

Summary
Perturbations of the composition of the symbiotic intestinal microbiota can have profound consequences for host metabolism and immunity. In mice, segmented filamentous bacteria (SFB) direct the accumulation of potentially pro-inflammatory Th17 cells in the intestinal lamina propria. We present the genome sequence of SFB isolated from mono-colonized mice, which classifies SFB phylogenetically as a unique member of Clostridiales with a highly reduced genome. Annotation analysis demonstrates that SFB depends on its environment for amino acids and essential nutrients and may utilize host and dietary glycans for carbon, nitrogen, and energy. Comparative analyses reveal that SFB is functionally related to members of the genus Clostridium and several pathogenic or commensal “minimal” genera, including Finegoldia, Mycoplasma, Borrelia, and Phytoplasma. However, SFB is functionally distinct from all 1,200 examined genomes, indicating a gene complement representing biology relatively unique to its role as a gut commensal closely tied to host metabolism and immunity.
Introduction
The composition of the commensal microbiota at mucosal surfaces normally reflects mutually beneficial co-evolution of both the host and the colonizing microbes. In the intestine, the microbial community influences host nutrition and metabolism as well as immune system functions, and the bacterial makeup is, in turn, regulated by the host, the environment and inter-microbial interactions (Elinav et al., 2011; Turnbaugh et al., 2006). Changes in the commensal bacterial balance have been associated with onset or severity of multiple diseases, and may reflect outgrowth of potentially pathogenic bacteria or loss of beneficial ones (Elinav et al., 2011; Garrett et al., 2010; Sokol et al., 2008). Alterations in the composition of the commensal microbiota can affect immunological fitness, by influencing the balance of different cell types in the host immune system and, by extension, altering the integrity of the intestinal barrier and the abundance of other luminal bacteria (Atarashi et al., 2011; Ivanov and Littman, 2011).
The segmented filamentous bacteria (SFB) were the first example of a commensal species that modulates host adaptive immune cell homeostasis. SFB induce accumulation of Th17 cells in the terminal ileum of mice, and thus influence future immune responses during infection or autoimmune inflammation (Gaboriau-Routhiau et al., 2009; Ivanov et al., 2009). Th17 cells are critical for maintenance of the integrity of mucosal barriers and have been implicated in a large number of autoimmune diseases in mice and humans. Cytokines produced by Th17 cells, including IL-17A, IL-17F, and IL-22, mediate host protection against pathogenic bacteria and fungi. IL-17A and IL-17F induce neutrophil recruitment while IL-22 induces production of anti-microbial peptides by intestinal epithelial cells. Production of these cytokines likely contributes to SFB-mediated enhanced protection of the host from pathogenic enteric bacteria such as Citrobacter rodentium, but also renders mice more susceptible to autoimmune disease (Ivanov et al., 2009). Thus, SFB-colonized mice are more likely to develop disease in a model of spontaneous arthritis and in myelin protein-induced autoimmune encephalopathy (Lee et al., 2011; Wu et al., 2010).
Segmented filamentous bacteria, first described more than four decades ago, are spore forming gram-positive anaerobic commensals that colonize the terminal ileum of mice and multiple other species around the time of weaning, forming long filaments and growing in close association with epithelial cells (Blumershine and Savage, 1978). The bacteria appear to bind tightly to epithelial cells and to induce cytoskeletal reorganization in these cells at the site of contact. SFB are currently unculturable ex vivo, which has impeded the development of genetic tools and studies of their biology. Nevertheless, SFB-monocolonized mice have been generated and studies in these mice have shown that SFB have major immunomodulatory effects (Talham et al., 1999). In addition to its effect on Th17 cells, SFB colonization induces secretory IgA production and recruitment of intraepithelial lymphocytes (Talham et al., 1999; Umesaki et al., 1995). Mice colonized with SFB have extensive changes in gene expression within the mucosa, e.g. induction of serum amyloid A gene expression in the epithelium (Ivanov et al., 2009), but the bacterial components responsible for this and the host signaling pathways involved in shaping the intestinal immune response are yet to be identified.
As a first step towards elucidating the mechanism by which SFB influences the immune system, we have sequenced the genome of SFB isolated from feces of SFB-monocolonized mice. The SFB genome classifies SFB phylogenetically as a unique member of Clostridiales, although it is distinct from all other sequenced Clostridial genomes and forms a unique phylogenetic unit within the group. The 1.57 Mb SFB genome is one of the smallest sequenced Clostridial genomes to date. Additionally, annotation and comparative functional analysis demonstrated that SFB occupies a metabolically intermediate position between Clostridial and highly reduced host-dependent genomes, such as Mycoplasmas. Specifically, the SFB genome lacks a number of basic metabolic pathways of free-living bacteria, but it is enriched in systems mediating interactions with the environment or the host, such as transporters and two-component systems. In spite of these similarities to Clostridia and Mycoplasma, however, the functional genomic complement of SFB did not cluster closely with any of 1,200 complete bacterial genomes, suggesting that SFB are metabolically unique symbionts, which is likely related to their immunomodulatory functions. Genome-wide queries across 263 human gut metagenomes - 124 MetaHIT (Qin et al., 2010) and 139 HMP (www.hmpdacc.org) - did not identify evidence for SFB presence in any of the tested human fecal samples.
SFB represents one of the very few microbial genomes of an uncultured bacterium sequenced directly from an intestinal or environmental sample, and its genomic sequence described here, and independently by Prakash et al. in this issue (Prakash et al., in this issue), will provide an invaluable tool for the identification of bacterial products mediating its immunomodulatory effects.
Results and Discussion
General Genome Features
The SFB genome sequence was obtained using genomic DNA from feces of SFB-monocolonized mice (Umesaki et al., 1995), as described in Methods. The order and orientation of the five contigs, which totaled 1,569,870 bp, were predicted based on similarity analysis of k-mers at contig ends, G-C skew and ORF strand bias (see Supplemental Material). The general features of the SFB genome are listed in Table 1. Consisting of a single chromosome with an average G-C content of 27.9%, SFB is unlike other intestinal Clostridia, which have markedly higher G-C content (40-50%) (Bruggemann and Gottschalk, 2008), and is more similar to environmental Clostridia belonging to cluster I (Table 1). Deviant G-C content was confined almost exclusively to two rRNA operons occupying a single small contig (contig 4), which had approximately 3x the average sequencing coverage, consistent with a collapse of additional repetitive rRNA operons during assembly (Figure 1 and Table S1). The SFB genome contains 38 tRNA genes, a relatively low number compared to the average Clostridium, but a complement sufficient to provide specificity for all 20 canonical amino acids and selenocysteine. A single origin of replication was assigned to a region showing a clear inflection in G-C skew and coding strand bias, adjacent to the dnaA gene, and containing 5 DnaA boxes (see Supplemental Material and Figures 1 and S1).
Table 1. General genome features of SFB genome as compared to the full genomes of 30 Clostridium species.
Signal peptides were predicted by LipoP, while localization was predicted by PSort (see Methods). The KEGG Automated Annotation Server (KAAS) was used to assign predicted coding sequences to orthologous groups.
| SFB | Clostridium AVG | Clostridium SD | |
|---|---|---|---|
| Genome Size | 1,569,870 | 3,974,341 | 739,004 |
| G+C % | 27.9 | 30.57 | 4.04 |
| Features | 1589 | 3678 | 639 |
| ORFs | 1533 | 3525 | 612 |
| ORF Density | 1024 | 1127 | 57 |
| tRNAs | 38 | 77 | 15 |
| rRNA Operons | 2 | 9 | 2 |
| Signal Peptidase I | 80 | 192 | 50 |
| Signal Peptidase II | 46 | 84 | 26 |
| Cell Wall | 20 | 29 | 9 |
| Membrane | 326 | 862 | 153 |
| Extracellular | 34 | 67 | 26 |
| Annotated (All) | 1137 | 2424 | 539 |
| Annotated (KAAS) | 792 | 1481 | 178 |
| Hypothetical | 396 | 1101 | 454 |
Figure 1. Circular representation of the SFB genome.
