genetic counseling regarding a diagnosis of autism and an abnormal microarray. He was the 8 lb 5 oz product of a full term, uncomplicated pregnancy born to a 20 year old gravida 2 para 1 to 2 female. The parents of the patient first became concerned at 2 years of age because he had no intelligible speech. His motor milestones were normal; he sat at 5 months and walked at 1 year of age. He had some babbling at one year of age, but no further progression of his speech.
At the time of his evaluation at the NCH Autism Center at age 3 ½ years, A154 exhibited echolalia, hand flapping, and fascination with lights. He would line up toys, and he also had daily tantrums, with episodes of aggression. His parents had to remove furniture from the house and put locks on doors and windows so he wouldn’t leave the house unattended. During his evaluation, psychology testing demonstrated a Mullen Early Scales of Learning Composite Score of 49 (<1%), with delays in all areas. A brief IQ score on the Leiter-R was 109 (73%). The total score on the ADOS Module 1 was 19, well above the cutoff for autistic disorder. Genetic testing performed following that evaluation included a normal male 46,XY blood karyotype and normal DNA testing for Fragile X syndrome. A BAC microarray (SignatureChip v4.0) demonstrated a ~535 kb single copy loss of 3 BACs from 3p26.3 that included the 5′ end of CNTN4 and part of the intergenic region between CNTN4 and the more telomeric CNTN6 locus (see Fig. 1A, B). The presence of this interstitial deletion was confirmed by FISH (Fig. 1C). Due to gaps in probe coverage, the nearest distal and proximal BAC clones that were not deleted were 500 kb and 700 kb away, respectively. Thus, the actual size of the deletion could have been considerably larger.
Figure 1.
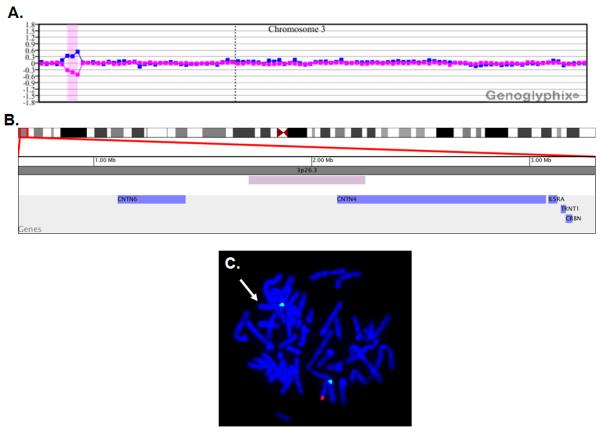
Microarray and FISH characterization of 3p26.3 deletion in patient A154. (A) Microarray analysis using a bacterial artificial chromosome (BAC)-based microarray (SignatureChip v4.0, Signature Genomic Laboratories, Spokane, WA) was performed using previously published methods [Bejjani et al., 2005] and showed a single-copy loss of 3 BAC clones from the short arm of chromosome 3 at 3p26.3 (chr3:1,711,180-2,245,625, UCSC March 2006 hg18 assembly), approximately 535 kb in size. Probes are arranged with the most distal p-arm clones on the left and the most distal q-arm clones on the right. The blue line is a plot of the array CGH data from the first microarray experiment (reference Cy5/patient Cy3). The pink line is a plot of the array CGH data from the second microarray experiment in which the dyes have been reversed (patient Cy5/reference Cy3). (B) A zoomed-in image of the deletion shown in (A). The minimal deletion size is represented by the purple bar, and the blue bars represent genes. Results are visualized using Genoglyphix (Signature Genomic Laboratories, Spokane, WA). (C) Metaphase FISH analysis showing deletion of the region shown by microarray analysis in (A). BAC clone RP11-96G4 from 3p26.3 is labeled in red, and the chromosome 3 centromere probe is labeled in green as a control. Missing red signal and retained green signal indicates deletion at 3p26.3 on one homologue (arrow).
At the time of his evaluation in genetics clinic, A154’s weight was 85th, height 35th, and head circumference 75-90th centile for age. The patient had obvious developmental delays but was not dysmorphic. He was extremely uncooperative, climbing on furniture and throwing objects in the room, and, thus, a detailed physical exam could not be performed.
The patient’s 6 year old older sister was diagnosed with a mixed expressive/receptive language disorder. Her ADOS score (Module 3) was 9, above the cutoff for an ASD, but below that for autistic disorder. Scores on the CARS (32) and GARS (70) did not support a diagnosis of ASD. The clinical psychologist and developmental pediatrician completing her multidisciplinary evaluation did not feel that she had an ASD, based on very good social skills.
The submicroscopic deletion at 3p26.3 was subsequently confirmed to be present in the patient’s sister, as well as in their phenotypically normal mother. Genetic counseling was performed with the family. We suggested that the deletion was likely contributing to, but unlikely to be the sole cause of, the developmental and behavioral abnormalities in the patient and his sister. As part of this research, we performed genomic sequencing of the coding exons of CNTN4 in A154 to exclude the presence of a second mutation on the paternal allele. No additional nonsynonomous substitutions, nor other deleterious mutations, were detected (data not shown).
Sequencing of the Coding Exons of CNTN4 in ASD Patients and Controls
We next performed genomic sequencing of the CNTN4 coding exons in a local cohort of 87 individuals with an ASD and 107 unrelated controls (see Methods). As shown in Table 1, four probands with an ASD were found to have a heterozygous nonsynonomous sequence variant within the coding region of CNTN4. The sites of the amino acid substitutions within CNTN4 domains are shown in Fig. 2A. Relevant clinical features of members of each of the families are summarized in Table 2. Each of these variants was inherited from an unaffected parent and altered a highly conserved amino acid residue. In addition, in 3 of the families, DNA was available from a sibling with a neurodevelopmental phenotype: ADHD and speech delay in family 2; autistic disorder in family 3; and mild MR, ADHD, and mental health issues in family 4 (see Table 2). The sibling in family 4 carried the same variant as his brother diagnosed with PDD-NOS and his unaffected father. Siblings in the other families did not inherit the variant allele. A single distinct coding variant was detected in one individual in our control population that was also found to alter a highly conserved amino acid (see Table 1). Using the method of Li and Leal [Li & Leal, 2008], there was no significant difference between the rates of rare variants detected in affecteds vs controls in CNTN4 by Fisher’s exact test (p=0.16)..
Table 1.
Coding Variants Identified in CNTN4
| Family | Relationship | Coding Change |
AA Change | Exon | PolyPhen Prediction | SIFT Prediction (Sequence Mode) |
Evolutionary Conservation |
|---|---|---|---|---|---|---|---|
| 1 | Proband Mother Father |
c.533A>G c.533A>G Wild type |
p.N178D p.N178D |
Exon 7 | Probably Damaging | Affects Protein Function | Conserved |
| 2 | Proband Mother Sibling |
c.662G>A c.662G>A Wild type |
p.G221D p.G221D |
Exon 8 | Probably Damaging | Affects Protein Function | Conserved |
| 3 | Proband Mother Father Sibling |
c.992A>G Wild type c.992A>G Wild type |
p.E331G p.E331G |
Exon 10 | Benign | Tolerated (“Not tolerated” using only specific CNTN4 protein sequences, see text) |
Conserved |
| 4 | Proband Mother Father Sibling |
c.1889A>G Wild type c.1889A>G c.1889A>G |
p.Y630C p.Y630C p.Y630C |
Exon 16 | Probably Damaging | Affects Protein Function | Conserved |
| Control | c.167C>G | p.P56R | Exon 4 | Probably Damaging | Affects Protein Function | Conserved |
Figure 2.
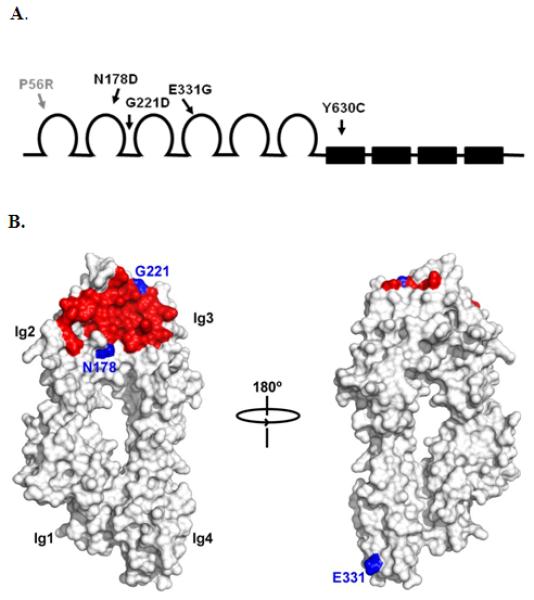
Schematic of domains of CNTN4 and approximate sites of variant amino acids. (A) The 6 Ig-like domains of CNTN4 are shown as open loops and the 4 fibronectin II-like domains as filled boxes. The sites of the variant amino acids are shown as gray (control) or black (ASD patients) arrows. (B) Surface representations of the 4 Ig-like domains of mouse CNTN4 (CNTN4Ig1-4) with binding site of PTPRG shown in red. The altered amino acids in the ASD patients within the Ig-like domains are shown in blue. The variant Y630C is not shown since this mutation lies within the FNIII region of CNTN4, the structure of which has not yet been determined.
Table 2.