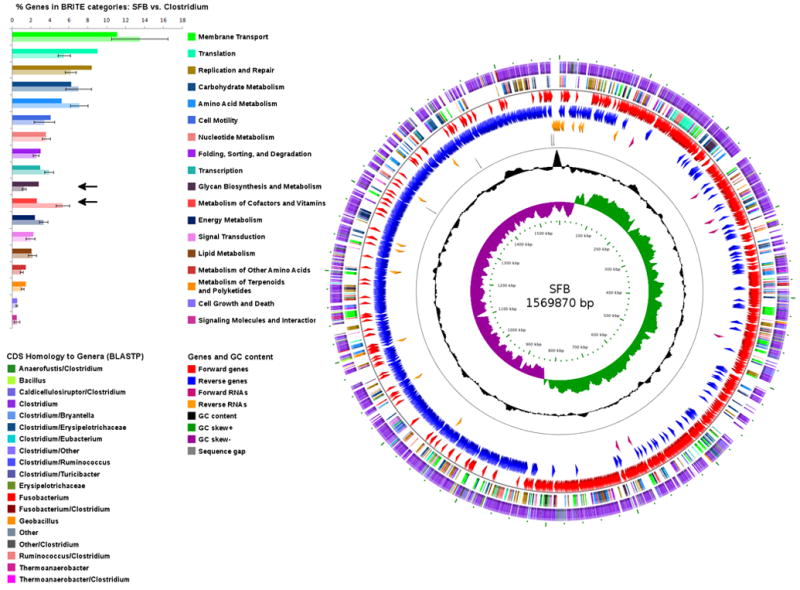
Wheel: The 5 contigs were arranged in order in a circular pseudochromosome (see Methods). Circles from outside in are: (i) ORF homology. Each ORF was color coded according to the genera most prevalent in its top ten PSI-BLAST hits (see left bottom corner for color legend). The SFB genome is dominated by ORFs homologous to Clostridium spp.; (ii) KEGG BRITE functional categories of annotated coding sequences (CDS) colored by type of category (listed on the left side of the figure); (iii) CDS on the forward strand; (iv) CDS on the reverse strand; (v) non-coding RNAs; (vi) markers delineating end of contigs; (vii) G-C content, and (viii) G-C skew, defined as (G − C) / (G + C).
Top Left: KEGG BRITE categories in SFB and the 30 available Clostridium spp. genomes. Percentage of genes in each BRITE category. Top column in each category - percentage in the SFB genome, bottom column - average percentage in Clostridium spp. Arrows indicate the two categories that differ in SFB.
Computational analysis did not identify any insertion sequences (IS), extrachromosomal sequences (e.g. plasmids or phages), or significant genomic islands, suggesting that the SFB genome is relatively stable, and has undergone little, if any, recent horizontal gene transfer (Table S2). We were, however, able to identify a 45 kb prophage cluster (SFBNYU_013700 through SFBNYU_014260) and a small 7 kb cluster of phage genes (SFBNYU_012100 through SFBNYU_012200). Three arrays of CRISPR sequences were identified, along with seven CRISPR-associated proteins (SFBNYU_008790 through SFBNYU_008850) (Table S3). CRISPR loci are found in certain bacteria where they confer resistance to exogenous genetic elements (Horvath and Barrangou, 2010).
We found virtually no evidence of polymorphisms in our reads, suggesting that the examined intestinal SFB population of mono-colonized mice is genetically homogenous. This may reflect the highly adapted nature of SFB to their host and environment. Due to the host-specific nature of SFB (Tannock et al., 1984), comparison of genomic SFB sequences from different mouse strains and host species, such as the rat-derived SFB genome reported in this issue by Prakash et al. (Prakash et al., in this issue), will potentially reveal important host-specific adaptation.
Coding Sequences And Phylogeny
A total of 1,533 coding sequences (CDS) were predicted from the SFB genomic sequence (Table 1) with an average length of 934 bp (Table 1 and Figure S2). There was a definite strand bias as 81.3% of predicted ORFs were encoded on the leading strand of DNA replication (Figure 1). Annotation was performed using multiple pipelines and manual curation (see Methods). 792 CDS were assigned to 718 KOs by KEGG (Moriya et al., 2007) and another 214 CDS were assigned to orthologous groups in the Microbial Genome Database for Comparative Analysis (MBGD) (Uchiyama et al., 2010). This level of annotation provided moderate coverage (52% in KO; 66% in MBGD) of the SFB genome at the gene level, on par with the 30 Clostridium genomes (45% in KO) included in KEGG. An additional 178 CDS were annotated by BLASTP or Pfam. In total, 1,184 or 77% of the CDS were assigned annotation, function or domain. Another 136 CDS were homologous to other genomes using “relaxed” criteria (see Supplemental Information), and finally, 213 (14% of total) CDS were unique to SFB.
To determine the overall similarity of the SFB proteome to previously identified proteins, we used PSI-BLAST to compare all putative SFB CDS to amino acid sequences deposited in NCBI (see Methods). We found 78% of SFB CDS significantly homologous (using “relaxed” criteria) to CDS from other genomes. Of these, 76% were most homologous to Clostridium spp. and Clostridium was among the top hits in another 10%. Therefore, the SFB genome is dominated by Clostridium-like CDS (Figures 1 and S3A). The homology to Clostridium was also evident at the nucleotide sequence level, as demonstrated by the similarity in codon usage bias (Figure S3B). Nevertheless, 24% of SFB CDS with significant homology were most similar to CDS from other genera, such as Bacillus, Thermoanaerobacter and Ruminococcus (Figure S3A).
To investigate the phylogenetic relationship of SFB to other bacteria we performed a phylogenomic analysis based on 28 conserved protein markers using AMPHORA (Wu and Eisen, 2008). This analysis positioned SFB nearest to members of cluster I Clostridia (belonging to the family Clostridiaceae of the order Clostridiales,), though at a significant distance from these species, and from any of the currently available bacterial genomes (Figure S4). This strongly suggests that SFB is a unique member of a novel cluster of Clostridia.
Comparative Functional Genomics Of SFB
To assess the SFB genome’s functional potential, we first collapsed all 718 annotated KOs into 219 metabolic modules (MO; small 5-20 gene pathways defined by KEGG). We then compared these and the SFB gene repertoire as annotated by both KEGG and MBGD to over 1,100 finished microbial genomes (1,209 in KEGG; 1,153 in MBGD). This allowed us to generate clustering networks (Figure 2) based on overall genomic metabolic potential, to identify the closest functionally related organisms, and to compare these results to the phylogenetic analysis above.
Figure 2. Genome-wide metabolic comparison between SFB and all sequenced genomes.

Analysis of microbial functional similarities based on shared orthologous gene families (A,B) and modules (C). A/B. The 1,209 genomes in KEGG and the 13,118 KEGG Orthology gene families (KOs) are reported as circles and small cyan triangles, respectively. Organisms are connected by edges to all gene families contained within their genome. A) Global network of all genomes for visual overview. SFB (large red circle) lies outside any cluster but is close to groups of several Firmicutes genera and in particular Clostridium, Thermoanaerobacter, Staphylococcus, and Streptococcus. Other genera, including Mycoplasma, Borrelia, Treponema, Finegoldia and Gardnerella are quantitatively similar to SFB (see text) but located in the network periphery due to overall reduced gene content. Despite their similarity to SFB in terms of genome size and host environment, Helicobacter and Campylobacter are located in different regions of the network suggesting different functional specialization. B) Sub-network of SFB and the 20 most similar organisms (Tversky index 0.75, Table 2); KOs included in at least two organisms are depicted. SFB is functionally distinct from both the cluster of Clostridia (further differentiated as C. botulinum, C. perfringens, and “other”) and the Thermoanaerobacteria. C) Metabolic comparison based on functional genomic potential (Table 2) highlights SFB’s similarity to several Myco/Phyto/Acholeplasma and to Finegoldia magna (for which only one genome is available). The width and color (white = low, green = medium, red = high) of edges reflect relationship strength. Again, despite considering only the most functionally similar organisms, SFB is not directly included in the clusters they form.