Clinical Features of CORA Families with Coding Variants in CNTN4
| Family | Variant | Gender | Age at Diagnosis of ASD |
Race | Diagnostic/Clinical Information1 | |
|---|---|---|---|---|---|---|
| 1. | Proband | N178D | M | 2 y 8 mo | Asian/Caucasian | Autistic Disorder Full scale IQ Stanford Binet 44; Leiter-R Composite IQ 72 (6 y 3 mo) ADOS total score 14 (Module 1; age 9 y 10 mo); ADI-R consistent with ASD |
| Mother | N178D | F | Caucasian | |||
| Father | wild type | M | Asian | |||
| 2. | Proband | G221D | M | 4 y | African American | Autistic Disorder ADOS total score 11 (Module 4; 18 y); ADI-R consistent with ASD 8th grade with IEP; adderall for ADHD, trileptal for aggression |
| Mother | G221D | F | African American | Reading difficulty 6th – 10th grade | ||
| Father | Not available | M | High school dropout; History of violence, incarceration | |||
| Sibling | wild type | M | African American | ADOS (Module 3; 9 y 6 mo) not consistent with ASD ADHD; speech therapy; IEP |
||
| 3. | Proband | E331G | M | 2 y 7 mo | Caucasian | PDD-NOS ADOS total score 7 (Module 2; 4 y 11 mo) History of hypospadias. |
| Mother | wild type | F | Caucasian | Depression. Celiac disease. Fibromyalgia. | ||
| Father | E331G | M | Caucasian | History of ADHD, juvenile detention. PhD in psychology | ||
| Sibling | wild type | M | 3 y | Caucasian | Autistic Disorder ADOS total score 19 (Module 3; 6 y 10 mo) WPPSI-R IQ full scale 71 (4 y 11 mo) |
|
| 4. | Proband | Y630C | M | 3 y | Caucasian | PDD-NOS ADOS total score 7 (Module 2; 5 y 4 mo)2; ADI-R consistent with ASD Dysmorphic, hypotonia, strabismus, undescended testes |
| Mother | wild type | F | Caucasian | History of eating disorder, alcoholism, depression. | ||
| Father | Y630C | M | Caucasian | Multiple sclerosis, diagnosed age 30 y | ||
| Sibling | Y630C | M | Caucasian | ADHD, bipolar disorder, generalized anxiety disorder, mild MR, motor tics ADOS (Module 4; 13 y), ADI-R, Krug Asperger’s Disorder Index not consistent with an ASD |
Parental information was reported by the individual or spouse. Clinical information on affecteds and siblings was obtained from medical records and, in some cases, confirmed by parental report.
The social affect and restricted behavior total score under the revised ADOS algorithm met the ASD cut-off of 8.
To our knowledge, none of these variants has been previously reported. We further characterized the evolutionary conservation at each affected position in CNTN4 and in the CNTN protein family. All five alterations change amino acids that are conserved in CNTN4 from humans to fish (see Table 3 and Supplemental Fig. S1). The phylogenetic tree of CNTNs has two main branches, invertebrate and vertebrate. The latter has two main sub-branches – CNTN1/2 and CNTN3/4/5/6. Two of the variant positions are conserved in the CNTN3/4/5/6 subfamily (G221D, E331G); one in all vertebrate CNTNs except for CNTN3 (N178D); and two in all vertebrate CNTNs as well as invertebrate CNTNs (P56R, Y630C). For comparison, four of the five variants (all but E331G) were predicted to alter protein function by the programs SIFT and Polyphen (Table 1) [Ng & Henikoff, 2001; Ramensky et al., 2002]. Given our evolutionary conservation results, and the fact that SIFT and Polyphen predictions are largely based on sequence homology, we considered that the classification of E331G as benign or tolerated might be incorrect. A simple explanation is that these programs failed to specifically identify (and conduct their analysis on) CNTN4 orthologs and included paralogs (i.e., other CNTN family members). We confirmed this to be the case for SIFT “misclassification” by running the program on the aligned CNTN4 proteins from Supplemental Fig. S1 (i.e., SIFT Aligned or Related Sequences modes), which resulted in E331G being called “not tolerated”.
Table 3.
Evolutionary Conservation of Variant Amino Acids
| CNTN protein1 | 1 | 2 | 3 | 4 | 5 | 6 | Dros |
|---|---|---|---|---|---|---|---|
| P56 (R; Ig1 domain) | + | + | + | + | + | + | + |
| N178 (D; Ig2) | + | + | − | + | + | + | − |
| G221 (D; loop Ig2/Ig3) |
− | − | + | + | + | + | − |
| E331 (G; Ig4) | − | − | + | + | (+/−)2 | + | − |
| Y630 (C; Fn3-1) | (+)3 | + | + | + | + | (+/−)4 | + |
The evolutionary conservation of the amino acid sequence of CNTN proteins 1-6 was compared among 30 vertebrates (23 mammals, chicken, frog, and 4 fish species), as well as the single CNTN protein in Drosophila melanogaster (Dros).
Asp/Glu are conserved in chick/frog (Gallus gallus, Xenopus tropicalis)/fish (zebrafish, Brachydanio rerio) CNTN5, but not in mammals.
Tyr is conserved in CNTN1 from at least three fish (medaka, Oryzias latipes; stickleback, Gasterosteus aculeatus; green puffer, Tetraodon nigroviridis), but not zebrafish (His, conservative substitution, aromatic, hydrophobic);
Tyr is conserved in CNTN6 from chick, fish (medaka, green puffer), and, of mammals, only in opossum (Monodelphis domestica) – a marsupial; CNTN6 of other mammals have Phe, a conservative substitution (aromatic, hydrophobic); CNTN6 from frogs or zebrafish is not known. The symbols (+) and (−) indicate the amino acid is conserved or not conserved, respectively.
Case-Control Association Analyses
A total of 30 SNPs with a minor allele frequency >0.01 were included in a case-control association analysis (see Supplemental Table S2). The minor T allele of SNP rs9820464 was present in a higher proportion of cases (probands; p=0.0086), and the associated haplotype in which it resided was also over represented among cases (p=0.0086). However, neither the SNP nor the haplotype remained statistically significant after permutation testing (p=0.16).
Analysis of the Binding of Variant CNTN4 Fragments to PTPRG in vitro
Numerous proteins have been shown to interact with one or more CNTNs. Among these are receptor protein tyrosine phosphatases (RPTPs) that combine cell adhesion molecule-like ectodomains as well as one or more intracellular tyrosine phosphatase domains. Recently, one of us demonstrated that the extracellular CA-like domain of the receptor tyrosine phosphatase PTPRG interacts with the second and third Ig domains of CNTN3, 4, 5 and 6 [Bouyain & Watkins, 2010]. Specifically, an affinity resin that included the CA-like domain of mouse PTPRG was used to isolate secreted fragments of murine CNTN4 tagged with hGH. Because 2 of the 4 variants in CNTN4 identified in the ASD probands are located within Ig repeats 2 and 3 and more specifically in the immediate vicinity of the PTPRG-binding site (N178D and G221D; see Fig. 2B), it was of interest to determine to what extent these mutations would affect PTPRG binding. We used site-directed mutagenesis to introduce each variant into a cDNA encoding a fusion of hGH and mouse CNTN4Ig1-4. Each variant protein was expressed transiently in HEK293 cells and incubated with a PTPRG-affinity resin. These experiments demonstrated that neither mutation had a detectable effect on PTPRG binding as the wild type and mutated forms of CNTN4Ig1-4 bound to the CA-like domain of PTPRG similarly (not shown).
Discussion
Several studies have implicated deletions in CNTN4 in the intellectual disabilities associated with 3p− syndrome, as well as in the pathogenesis of ASDs [Fernandez et al., 2008; Malmgren, Sahlen, Wide, Lundvall, & Blennow, 2007; Roohi et al., 2009]. Recently, we identified a ~535 kb deletion within the 5′ UTR of CNTN4 in a patient with a severe autistic phenotype. The identical deletion was found in the patient’s phenotypically normal mother, as well as his older sister who was diagnosed with a mixed expressive/receptive language disorder and some autistic behaviors. Based on this case, as well as the accumulating evidence implicating contactins and contactin associated proteins in the pathogenesis of ASDs, we undertook sequencing of the coding regions of CNTN4 in a local cohort of ASD families in comparison with a set of controls. To our knowledge, this is the first report of genomic sequencing of coding regions of the locus in individuals with ASDs. We identified unique coding variants in 4/75 unrelated individuals affected with an ASD (5.3%), as well as in 1/107 samples from a control population (0.9%). All five variants are nonsynonomous substitutions at amino acid positions conserved from humans to fish. However, the data do not reach statistical significance (p<0.05). Each of the CNTN4 variants within the ASD group was inherited from a clinically unaffected parent, although 2 of the parents exhibited some learning or mental health issues (see Table 2). There were no parent of origin effects noted in the small sample set (2 maternal/2 paternal). In 3 of the 4 families, there was a full sibling with a neurodevelopmental phenotype. In one family (family 4), the sibling carried the same variant as his brother with an ASD; the sibling in each of the other families did not. The inheritance of a variant allele from an apparently unaffected parent, as well as discordance within some sibships, has been found for multiple CNVs causally associated with ASDs, including 16p11.2 [Kumar et al., 2008; Marshall et al., 2008; Weiss et al., 2008] and 1q21 [Mefford et al., 2008], and single genes, such as SHANK3, PTEN, and CNTNAP2, among others [Bakkaloglu et al., 2008; Moessner et al., 2007; Varga, Pastore, Prior, Herman, & McBride, 2009].