We used the Tversky index (Tversky, 1977) with α=0.75 to identify organisms sharing most of the SFB gene complement (see Methods). This analysis identified Clostridia from the Clostridium and Thermoanaerobacter genera as most metabolically similar to SFB at the gene level (Table 2 and Figure 2A,B), providing functional support for their phylogenetic relationship (Figure S4). At the level of metabolic modules, three strains of Lactococcus lactis and nine strains of Streptococcus pyogenes, both members of the class Bacilli, were identified in addition to the aforementioned Clostridia. SFB possess few metabolic modules not present in these organisms (Figure 3A – green circles and Table S4A), with one notable exception being M00223 (phosphonate transport system), fully present in SFB and Lactococcus lactis, yet absent in Streptococcus pyogenes and most Clostridium spp. More striking are the modules absent from SFB in comparison to relatives with larger genomes (Figure 3A – red circles). Like these organisms, SFB have a complete EMP glycolysis pathway (M00001), have similar components of the pentose phosphate shunt (M00004), possess the ability to synthesize (M00082-3), but not degrade (M00087), fatty acids, and lack nearly all components of the TCA cycle (M00009) (Figures 4 and S5 and Table S5A). Additionally, amino acid metabolism is almost entirely absent from SFB, except for lysine biosynthesis (M00016), aspartate, glutamate, asparagine, and glutamine interconversion, and glycine hydroxymethyltransferase catalyzing the interconversion of glycine and serine (Figure S5 and Table S5A). Present in SFB’s functional relatives but not in SFB (z-score lower than -1.0) are pathways for proline (M00015) and cysteine (M00021) biosynthesis (Figure 3A and Table S4A) as well as tryptophan (M00023), threonine (M00018), histidine (M00026), and leucine (M00019) biosynthesis although with a smaller effect (z-score lower than -0.5).
Table 2. Top organisms functionally similar to SFB based on shared gene families and metabolic modules.
1,191 microbial reference genomes were sorted by the specific orthologous gene families (using MBGD (Uchiyama et al., 2010)), general gene families (KO) (using the KEGG Orthology (Kanehisa et al., 2010)), or metabolic modules (MO) (small ~5-20 gene pathways from KEGG) shared with SFB. Genera appearing at least twice among the 20 most similar organisms are shown here, using the Tversky similarity index with α=0.25 (emphasizing organisms with few pathways not carried by SFB) and with α=0.75 (emphasizing organisms missing few pathways carried by SFB). Percentages in parentheses refer to the fraction of the top 20 genomes that fall within the respective genus. In addition, the catalogs were split into core (present in at least 75% of available genomes) and variable (present in 5-25%) subsets, and the reference genomes most similar to SFB in these subsets are shown here. SFB carries core subsets similar to both Firmicutes and minimal organisms, but its variable subsets are most similar to Clostridia and Streptococci.
| Genus | Total genomes in genus | MBGD | KO | MO | |||
|---|---|---|---|---|---|---|---|
| α = 0.25 | α = 0.75 | α = 0.25 | α = 0.75 | α = 0.25 | α = 0.75 | ||
| # of organisms within the 20 genomes sharing the most gene families | |||||||
|
| |||||||
| Borrelia | 8 | 6 (30%) | 7 (35%) | ||||
| Clostridium | 31 | 2 (10%) | 16 (80%) | 12 (60%) | 2 (10%) | ||
| Lactococcus | 4 | 3 (15%) | |||||
| Mycoplasma | 26 | 10 (50%) | 4 (20%) | 11 (55%) | |||
| Streptococcus | 52 | 9 (45%) | |||||
| Thermoanaerobacter | 7 | 4 (20%) | 5 (25%) | 2 (10%) | |||
| Ureaplasma | 3 | 3 (15%) | |||||
|
| |||||||
| # of organisms within the 20 genomes sharing the most CORE gene families | |||||||
|
| |||||||
| Borrelia | 8 | 7 (35%) | 7 (35%) | 2 (10%) | |||
| Clostridium | 31 | 11 (55%) | |||||
| Lactobacillus | 25 | 5 (25%) | |||||
| Mycoplasma | 26 | 5 (25%) | 2 (10%) | ||||
| Propionibacterium | 3 | 3 (15%) | |||||
| Streptococcus | 52 | 6 (30%) | 8 (40%) | 10 (50%) | |||
| Thermoanaerobacter | 7 | 2 (10%) | 2 (10%) | ||||
| Ureaplasma | 3 | 3 (15%) | |||||
|
| |||||||
| # of organisms within the 20 genomes sharing the most VARIABLE gene families | |||||||
|
| |||||||
| Clostridium | 30 | 15 (75%) | 18 (90%) | 6 (30%) | 12 (60%) | 6 (30%) | |
| Streptococcus | 50 | 5 (25%) | 4 (20%) | 9 (45%) | |||
| Thermoanaerobacter | 7 | 4 (20%) | 2 (10%) | 6 (30%) | 6 (30%) | 2 (10%) | 4 (20%) |
Figure 3. KEGG modules over- and under-represented in the SFB genome compared to its 20 most similar organisms.

Overview of the KEGG functional hierarchy; highlighted leaf nodes represent metabolic modules over- (green) or under-enriched (red) in SFB relative to the 20 most functionally similar organisms. Black circles represent modules present at a similar level in SFB and these organisms (absolute z-score <1) and white circles are absent in both. Similar organisms were computed using A) Tversky index α = 0.75 (representing larger related genomes) and B) Tversky index α = 0.25 (smaller, more minimal related organisms) and are listed in Table 2. A complete list of KEGG modules differentiating SFB from functionally similar organisms can be found in Table S4A and S4B.
Figure 4. Predicted SFB metabolic pathways.
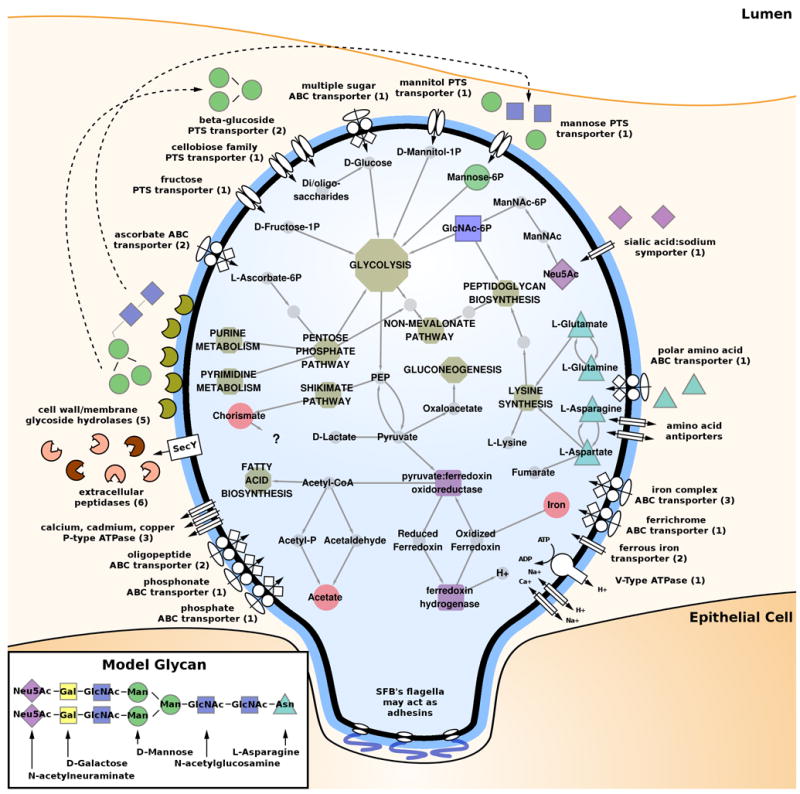
Overview of SFB metabolic pathways. SFB are highly auxotrophic and have a few complete essential pathways mostly for utilization of glycans and monosaccharides. They have complete glycolysis and pentose phosphate pathways, but lack the TCA cycle. Although fatty acid biosynthesis pathways are present, fatty acid metabolism pathways are absent. Absent as well are most pathways for co-factor and amino acid biosynthesis with the exception of the interrelated pathways for lysine, aspartate, glutamate, asparagine, and glutamine as noted in the figure. In contrast, multiple oligosaccharide and metal ion (in particular iron) transport and utilization mechanisms are present in SFB, including PTS, ABC, and other transporters, as well as extracellular peptidases and glycosyl hydrolases, which are shown interacting with extracellular glycans. SFB appear to be able to digest the glycoprotein components of the mucus layer and import sugars released in the process via ABC transporters and TCS. Once imported, several enzymes prepare these substrates for glycolysis. All these pathways provide SFB with the ability to acquire multiple metabolites from the surrounding environment and the host. Within the cell, essential pathways leading from import of polysaccharides through production of peptidoglycan, fatty acids, reduced ferredoxin, and acetate are shown.
Compared with the Clostridia, SFB have a greatly reduced complement of enzymes involved in co-factor and vitamin metabolism (Figure 1). For example, SFB cannot synthesize coenzyme-A (CoA) from pantothenate (vitamin B5), but possess the enzymes necessary to convert pantetheine (a vitamin B5 metabolite) to CoA, which may indicate the dependence on environmentally derived vitamin B5 metabolites. This suggests that compared to functionally related organisms SFB have unusual auxotrophic needs to obtain nearly all amino acids and many co-factors from the environment. Interestingly, SFB possess the V-type (M00159), while functional relatives possess the F-type (M00157), ATPase. While both couple substrate translocation across a membrane to phosphorylation of adenosine, they differ slightly in function. The F-type, when running in the forward direction, couples a proton-motive force to the generation of ATP during respiration. In contrast, the V-type translocates both protons and sodium, and in eukaryotes runs in the reverse direction, acidifying vacuoles with concomitant hydrolysis of ATP (Yokoyama and Imamura, 2005). As fermentative obligate anaerobes, SFB cannot create, and do not utilize, a proton gradient for ATP generation. They do, however, possess a complete set of flagellar genes and several proton- and sodium-coupled transporters, all of which rely on a concentration gradient (Minamino et al., 2008). This suggests that SFB have retained this ATPase to generate such gradient.
Although this analysis identified Clostridia as metabolically comparable to SFB, even the most similar organisms (top 20 genomes for Tversky index with α=0.75) share at best a fraction of the SFB gene families (570 ± 18 of 718 or 79% KOs and 628 ± 27 of 1,003 or 63% MBGD). This is despite the fact that these organisms tend to carry much larger gene complements than SFB (547 ± 76 or 76% additional distinct genes in KO and 1,199 ± 181 or 120% in MBGD). We therefore turned to a lower Tversky index (α=0.25) in order to identify organisms with gene complements closer to a strict subset of those carried by SFB. This identified several “minimal” genera (Mycoplasma, Ureaplasma, and Borrelia) as functional relatives of SFB (Figure 2C and Table 2). These are all endosymbionts, obligate or pathogenic, most with reduced genome sizes. SFB has similar characteristics, and this may reflect its evolution as an obligate commensal (Kilian et al., 2008; Toft and Andersson, 2010; Yus et al., 2009). Modules present in SFB but not in related organisms with small genomes were mostly transporters, e.g. for mannose (M00276), cellobiose (M00275), beta-glucoside (M00271), iron (M00240), zinc (M00242), and amino acids (M00236) (Figure 3B and Table S4B). Therefore, SFB is more enriched in transporter pathways than highly auxotrophic minimal organisms. Again, even the most similar organisms with small genomes identified here show only partial overlap with the SFB gene complement (328 ± 65 of 718 or 47% different KO and 288 ± 86 of 1,003 or 29% in MBGD) and tend to carry a number of additional genes (153 ± 76 or 21% in KO and 238 ± 175 or 24% in MBGD).
The above analyses show that, by both the KEGG and MBGD definitions of orthologous gene families, SFB carry a strikingly distinct gene complement as compared to almost 1,200 currently sequenced archaea and bacteria. As a result, SFB do not form a functional cluster with any other genome (Figure 2A) but, instead, occupy a middle ground between minimal relatives identified using the Tversky index with α=0.25 and relatives (mainly Clostridia) with larger genomes identified with α=0.75.
To determine which genes in SFB are essential and which are likely to represent niche-specific adaptations, we further grouped all gene families (KO and MBGD) and modules (MO) into “core” members, present in at least 75% of the 1,100+ finished microbial genomes, and “variable” members, present in at least 5% but at most 25% (Table S6A,B). We then identified the sequenced genomes most similar to SFB, according to the two groups (Table 2). SFB carry an expected complement of core genes and a reduced number of variable genes and modules (Table S6A). Furthermore, their 13 core modules represent basic essential pathways and many are shared with a variety of minimal and non-minimal organisms (Table 2 and S6B). In contrast, their variable complement is most similar only to Clostridial and Streptococcal Firmicutes, at both the gene and module level (Table 2). Therefore, although the core SFB genome is similar to both Clostridial and minimal genomes, its variable genome is again closest to Clostridia (Table 2). Of the variable modules identified in SFB (Table S6B), five are substrate-specific components of the phosphotransferase system (PTS) (M00271, M00274-6, M00283), suggesting that SFB’s specific set of sugar-importing PTS transporters may represent adaptation to life as a gut commensal.
Finally, we compared the SFB genome’s functional capacity to a set of bacteria of specific interest due to their phylogenetic relatedness, functional similarity, or related genome size and/or mucosal habitat (Table S7). In terms of the well-characterized biology represented in KEGG orthologous families and modules, SFB remains moderately similar to Clostridium spp. and minimal pathobionts, including F. magna (sharing e.g. M00256 and several other transport systems), T. denticola (sharing iron/zinc/cobalt transporters M00240/2 and M00245/6), G. vaginalis (sharing sugar/ribose transporters), and B. burgdorferi (sharing the cellobiose PTS). In contrast, Helicobacter and Campylobacter spp. possess very different gene complements from SFB and form distinct functional clusters (Figure 2A and Table S7), despite living in the mammalian gastrointestinal tract and having comparably reduced genomes. In terms of the largely uncharacterized orthologous families in MBGD, SFB remains overall genomically dissimilar even with the most phylogenetically related Clostridia, suggesting that a wealth of minimal gut commensal biology remains to be characterized from this unique organism (Table S7).
Putative Factors Mediating The Interaction Of SFB With The Host Environment
Microbe-host interactions may be achieved through, among other mechanisms, secretion of immunomodulatory substances, production of surface molecules that mediate adhesion and interaction with epithelial cells, or production of enzymes that modify the host extracellular matrix or cell surface receptors. As SFB has the ability to modulate intestinal immune responses, we searched the SFB proteome (defined as the translated SFB genome) for factors potentially involved in host-microbe interaction.
The SFB proteome has 46 (3% of total) flagellum- and chemotaxis-related proteins. These include a complete set of flagellum biosynthesis proteins, organized in three genomic clusters, and three copies of the key filament protein, flagellin (Table S8A and Figure S6). The presence of an intact flagellar apparatus suggests that SFB may be motile at some point in its lifecycle or utilize flagella for other purposes. Homologs of these proteins can be found in all sequenced Clostridium spp., except C. perfringens, and were not identified as over- or under-represented in our comparative genomic analysis. Although SFB have been observed by electron microscopy in a number of species, no study has yet reported the presence of flagella. In addition to motility, intestinal bacteria may use flagella for penetrating the mucus or attaching to epithelial cells (Celli et al., 2009; Guerry, 2007). For example, nonflagellated strains of C. difficile have a 10-fold reduction in adherence to the cecum of mice, as compared to flagellated strains (Tasteyre et al., 2001). Flagella have also been shown to mediate adhesion to an environmental surface in other species (Tyson et al., 2004). Negative regulation of flagellum expression has been proposed as part of a mechanism for establishing intestinal colonization by E. coli (Giraud et al., 2008). Therefore, although present in the SFB genome, flagellum genomic clusters may be negatively regulated and lack of flagellum expression may be part of the adaptation of SFB as an intestinal colonizer. However, in contrast to E. coli, SFB is a highly adapted commensal symbiont and is not observed outside the host. Moreover, genome reduction and loss of presumably mutualism-preventing genes is a major feature of the SFB genome. The preservation of multiple flagellum loci is therefore likely to be functionally important.