The six member contactin gene family is highly conserved throughout vertebrate evolution, suggesting the encoded proteins perform key functions in neurodevelopment. As detailed below, mutations identified in selected human contactins and genetically engineered mouse mutations in several of the loci further support the key roles of this gene family in developmental neurobiology. Mutations in CNTN1 have been identified in a single consanguineous family with an autosomal recessive lethal congenital myopathy [Compton et al., 2008], while mice with a targeted null allele in the orthologous gene demonstrate ataxia by postnatal day (P) 10 with subsequent poor growth and death by P18 [Berglund et al., 1999]. Targeted mouse mutations for Cntn2,4,5, and 6 have been developed; all of these homozygous mutants are viable, fertile, and without any obvious phenotypic or anatomic abnormalities. However, homozygous Cntn2 deficient animals show altered clustering of juxtaparanodal potassium channels and impaired learning and memory, with reduced spontaneous motor activity and abnormal coordination of their gait [Poliak et al., 2003; Savvaki et al., 2008]. Mice with a homozygous targeted mutation in Cntn4 (Big-2) demonstrate abnormal projections and mistargeting of olfactory axons, consistent with a role for the protein in axon guidance [Kaneko-Goto, Yoshihara, Miyazaki, & Yoshihara, 2008]. Murine Cntn4 is also expressed in selected cerebellar granule and Purkinje cells, and the authors state that they are pursuing behavior studies of their knockout mice to assess subtle deficits in cortical and/or cerebellar function. Cntn5 (NB-2) is preferentially expressed in central auditory pathways, and mice with a homozygous Cntn5 targeted mutation demonstrate an abnormal audiogenic seizure response with defective synapse formation and increased apoptosis of auditory neurons [Takeda et al., 2003; Toyoshima et al., 2009]. Cntn6 (NB-3) homozygous knockout mice demonstrate impaired motor coordination, with poor performance in the rotarod and wire hang assays [Takeda et al., 2003]. Finally, a previous report of a single base change in the 3′-UTR of human CNTN4 in a Japanese family with autosomal dominant spinocerebellar ataxia [Miura et al., 2006] was subsequently determined to be due to mutations in a nearby gene, ITPR1 [Iwaki et al., 2008].
The varied phenotypes of mice and humans with contactin gene family member mutations likely reflect differing patterns and sites of expression of each protein within the brain, as well as interactions with different subsets of proteins at neuronal synapses. Such interactions could occur between proteins on the same cell (in cis) or on the surface of different cells (in trans), such as neurons and surrounding glia. In addition to complexing with one or more contactin associated proteins, specific contactin family member proteins interact with different members of the L1 family of Ig superfamily proteins. Associations of CNTN1 and CNTN2 with neurexin family members CASPR1 and CASPR2, respectively, are essential for proper propogation of action potentials along myelinated neurons (reviewed in [Shimoda & Watanabe, 2009]). Finally, contactins interact with receptor protein tyrosine phosphatases (RPTPs) that contain extracellular cell adhesion domains, as well as one or more intracellular tyrosine phosphatase domains. In particular, neuronal CNTN1 binds to the CA-like domain found in the extracellular region of the tyrosine phosphatase PTPRZ that is expressed on the surface of glial cells. The interaction between CNTN1 and PTPRZ promotes neurite outgrowth and induces bidirectional cell signaling [Peles et al., 1995].
Recently, it was demonstrated that the CA-like domain of the closely related phosphatase PTPRG binds to the second and third Ig domains of CNTNs 3-6 [Bouyain & Watkins, 2010]. Using site-directed mutagenesis and affinity isolation assays, we could detect no difference in binding efficiencies of the murine CNTN4Ig1-4 fragments containing conserved variants found in our ASD families at amino acid positions 178 or 221. There are several possible reasons why each amino acid substitution could be deleterious in vivo and yet function normally in the assay employed here. Indeed, these mutations are located at the periphery of the PTPRG-binding site in CNTN4 so that the effect of each change may be too subtle to be detected using affinity-isolation assays. A decrease or increase in affinity of only 2 fold would be unlikely to be detected in these assays yet could have significant effect on intercellular adhesion and/or signaling. The effects of the mutations on binding could be evaluated by comparing the binding of the CA-like domain of PTPRG to immobilized CNTN4 or its variants by surface plasmon resonance. In addition, each mutation could affect the folding and/or the trafficking of CNTN4 to the cell surface. On the other hand, it is possible that additional cell surface receptors, such as CNTN-associated proteins, participate in the interactions between CNTN4 and PTPRG to form active signaling complexes. A clearer picture will emerge as the physiological function of the interactions between CNTN4 and PTPRG is defined in more detail.
There are several possible explanations for the lack of substantive evidence supporting the hypothesis of CNTN4 as an ASD susceptibility locus. The frequency of CNVs within CNTN4 in normal individuals can be estimated as 0.5% (12 exon-spanning CNVs/2382 controls) based on data in the Database of Genomic Variants [Iafrate et al., 2004], and there are reports of phenotypically normal individuals with heterozygous deletions of the entire locus [Pohjola et al., 2010; Shuib et al., 2009]. The large size of the gene (almost 1 Mb) could make it a frequent site for small genomic rearrangements, similar to the DMD or MACROD2 locus [Bradley et al., 2010]. Thus, it is possible that genomic rearrangements in CNTN4, and rare sequence variants, are incidental and not associated with a disease phenotype. It is also possible that CNTN4 is a low frequency ASD susceptibility locus that, in combination with other genetic and/or environmental factors, can contribute to an autism spectrum phenotype. Establishing significance for rare variants can be extremely difficult and may require large numbers (>1000) of affected individuals and controls. Differing ethnicities in the affected and control populations studied may further complicate interpretations, if there are racial or ethnic differences in frequency of the variants. Data from the 1000 Genomes Project may help clarify issues concerning selection of appropriate control populations [Durbin et al., 2010].