To explain the tight attachment of SFB to epithelial cells, we searched for additional adhesion proteins. In gram-positive bacteria, proteins may be exported from the cell via several mechanisms: signal recognition particle (SRP), Sec, and twin-arginine translocation (Tat) pathways, as well as type IV secretion systems. The SFB genome encodes a complete set of Sec proteins, signal peptidases I and II, and SRP, but no Tat proteins, nor type IV secretion systems. Using PSort and LipoP (Juncker et al., 2003; Yu et al., 2010), we located signal peptides in 126 proteins, of which 20 were predicted to be cell wall-localized and another 34 to be extracellular (Table S8B). As cell wall-attached proteins have been associated with pathogenicity in Clostridia, and many are adhesins, we searched the SFB proteome for sortase enzymes, which catalyze the attachment of LPxTG motif-containing proteins to the cell wall (Hendrickx et al., 2011), and for domains found in other cell wall-attached proteins. The latter include S-layer homology (SLH), cell wall-binding (CWB), and LysM domains (Bruggemann and Gottschalk, 2008). The SFB genome does not encode any sortase homologs, and we were unable to locate LPxTG-containing proteins in SFB using the criteria described by Boekhorst et al. (Boekhorst et al., 2005). Similarly, using the Pfam database, we did not identify any cell wall attachment domains, with the exception of two peptidoglycan-binding domain-containing proteins with uncharacterized function (Table S8C). The absence of SLH domains in SFB is in contrast to other Clostridia, such as C. difficile, in which S-layer and associated proteins are important virulence determinants (Ausiello et al., 2006). Though SFB lack most known adhesion-related motifs, we were able to locate a fibronectin-binding domain-containing protein (SFBNYU_005870) with homology to clostridial proteins that bind soluble and immobilized fibronectin and play a role in intestinal colonization (Barketi-Klai et al., 2011). This protein, along with the flagella discussed above, may aid SFB adherence to the host epithelium.
In the gut, glycans in various states of degradation, from host, dietary, and microbial sources, are abundant energy resources for commensal microbes. Gut-colonizing species, such as Bacteroides thetaiotaomicron and Bifidobacterium bifidum, have evolved substantial mechanisms for degrading, importing, and utilizing glycans. In addition, mucosa-associated bacteria, such as SFB, have to overcome the mucus layer barrier that excludes most other commensals from interacting with intestinal epithelial cells. Among the 16 glycosyl hydrolases encoded by the SFB genome (Table S5B and Table S9), 11 are homologous to enzymes that may cleave glycosidic linkages in glycans, and, among these, three are not present in minimal organisms most functionally similar to SFB. In addition, although SFB does not encode nearly as many genes for glycan foraging activity as major gut symbionts, such as B. theta, genes involved in glycan metabolism were the only BRITE gene category that was overrepresented in SFB in comparison to the rest of the sequenced Clostridia (Figure 1 and Table S5B). Therefore the presence of glycan utilization pathways separates SFB from its close functional minimal and Clostridial relatives. These included genes for the cleavage, import and utilization of glycan sugar moieties, such N-acetylglucosamine, mannose, and sialic acids, as well as six extracellular peptidases (Table S5B). For example, we located a complete set of genes for uptake and utilization of sialic acid, a common terminal modification on eukaryotic glycoproteins which can be used as a carbon and energy source by gut bacteria. In contrast to other gut bacteria, however, the SFB genome lacks a sialidase gene and therefore SFB itself cannot liberate this sugar from host glycoproteins. This suggests that the pathway requires the activity of exogenous sialidases, underscoring SFB’s nature as an obligate symbiont. The increased ability of SFB over functional relatives to degrade and utilize only certain glycan residues (e.g., genes utilizing fucose, galactose, or N-acetylgalactosamine are absent) suggests that in the competitive environment of a complex gut microbiota it may use intermediate products of glycan degradation produced by other species, representing context-dependent auxotrophy.
A notable feature of the SFB genome is the abundance of genes involved in interaction with and acquisition of metabolites from the environment, including those encoding transporters and transcriptional regulators responsive to external cues. Using the Pfam database, we identified 63 putative transcriptional regulatory proteins (excluding DNA replication and ribosomal proteins) (Table S10), which represent 4% of the SFB CDS. These include transcriptional initiators for the flagellar and sporulation operons and nine pairs of two-component system histidine kinase and response regulator genes, most of which seem to have unique unassigned functions (Table S11). These genes also represented a large fraction (17 of 109 KOs or 16%) of the gene families present in SFB but not in the top 20 most similar organisms using the Tversky index with α=0.25 (Table S4C).
Among 1,184 annotated CDS, 117 or 10% are predicted components of ABC, PTS, and other transporters, a large fraction of which are specific for metal ion uptake and efflux (Table S8D). SFB has multiple P-type ATPase genes implicated in the efflux of toxic heavy metals, such as cadmium, and several putative transporters for zinc, iron, cobalt, nickel, magnesium, and chromate. Iron is a key component of SFB metabolism and is the most highly represented among the metal utilization genes (Table S8D). In the gut, SFB must compete with both the host and other members of the microbiota for iron. As environmental iron is poorly soluble, bacteria and fungi have evolved to produce and export small, iron-chelating molecules known as siderophores. During infection, pathogens secrete these molecules to capture host iron, and as part of the inflammatory response the host increases production of its own iron-sequestering proteins in an effort to out-compete the invading microorganisms (Flo et al., 2004). SFB possess a great variety of iron uptake systems for all types of extracellular iron, which may be an adaption to the iron-scarce gut environment. These include a complete fhu hydroxamate siderophore ABC transporter, two sets of feoAB ferrous iron transporters, and three uncharacterized iron complex ABC transporters with homology to vibrioferrin and heme importers (Table S8D). Although SFB is capable of utilizing a variety of siderophores, it is unable to synthesize any of its own, and may utilize the plentiful siderophores produced by neighboring species. FeoB homologs are common in intestinal bacteria, such as H. pylori, where FeoB is thought to provide the major pathway for Fe2+ uptake and is essential for colonization of the murine gastric mucosa (Velayudhan et al., 2000). Finally, the SFB genome encodes three ferric uptake regulator (FUR) family proteins, which have been shown to control the intracellular concentration of iron in many bacteria. Interestingly, each FUR protein is most homologous to proteins in different groups of bacteria (Acetovibrio, Thermoanaerobacter, and Clostridium respectively) underscoring the importance of iron uptake in SFB.
SFB Are Not Present In Human
SFB have not yet been identified in humans. No SFB 16S rRNA sequence is present in any of the published human 16S metagenomic databases. However, almost all of the published human intestinal 16S sequences are derived from fecal samples. Because SFB is endemic for the small intestine in rodents it may be relatively less represented in human feces and examination of the 16S gene may be insufficient to detect it. We therefore looked for the presence of any part of the SFB genome in human metagenomic datasets. We utilized the MetaHIT database (Qin et al., 2010), which is the largest published metagenomic database, containing 576.7 Gb of sequence from 124 individuals. The reads in the MetaHIT database were compared to the genome of SFB. As a control for the sensitivity of the assay, the reads were also compared to the genomes of six control intestinal organisms. The percentage of reads with >95% identity to any part of the genomic sequence represents the abundance of the organism in each of the 124 MetaHIT samples (Figure 5A). We also calculated the coverage of each of the genomes in the individual human samples (Figure 5B). The control organisms were chosen as present in human fecal samples with variable, but usually low, abundance. For example only one of the controls, E. faecalis, was among the 57 frequent microbial genomes with >1% coverage in most samples as reported by (Qin et al., 2010). To increase confidence that the detected reads come from the chosen control genome, we used a higher threshold of identity from that previously reported (95% vs 90% in (Qin et al., 2010)).
Figure 5. Presence of genomes of intestinal microorganisms in the MetaHIT human metagenome database.

The WGS Illumina reads of 124 individual fecal samples in the MetaHIT database (Qin et al., 2010) were aligned to the SFB genome and six other reference genomes (Clostridium perfringens ATCC13124, Enterococcus faecalis V583, Enterococcus faecium TX1330, Escherichia coli MG1655, Lactobacillus johnsonii NCC533, and Methanobrevibacter smithii ATCC35061). Reads with alignment identity of 95% or higher were used to calculate the relative abundance (the percentage of mapped reads in total reads) and genome coverage (percentage of genome bases aligned to reads) for each genome. Heatmaps representing the relative abundance (A) and coverage (B) in each human sample for each of the seven organisms are shown. All organisms were detected in multiple (albeit not all samples) with the exception of SFB and E. faecium. SFB was the only organism not detected in any sample at thresholds of 0.02% abundance and 0.5% coverage (see text).
As shown in Figure 5, with the exception of E. faecium, the average abundance of the control genomes in all 124 samples ranged from 0.02 to 0.26% (Figure 5A). The average coverage of the control genomes ranged from 0.7 to 18% (Figure 5B) with a maximum coverage in individual samples from 9 to 91% (Table S12). In contrast, the average abundance of SFB genome reads was 5-65 fold lower at 0.004% and the maximum coverage in individual samples was only 0.4% (Table S12). At a threshold of 0.5% coverage the presence of 5 of the 6 control organisms ranged from 28 out of 124 samples for L. jonhsonii to 100 out of 124 for E. coli. E. faecium was generally not well detected in the MetaHIT samples. It is possible that the relative abundance and representation of E. faecium, which is the cause of some VRE nosocomial infections occurring after antibiotic treatment, is extremely low in healthy individuals, which may explain our inability to detect it. Still, at a coverage threshold of 0.5%, E. faecium genome sequences were present in 4 of the 124 samples. In contrast, SFB was not detected in any of the samples at this threshold (Table S12). Moreover, the SFB genome was not detected in any of 139 human fecal samples of the recently released human microbiome project (HMP) metagenomic database (hmpdacc.org). We therefore conclude that the MetaHIT and HMP datasets do not contain SFB genomic sequences and that SFB is either absent or below the level of detection in these samples.
Finally, we attempted to directly detect SFB in a set of human fecal samples. We generated primers for five SFB-unique genes and examined their representation in eight human samples. Although all genes were detected in purified SFB DNA, as well as fecal genomic DNA from Taconic C57BL/6 mice, none were detected in feces of conventionally-raised Jackson C57BL/6 mice (which do not contain SFB (Ivanov et al., 2008)) or in any of the human samples (Figure S7).
The absence of SFB in human samples may reflect a sampling bias of fecal samples from adult individuals in the MetaHIT and HMP databases. In rodents, SFB are known to colonize the terminal ileum, where they have been reported to decrease in abundance in older animals (Snel et al., 1998). Moreover, the description of SFB in multiple mammalian species has almost exclusively been based on morphological identification in terminal ileum samples. Nevertheless, the decrease of SFB in mice is not absolute and is not observed in cecal contents (Snel et al., 1998), and SFB are easily detected in mouse feces throughout life. Therefore, we expect to find traces of SFB DNA in fecal adult human samples if the organism colonizes humans.
Alternatively, SFB may have co-evolved with other mammalian species, but may be incompatible with the human gut environment, where its immunomodulatory functions may be performed by other commensal species. Further sequencing of the microbiota of other animal species and of healthy and patient human populations with different demographic profiles as well as the examination of mucosa-associated bacteria from human terminal ileum will be required to determine in detail the host range of SFB and its contribution to mucosal immunity in humans. Such studies will also provide insight into conserved genes that may influence development of Th17 cells.
Conclusions
The genome sequence of SFB obtained from monoassociated mice provides the opportunity to learn how a commensal bacterium can influence the balance of T cells not only locally in the intestinal lamina propria, but also systemically. Examination of the encoded proteins and pathways reveals SFB to be highly dependent on nutrients and co-factors derived from the host environment. SFB colonize and expand well in germ-free animals, which indicates that host factors rather than extrinsic bacterial factors are sufficient to promote their growth. However, because of the increased dependence on external metabolites, SFB functionality in vivo is likely to be affected by the activity of other commensal bacteria as well. It is therefore not surprising that the conditions for culturing SFB have remained elusive since the first attempts several decades ago. The highly reduced nature of the SFB genome may represent not only the establishment of host dependence to occupy an environmental niche, but also a host-directed evolutionary adaptation to secure mutualism with an important immunomodulatory microbe. Information derived from the SFB genome sequence will hopefully provide clues as to auxotrophies that will facilitate culturing SFB as well as establishing of assay systems to elucidate the mechanisms by which SFB or its products induce accumulation of Th17 cells and, potentially, also contribute to maintenance of the integrity of the epithelial barrier. Although SFB has yet to be identified as a human commensal, the insights gained from the analysis of its genome and biology will contribute to a better understanding of how bacteria affect host inflammatory processes.
Methods
Genome sequencing and assembly
Genomic DNA was isolated from fecal pellets of mono-colonized mice as previously described (Ivanov et al., 2009) and was further purified using a genomic DNA purification kit from Qiagen. 454 XLR mate paired and SOLiD sequencing generated 1.3 million 331±102 bp reads and 22.5 million 50 bp reads. 454 reads were filtered by aligning to the Mus musculus genome and assembled with Newbler (2010-04 pre-release). SOLiD reads were aligned to the assembly using BWA (Li and Durbin, 2010). The remaining contigs were aligned to NCBI’s NT nucleotide database using BLASTN, and removed if found likely to be from a contaminant source. The final assembly consists of 5 contigs totaling 1,569,870 bp with an N50 contig length of 1,317,732 bp, and an average sequence depth of 341x.
Gene annotation
The prediction of protein coding genes was accomplished by Glimmer 3 (Delcher et al., 2007) and GeneMark (Besemer and Borodovsky, 1999). tRNAScan (Lowe and Eddy, 1997) was used for tRNA prediction, RNAmmer (Lagesen et al., 2007) for rRNA prediction, and RFAM/infernal for other non-coding RNA genes (Griffiths-Jones et al., 2005). Gene annotation was accomplished by submission to RAST (Aziz et al., 2008), KAAS (Moriya et al., 2007), IMG/ER (Markowitz et al., 2009), and a prokaryotic annotation pipeline created by the Human Genome Sequencing Center at the Baylor College of Medicine. Domain families for each called ORF were found using Pfamscan v1.3 (Finn et al., 2010) with HMMer v3.0b3 and database Pfam-A v24.0. Differing annotations for the same ORF were resolved manually by choosing the annotation most consistent with PSI-BLAST hits to NCBI’s NR database and Pfam domains present in the ORF. In instances where there was a convincing alignment (>50% identity across >70% of the ORF) to multiple proteins annotated differently, the ORF was labeled a “hypothetical protein” or according to the conserved domains it contained.
Annotation of SFB ORFs with KEGG and MBGD orthologous families
The 1,533 ORFS detected for the SFB genome were mapped into the KEGG Orthology (KO) database (as of March, 2011) as determined by the KEGG Automatic Annotation Server (KAAS) (Moriya et al., 2007) and into the MBGD database (Uchiyama et al., 2010) (version 2010-02) of orthologous families using blastn (best-hit approach, e-value cutoff at 1E-10, minimum alignment of 100bp). KO and MBGD include 1,191 and 1,153 microbial genomes, respectively. We found a total of 792 SFB ORFs (52%) with homologs in one of the 13,118 KOs (corresponding to 718 distinct KO families), whereas 1,003 ORFs (66%) had a confident match in at least one of the ~4M genes in MBGD belonging to one of the 236,073 gene families with at least 5 orthologs. This information was analyzed as two matrixes reporting the abundance of each gene family in each genome (all 1,153 organisms in MBDG, 1,186 high-quality KEGG microbial genomes, and a set of five eukaryotic outgroups including Homo sapiens, Mus musculus, Caenorhabditis elegans, Drosophila melanogaster, and Arabidopsis thaliana).
Assessing functional similarity of SFB with microbial reference genomes
To assess the functional similarity between SFB and other microbes, we compared the corresponding functional profiles in the above two matrixes. Specifically, in order to consider not only the number of shared gene families between SFB and another genome X, but also the number of genes lacking in one of the two compared organisms, we used the Tversky index (Tversky, 1977), which is a generalization of the Jaccard index (Real and Vargas, 1996) defined as:
where |SFB ∩ X| is the number of gene families shared between SFB and X, |SFB − X| the number of gene families in SFB but not X, |X − SFB| the number of gene families in X but not SFB, and the parameter α is a positive value weighting the relative importance of “extra” genes versus “missing” genes. α=0.75 encourages greater similarity to genomes containing a high fraction of SFB genes (i.e. X tends to be a superset of SFB), and α=0.25 upweights genomes with few genes not in the SFB genome (i.e. X tends to be a subset of SFB). Ranking all genomes in MBGD and KEGG according to their Tversky similarity with respect to SFB, we identified the 20 closest organisms both at α=0.75 and at α=0.25.