Finally, with the availability of commercial oligonucleotide microarray analysis at our institution, subsequent to completing sequencing of CNTN4 in ASD patients and controls, we repeated array studies on patient A154 to clarify the exact extent of his deletion. The deletion, in fact, spans 1.09 Mb, with centromeric and telomeric gaps of 67.26 and 57.8 kb, respectively. With the higher resolution on the oligo array, the deletion includes part of exon 14 through exon 23 of the telomeric CNTN6 gene as well as the previously reported 5′ untranslated region of CNTN4. Sequencing of coding exons of CNTN6 in our ASD families is in progress.
Supplementary Material
Acknowledgements
We are grateful to all of the families participating in the CORA registry. We thank Signature Genomic Laboratories for providing array and FISH images. This research was supported in part by USAF Department of Defense grant/Cooperative Agreement FA7014-09-2-0004 (G.E.H.), and NIH R21 HG004663 (C.E.A.) and R01 GM088806 (S.B.).
This research was supported in part by NIH R21 HG004663 (C.E.A.) and HQ AF Surgeon General under Award No. FA7014-09-2-0004 (G.E.H.). Opinions, interpretations, conclusions and recommendations are those of the author and are not necessarily endorsed by the U.S. Air Force.
Footnotes
Publisher's Disclaimer: Disclaimers The views and opinions expressed in this article are those of the author(s) and do not reflect official policy or position of the United State Air Force, Department of Defense, or US Government. The use of product(s) and/or manufacturer name(s) is added for clarification only; in no way implies endorsement by the authors, USAF, or DoD of the product(s) or manufacturer(s).
References
- Abrahams BS, Geschwind DH. Advances in autism genetics: on the threshold of a new neurobiology. Nat Rev Genet. 2008;9:341–355. doi: 10.1038/nrg2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alarcon M, Abrahams BS, Stone JL, Duvall JA, Perederiy JV, Bomar JM, et al. Linkage, association, and gene-expression analyses identify CNTNAP2 as an autism-susceptibility gene. Am J Hum Genet. 2008;82:150–159. doi: 10.1016/j.ajhg.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Arking DE, Cutler DJ, Brune CW, Teslovich TM, West K, Ikeda M, et al. A common genetic variant in the neurexin superfamily member CNTNAP2 increases familial risk of autism. Am J Hum Genet. 2008;82:160–164. doi: 10.1016/j.ajhg.2007.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakkaloglu B, O’Roak BJ, Louvi A, Gupta AR, Abelson JF, Morgan TM, et al. Molecular cytogenetic analysis and resequencing of contactin associated protein-like 2 in autism spectrum disorders. Am J Hum Genet. 2008;82:165–173. doi: 10.1016/j.ajhg.2007.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bejjani BA, Saleki R, Ballif BC, Rorem EA, Sundin K, Theisen A, et al. Use of targeted array-based CGH for the clinical diagnosis of chromosomal imbalance: is less more? Am J Med Genet A. 2005;134:259–267. doi: 10.1002/ajmg.a.30621. [DOI] [PubMed] [Google Scholar]
- Berglund EO, Murai KK, Fredette B, Sekerkova G, Marturano B, Weber L, et al. Ataxia and abnormal cerebellar microorganization in mice with ablated contactin gene expression. Neuron. 1999;24:739–750. doi: 10.1016/s0896-6273(00)81126-5. [DOI] [PubMed] [Google Scholar]
- Bouyain S, Watkins DJ. The protein tyrosine phosphatases PTPRZ and PTPRG bind to distinct members of the contactin family of neural recognition molecules. Proc Natl Acad Sci U S A. 2010;107:2443–2448. doi: 10.1073/pnas.0911235107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley WE, Raelson JV, Dubois DY, Godin E, Fournier H, Prive C, Allard R, Pinchuk V, Lapalme M, Paulussen RJ, Belouchi A. Hotspots of large rare deletions in the human genome. PLoS ONE. 2010;5:e9401. doi: 10.1371/journal.pone.0009401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbach JP, van der Zwaag B. Contact in the genetics of autism and schizophrenia. Trends Neurosci. 2009;32:69–72. doi: 10.1016/j.tins.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Compton AG, Albrecht DE, Seto JT, Cooper ST, Ilkovski B, Jones KJ, et al. Mutations in contactin-1, a neural adhesion and neuromuscular junction protein, cause a familial form of lethal congenital myopathy. Am J Hum Genet. 2008;83:714–724. doi: 10.1016/j.ajhg.2008.10.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dijkhuizen T, van Essen T, van der Vlies P, Verheij JB, Sikkema-Raddatz B, van der Veen AY, et al. FISH and array-CGH analysis of a complex chromosome 3 aberration suggests that loss of CNTN4 and CRBN contributes to mental retardation in 3pter deletions. Am J Med Genet A. 2006;140:2482–2487. doi: 10.1002/ajmg.a.31487. [DOI] [PubMed] [Google Scholar]
- Durbin RM, Abecasis GR, Altshuler DL, Auton A, Brooks LD, Gibbs RA, et al. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]; Fernandez T, Morgan T, Davis N, Klin A, Morris A, Farhi A, et al. Disruption of contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am J Hum Genet. 2004;74:1286–1293. doi: 10.1086/421474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez T, Morgan T, Davis N, Klin A, Morris A, Farhi A, et al. Disruption of Contactin 4 (CNTN4) results in developmental delay and other features of 3p deletion syndrome. Am J Hum Genet. 2008;82:1385. doi: 10.1016/j.ajhg.2008.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez TV, Garcia-Gonzalez IJ, Mason CE, Hernandez-Zaragoza G, Ledezma-Rodriguez VC, Anguiano-Alvarez VM, et al. Molecular characterization of a patient with 3p deletion syndrome and a review of the literature. Am J Med Genet A. 2008;146A:2746–2752. doi: 10.1002/ajmg.a.32533. [DOI] [PubMed] [Google Scholar]
- Glessner JT, Wang K, Cai G, Korvatska O, Kim CE, Wood S, et al. Autism genome-wide copy number variation reveals ubiquitin and neuronal genes. Nature. 2009;459:569–573. doi: 10.1038/nature07953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iafrate AJ, Feuk L, Rivera MN, Listewnik ML, Donahoe PK, Qi Y, et al. Detection of large-scale variation in the human genome. Nat Genet. 2004;36:949–951. doi: 10.1038/ng1416. [DOI] [PubMed] [Google Scholar]
- Iwaki A, Kawano Y, Miura S, Shibata H, Matsuse D, Li W, et al. Heterozygous deletion of ITPR1, but not SUMF1, in spinocerebellar ataxia type 16. J Med Genet. 2008;45:32–35. doi: 10.1136/jmg.2007.053942. [DOI] [PubMed] [Google Scholar]
- Kaneko-Goto T, Yoshihara S, Miyazaki H, Yoshihara Y. BIG-2 mediates olfactory axon convergence to target glomeruli. Neuron. 2008;57:834–846. doi: 10.1016/j.neuron.2008.01.023. [DOI] [PubMed] [Google Scholar]
- Katidou M, Vidaki M, Strigini M, Karagogeos D. The immunoglobulin superfamily of neuronal cell adhesion molecules: lessons from animal models and correlation with human disease. Biotechnol J. 2008;3:1564–1580. doi: 10.1002/biot.200800281. [DOI] [PubMed] [Google Scholar]
- Kumar RA, KaraMohamed S, Sudi J, Conrad DF, Brune C, Badner JA, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17:628–638. doi: 10.1093/hmg/ddm376. [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, et al. Clustal W and Clustal X version 2.0. Bioinformatics. 2007;23:2947–2948. doi: 10.1093/bioinformatics/btm404. [DOI] [PubMed] [Google Scholar]; Li B, Leal SM. Methods for detecting associations with rare variants for common diseases: application to analysis of sequence data. Am J Hum Genet. 2008;83:311–321. doi: 10.1016/j.ajhg.2008.06.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malmgren H, Sahlen S, Wide K, Lundvall M, Blennow E. Distal 3p deletion syndrome: detailed molecular cytogenetic and clinical characterization of three small distal deletions and review. Am J Med Genet A. 2007;143A:2143–2149. doi: 10.1002/ajmg.a.31902. [DOI] [PubMed] [Google Scholar]
- Marshall CR, Noor A, Vincent JB, Lionel AC, Feuk L, Skaug J, et al. Structural variation of chromosomes in autism spectrum disorder. Am J Hum Genet. 2008;82:477–488. doi: 10.1016/j.ajhg.2007.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mefford HC, Sharp AJ, Baker C, Itsara A, Jiang Z, Buysse K, et al. Recurrent rearrangements of chromosome 1q21.1 and variable pediatric phenotypes. N Engl J Med. 2008;359:1685–1699. doi: 10.1056/NEJMoa0805384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura S, Shibata H, Furuya H, Ohyagi Y, Osoegawa M, Miyoshi Y, et al. The contactin 4 gene locus at 3p26 is a candidate gene of SCA16. Neurology. 2006;67:1236–1241. doi: 10.1212/01.wnl.0000238510.84932.82. [DOI] [PubMed] [Google Scholar]