Detection of pathways differentiating SFB from functionally similar organisms
We assessed the metabolic potential of SFB in comparison with the ~1,200 organisms in MBGD and KEGG by identifying pathways and small metabolic modules present in their genomes. Functional units of approximately 5 to 20 genes describing small pathways and structural complexes were defined using KEGG modules, which were organized into larger pathways and functional classes by the BRITE hierarchy. Coverage (presence/absence) of each of the 371 KEGG modules was determined based on the fraction of its KOs present in each genome, and the metabolic profiles obtained in this manner for all genomes were processed as above to identify the genomes closest to SFB. The over- or under-enrichment of single pathways in SFB compared to its 20 closest organisms (using both Tversky α =0.75 and α =0.25) and model organisms was calculated as the z-score of SFB module abundance with respect to their average abundance in the set of similar genomes and visualized on the BRITE hierarchy using an in-house tool for circular dendrogram visualization (Segata et al., 2011).
MetaHit WGS read mapping analysis
WGS reads from 124 individuals in the MetaHIT database (Qin et al., 2010) were aligned to SFB and six other reference genomes (Clostridium perfringens ATCC13124, Enterococcus faecalis V583, Enterococcus faecium TX1330, Escherichia coli MG1655, Lactobacillus johnsonii NCC533, Methanobrevibacter smithii ATCC35061; accession numbers: CP000246.1, NC_004668.1, NZ_ACHL00000000, U00096.2, AE017198.1, NC_009515.1, respectively) using BWA aligner (Li and Durbin, 2010). Reads with alignment identity of 95% or higher were considered good matches and used in relative abundance and genome coverage analysis. Relative abundance was defined as the percentage of mapped reads in total reads for each genome, and genome coverage was defined as percentage of genome bases aligned to reads.
Supplementary Material
Acknowledgments
We thank Agnes Viale, Juan Li, and Nicolas Socci (MSKCC) for sequencing and advice on computational analysis, Anthony Maresso and Richard Gibbs (Baylor College of Medicine) and Jonathan Foley (UC Berkeley) for helpful discussions, Ramnik Xavier (MGH and Broad Institute) for organizing the HMP analysis, Kenya Honda (Univ. of Tokyo) and Yoshinori Umesaki (Yakult) for SFB samples, Bernard Henrissat (AFMB) for running the CAZy pipeline and Jose Scher (NYU) for providing human fecal samples. The study was supported by NIH grant 5RC2AR058986 and the Howard Hughes Medical Institute (D.R.L.), Crohn’s and Colitis Foundation of America Award CDA#2388 and NIH grant 4R00DK85329-02 (I.I.I.), NIH 1R01HG005969 and NSF DBI-1053486 (C.H. and N.S.), NIH U54HG004969 (D.G.), and NIH/NHGRI 1U54HG004973-01 (X.Q.)
Footnotes
Accession Numbers The Candidatus Arthromitus sp. SFB-mouse-NYU genome sequence was deposited at NCBI under accession XXXXXX-XXXXXX. Future project data will be aggregated by NCBI BioProject accession PRJNA71495.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Atarashi K, Umesaki Y, Honda K. Microbiotal influence on T cell subset development. Seminars in immunology. 2011;23:146–153. doi: 10.1016/j.smim.2011.01.010. [DOI] [PubMed] [Google Scholar]
- Ausiello CM, Cerquetti M, Fedele G, Spensieri F, Palazzo R, Nasso M, Frezza S, Mastrantonio P. Surface layer proteins from Clostridium difficile induce inflammatory and regulatory cytokines in human monocytes and dendritic cells. Microbes Infect. 2006;8:2640–2646. doi: 10.1016/j.micinf.2006.07.009. [DOI] [PubMed] [Google Scholar]
- Aziz RK, Bartels D, Best AA, DeJongh M, Disz T, Edwards RA, Formsma K, Gerdes S, Glass EM, Kubal M, et al. The RAST Server: rapid annotations using subsystems technology. BMC Genomics. 2008;9:75. doi: 10.1186/1471-2164-9-75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barketi-Klai A, Hoys S, Lambert-Bordes S, Collignon A, Kansau I. Role of fibronectin binding protein A in Clostridium difficile intestinal colonization. J Med Microbiol. 2011;60:1155–1161. doi: 10.1099/jmm.0.029553-0. [DOI] [PubMed] [Google Scholar]
- Besemer J, Borodovsky M. Heuristic approach to deriving models for gene finding. Nucleic Acids Res. 1999;27:3911–3920. doi: 10.1093/nar/27.19.3911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumershine RV, Savage DC. Filamentous microbes indigenous to the murine small bowel: a scanning electron microscopic study of their morphology and attachment to the epithelium. Microb Ecol. 1978:95–103. doi: 10.1007/BF02014280. [DOI] [PubMed] [Google Scholar]
- Boekhorst J, de Been MW, Kleerebezem M, Siezen RJ. Genome-wide detection and analysis of cell wall-bound proteins with LPxTG-like sorting motifs. J Bacteriol. 2005;187:4928–4934. doi: 10.1128/JB.187.14.4928-4934.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggemann H, Gottschalk G. Comparative genomics of clostridia: link between the ecological niche and cell surface properties. Ann N Y Acad Sci. 2008;1125:73–81. doi: 10.1196/annals.1419.021. [DOI] [PubMed] [Google Scholar]
- Celli JP, Turner BS, Afdhal NH, Keates S, Ghiran I, Kelly CP, Ewoldt RH, McKinley GH, So P, Erramilli S, et al. Helicobacter pylori moves through mucus by reducing mucin viscoelasticity. Proc Natl Acad Sci U S A. 2009;106:14321–14326. doi: 10.1073/pnas.0903438106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delcher AL, Bratke KA, Powers EC, Salzberg SL. Identifying bacterial genes and endosymbiont DNA with Glimmer. Bioinformatics. 2007;23:673–679. doi: 10.1093/bioinformatics/btm009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elinav E, Strowig T, Kau AL, Henao-Mejia J, Thaiss CA, Booth CJ, Peaper DR, Bertin J, Eisenbarth SC, Gordon JI, et al. NLRP6 Inflammasome Regulates Colonic Microbial Ecology and Risk for Colitis. Cell. 2011;145:745–757. doi: 10.1016/j.cell.2011.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finn RD, Mistry J, Tate J, Coggill P, Heger A, Pollington JE, Gavin OL, Gunasekaran P, Ceric G, Forslund K, et al. The Pfam protein families database. Nucleic Acids Res. 2010;38:D211–222. doi: 10.1093/nar/gkp985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, Aderem A. Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature. 2004;432:917–921. doi: 10.1038/nature03104. [DOI] [PubMed] [Google Scholar]
- Gaboriau-Routhiau V, Rakotobe S, Lecuyer E, Mulder I, Lan A, Bridonneau C, Rochet V, Pisi A, De Paepe M, Brandi G, et al. The key role of segmented filamentous bacteria in the coordinated maturation of gut helper T cell responses. Immunity. 2009;31:677–689. doi: 10.1016/j.immuni.2009.08.020. [DOI] [PubMed] [Google Scholar]
- Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML, Punit S, Karlsson M, Bry L, Glickman JN, et al. Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe. 2010;8:292–300. doi: 10.1016/j.chom.2010.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giraud A, Arous S, De Paepe M, Gaboriau-Routhiau V, Bambou JC, Rakotobe S, Lindner AB, Taddei F, Cerf-Bensussan N. Dissecting the genetic components of adaptation of Escherichia coli to the mouse gut. PLoS Genet. 2008;4:e2. doi: 10.1371/journal.pgen.0040002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths-Jones S, Moxon S, Marshall M, Khanna A, Eddy SR, Bateman A. Rfam: annotating non-coding RNAs in complete genomes. Nucleic Acids Res. 2005;33:D121–124. doi: 10.1093/nar/gki081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerry P. Campylobacter flagella: not just for motility. Trends Microbiol. 2007;15:456–461. doi: 10.1016/j.tim.2007.09.006. [DOI] [PubMed] [Google Scholar]
- Hendrickx AP, Budzik JM, Oh SY, Schneewind O. Architects at the bacterial surface - sortases and the assembly of pili with isopeptide bonds. Nat Rev Microbiol. 2011;9:166–176. doi: 10.1038/nrmicro2520. [DOI] [PubMed] [Google Scholar]
- Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327:167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 2009;139:485–498. doi: 10.1016/j.cell.2009.09.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, Littman DR. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe. 2008;4:337–349. doi: 10.1016/j.chom.2008.09.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanov II, Littman DR. Modulation of immune homeostasis by commensal bacteria. Curr Opin Microbiol. 2011;14:106–114. doi: 10.1016/j.mib.2010.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juncker AS, Willenbrock H, Von Heijne G, Brunak S, Nielsen H, Krogh A. Prediction of lipoprotein signal peptides in Gram-negative bacteria. Protein Sci. 2003;12:1652–1662. doi: 10.1110/ps.0303703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilian M, Poulsen K, Blomqvist T, Havarstein L, Bek-Thomsen M, Tettelin H, Sorensen U. Evolution of Streptococcus pneumoniae and its close commensal relatives. PLoS ONE. 2008;3:e2683. doi: 10.1371/journal.pone.0002683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lagesen K, Hallin P, Rodland EA, Staerfeldt HH, Rognes T, Ussery DW. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007;35:3100–3108. doi: 10.1093/nar/gkm160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YK, Menezes JS, Umesaki Y, Mazmanian SK. Proinflammatory T-cell responses to gut microbiota promote experimental autoimmune encephalomyelitis. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(Suppl 1):4615–4622. doi: 10.1073/pnas.1000082107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowe TM, Eddy SR. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997;25:955–964. doi: 10.1093/nar/25.5.955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markowitz VM, Mavromatis K, Ivanova NN, Chen IM, Chu K, Kyrpides NC. IMG ER: a system for microbial genome annotation expert review and curation. Bioinformatics. 2009;25:2271–2278. doi: 10.1093/bioinformatics/btp393. [DOI] [PubMed] [Google Scholar]
- Minamino T, Imada K, Namba K. Molecular motors of the bacterial flagella. Curr Opin Struct Biol. 2008;18:693–701. doi: 10.1016/j.sbi.2008.09.006. [DOI] [PubMed] [Google Scholar]
- Moriya Y, Itoh M, Okuda S, Yoshizawa AC, Kanehisa M. KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res. 2007;35:W182–185. doi: 10.1093/nar/gkm321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prakash T, Oshima K, Morita H, Fukuda S, Imaoka A, Kumar N, Sharma VK, Kim S, Takahashi M, Saitou N, et al. Complete genome sequences of rat and mouse segmented filamentous bacteria, a potent inducer of Th17 cell differentiation. Cell Host Microbe. 2011 doi: 10.1016/j.chom.2011.08.007. in this issue. [DOI] [PubMed] [Google Scholar]
- Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature. 2010;464:59–65. doi: 10.1038/nature08821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Real R, Vargas JM. The probabilistic basis of Jaccard’s index of similarity. Systematic biology. 1996;45:380. [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, Huttenhower C. Metagenomic biomarker discovery and explanation. Genome Biol. 2011;12:R60. doi: 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snel J, Hermsen CC, Smits HJ, Bos NA, Eling WM, Cebra JJ, Heidt PJ. Interactions between gut-associated lymphoid tissue and colonization levels of indigenous, segmented, filamentous bacteria in the small intestine of mice. Can J Microbiol. 1998;44:1177–1182. doi: 10.1139/cjm-44-12-1177. [DOI] [PubMed] [Google Scholar]
- Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermudez-Humaran LG, Gratadoux JJ, Blugeon S, Bridonneau C, Furet JP, Corthier G, et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc Natl Acad Sci USA. 2008;105:16731–16736. doi: 10.1073/pnas.0804812105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talham GL, Jiang HQ, Bos NA, Cebra JJ. Segmented filamentous bacteria are potent stimuli of a physiologically normal state of the murine gut mucosal immune system. Infect Immun. 1999;67:1992–2000. doi: 10.1128/iai.67.4.1992-2000.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tannock GW, Miller JR, Savage DC. Host specificity of filamentous, segmented microorganisms adherent to the small bowel epithelium in mice and rats. Appl Environ Microbiol. 1984;47:441–442. doi: 10.1128/aem.47.2.441-442.1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasteyre A, Barc MC, Collignon A, Boureau H, Karjalainen T. Role of FliC and FliD flagellar proteins of Clostridium difficile in adherence and gut colonization. Infect Immun. 2001;69:7937–7940. doi: 10.1128/IAI.69.12.7937-7940.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toft C, Andersson SGE. Evolutionary microbial genomics: insights into bacterial host adaptation. Nature Reviews Genetics. 2010;11:465–475. doi: 10.1038/nrg2798. [DOI] [PubMed] [Google Scholar]
- Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI. An obesity-associated gut microbiome with increased capacity for energy harvest. Nature. 2006;444:1027–1031. doi: 10.1038/nature05414. [DOI] [PubMed] [Google Scholar]
- Tversky A. Features of similarity. Psychological review. 1977;84:327. [Google Scholar]
- Tyson GW, Chapman J, Hugenholtz P, Allen EE, Ram RJ, Richardson PM, Solovyev VV, Rubin EM, Rokhsar DS, Banfield JF. Community structure and metabolism through reconstruction of microbial genomes from the environment. Nature. 2004;428:37–43. doi: 10.1038/nature02340. [DOI] [PubMed] [Google Scholar]
- Uchiyama I, Higuchi T, Kawai M. MBGD update 2010: toward a comprehensive resource for exploring microbial genome diversity. Nucleic Acids Res. 2010;38:D361–365. doi: 10.1093/nar/gkp948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umesaki Y, Okada Y, Matsumoto S, Imaoka A, Setoyama H. Segmented filamentous bacteria are indigenous intestinal bacteria that activate intraepithelial lymphocytes and induce MHC class II molecules and fucosyl asialo GM1 glycolipids on the small intestinal epithelial cells in the ex-germ-free mouse. Microbiol Immunol. 1995;39:555–562. doi: 10.1111/j.1348-0421.1995.tb02242.x. [DOI] [PubMed] [Google Scholar]
- Velayudhan J, Hughes NJ, McColm AA, Bagshaw J, Clayton CL, Andrews SC, Kelly DJ. Iron acquisition and virulence in Helicobacter pylori: a major role for FeoB, a high-affinity ferrous iron transporter. Mol Microbiol. 2000;37:274–286. doi: 10.1046/j.1365-2958.2000.01987.x. [DOI] [PubMed] [Google Scholar]
- Wu HJ, Ivanov II, Darce J, Hattori K, Shima T, Umesaki Y, Littman DR, Benoist C, Mathis D. Gut-residing segmented filamentous bacteria drive autoimmune arthritis via T helper 17 cells. Immunity. 2010;32:815–827. doi: 10.1016/j.immuni.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu M, Eisen JA. A simple, fast, and accurate method of phylogenomic inference. Genome Biol. 2008;9:R151. doi: 10.1186/gb-2008-9-10-r151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yokoyama K, Imamura H. Rotation, structure, and classification of prokaryotic V-ATPase. J Bioenerg Biomembr. 2005;37:405–410. doi: 10.1007/s10863-005-9480-1. [DOI] [PubMed] [Google Scholar]
- Yu NY, Wagner JR, Laird MR, Melli G, Rey S, Lo R, Dao P, Sahinalp SC, Ester M, Foster LJ, et al. PSORTb 3.0: improved protein subcellular localization prediction with refined localization subcategories and predictive capabilities for all prokaryotes. Bioinformatics. 2010;26:1608–1615. doi: 10.1093/bioinformatics/btq249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yus E, Maier T, Michalodimitrakis K, van Noort V, Yamada T, Chen WH, Wodke JA, Guell M, Martinez S, Bourgeois R, et al. Impact of genome reduction on bacterial metabolism and its regulation. Science. 2009;326:1263–1268. doi: 10.1126/science.1177263. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.