- Moessner R, Marshall CR, Sutcliffe JS, Skaug J, Pinto D, Vincent J, et al. Contribution of SHANK3 mutations to autism spectrum disorder. Am J Hum Genet. 2007;81:1289–1297. doi: 10.1086/522590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrow EM, Yoo SY, Flavell SW, Kim TK, Lin Y, Hill RS, et al. Identifying autism loci and genes by tracing recent shared ancestry. Science. 2008;321:218–223. doi: 10.1126/science.1157657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res. 2001;11:863–874. doi: 10.1101/gr.176601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peles E, Nativ M, Campbell PL, Sakurai T, Martinez R, Lev S, et al. The carbonic anhydrase domain of receptor tyrosine phosphatase beta is a functional ligand for the axonal cell recognition molecule contactin. Cell. 1995;82:251–260. doi: 10.1016/0092-8674(95)90312-7. [DOI] [PubMed] [Google Scholar]
- Pohjola P, de Leeuw N, Penttinen M, Kaariainen H. Terminal 3p deletions in two families--correlation between molecular karyotype and phenotype. Am J Med Genet A. 2010;152A:441–446. doi: 10.1002/ajmg.a.33215. [DOI] [PubMed] [Google Scholar]
- Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, et al. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 2003;162:1149–1160. doi: 10.1083/jcb.200305018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramensky V, Bork P, Sunyaev S. Human non-synonymous SNPs: server and survey. Nucleic Acids Res. 2002;30:3894–3900. doi: 10.1093/nar/gkf493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roohi J, Montagna C, Tegay DH, Palmer LE, DeVincent C, Pomeroy JC, et al. Disruption of contactin 4 in three subjects with autism spectrum disorder. J Med Genet. 2009;46:176–182. doi: 10.1136/jmg.2008.057505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savvaki M, Panagiotaropoulos T, Stamatakis A, Sargiannidou I, Karatzioula P, Watanabe K, et al. Impairment of learning and memory in TAG-1 deficient mice associated with shorter CNS internodes and disrupted juxtaparanodes. Mol Cell Neurosci. 2008;39:478–490. doi: 10.1016/j.mcn.2008.07.025. [DOI] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Malhotra D, Troge J, Lese-Martin C, Walsh T, et al. Strong association of de novo copy number mutations with autism. Science. 2007;316:445–449. doi: 10.1126/science.1138659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaffer LG, McCaskill C, Han JY, Choo KH, Cutillo DM, Donnenfeld AE, Weiss L, Van Dyke DL. Molecular characterization of de novo secondary trisomy 13. Am J Hum Genet. 1994;55:968–974. [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Dies KA, Holm IA, Bridgemohan C, Sobeih MM, Caronna EB, et al. Clinical genetic testing for patients with autism spectrum disorders. Pediatrics. 2010;125:e727–735. doi: 10.1542/peds.2009-1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimoda Y, Watanabe K. Contactins: Emerging key roles in the development and function of the nervous system. Cell Adh Migr. 2009;3 doi: 10.4161/cam.3.1.7764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuib S, McMullan D, Rattenberry E, Barber RM, Rahman F, Zatyka M, et al. Microarray based analysis of 3p25-p26 deletions (3p- syndrome) Am J Med Genet A. 2009;149A:2099–2105. doi: 10.1002/ajmg.a.32824. [DOI] [PubMed] [Google Scholar]
- Szatmari P, Paterson AD, Zwaigenbaum L, Roberts W, Brian J, Liu XQ, et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat Genet. 2007;39:319–328. doi: 10.1038/ng1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeda Y, Akasaka K, Lee S, Kobayashi S, Kawano H, Murayama S, et al. Impaired motor coordination in mice lacking neural recognition molecule NB-3 of the contactin/F3 subgroup. J Neurobiol. 2003;56:252–265. doi: 10.1002/neu.10222. [DOI] [PubMed] [Google Scholar]
- Toyoshima M, Sakurai K, Shimazaki K, Takeda Y, Shimoda Y, Watanabe K. Deficiency of neural recognition molecule NB-2 affects the development of glutamatergic auditory pathways from the ventral cochlear nucleus to the superior olivary complex in mouse. Dev Biol. 2009;336:192–200. doi: 10.1016/j.ydbio.2009.09.043. [DOI] [PubMed] [Google Scholar]
- Varga EA, Pastore M, Prior T, Herman GE, McBride KL. The prevalence of PTEN mutations in a clinical pediatric cohort with autism spectrum disorders, developmental delay, and macrocephaly. Genet Med. 2009;11:111–117. doi: 10.1097/GIM.0b013e31818fd762. [DOI] [PubMed] [Google Scholar]
- Weiss LA, Shen Y, Korn JM, Arking DE, Miller DT, Fossdal R, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358:667–675. doi: 10.1056/NEJMoa075974. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.