

Abstract
Recent findings assessing the utility of biomarkers are reviewed that help identify the basis for disease in patients with frontotemporal lobar degeneration (FTLD) spectrum pathology. Biofluid studies identify about 15% of patients with FTLD due to a genetic mutation that is associated with the specific histopathologic features of TDP-43 or a tauopathy. Other genetically-based risk factors and targeted proteomic searches of plasma and cerebrospinal fluid have suggested additional markers that may be useful in sporadic cases of FTLD. While progress has been made in developing biomarkers for FTLD, additional work is needed to extend these advances so that the histopathologic abnormality causing FTLD can be specified in an individual patient.
Keywords: frontotemporal lobar degeneration, biomarker, cerebrospinal fluid, neuroimaging
Treatment trials for frontotemporal lobar degeneration (FTLD) are beginning to emerge. These trials are targeted at the histopathologic abnormality causing FTLD. The goal of these trials is to change the natural history of this disorder. Detailed descriptions have been developed that characterize the spectrum of FTLD pathology(1). One common abnormality is FTLD-TDP. This is due to the accumulation of ubiquitinated and hyperphosphorylated trans-active DNA-binding protein of ~43 kD (TDP-43)(2). A second family of conditions associated with the accumulation of the microtubule-associated protein tau constitutes another common cause of FTLD – although somewhat diverse biochemically, these pathologies are collectively known as FTLD-tau(3). Conditions contributing to the family of tauopathies include dementia with Pick bodies, corticobasal degeneration and progressive supranuclear palsy. A less common histopathologic finding is FTLD-FUS, related to fused-in-sarcoma immunoreactive inclusions(4).
CLINICAL AND IMAGING MARKERS OF DISEASE
Clinical assessment plays an important screening role in the initial identification of patients with a phenotype corresponding to this FTLD spectrum of pathology. This is reasonable because there is some agreement between a clinical FTLD syndrome and an underlying pathological abnormality. Patients with semantic dementia, also known as the semantic variant of primary progressive aphasia (svPPA), often have TDP-43 pathology(5). Patients with progressive non-fluent aphasia, also known as the non-fluent/agrammatic variant of PPA (naPPA), frequently have a tauopathy(6). Other PPA patients have an atypical, aphasic form of Alzheimer’s disease (AD), also known as the logopenic variant of PPA (lvPPA), and this may overlap with PPA due to FTLD spectrum pathology(7). A recent survey of unselected clinical-pathological series of PPA patients confirmed this general picture, but emphasized that the association of a clinical phenotype with histopathology at autopsy is less reliable in individual patients(8). The situation is more confusing for behavioral-variant FTD (bvFTD), where rare studies attempt to distinguish the clinical characteristics of a social disorder related to FTLD-tau or FTLD-TDP(9). These patients may be indistinguishable from cases with a so-called frontal variant of autopsy-proven AD (fvAD)(10).
Quantitative neuropsychological measures of language and cognition in autopsy-proven cases of FTLD provide another approach to identifying the histopathology underlying patients in this clinical spectrum of disease. Patients with a TDP-43 proteinopathy are significantly more impaired than patients with a tauopathy or AD pathology on measures of confrontation naming and category naming fluency guided by the letters F, A, and S(11–13). By comparison, patients with tau-positive pathology are significantly more impaired than patients with a TDP-43 proteinopathy and patients with AD pathology on non-linguistic measures of executive functioning involving visual constructions and reverse digit span(12, 13). Within the PPA spectrum, episodic memory is relatively impaired in lvPPA patients with AD pathology compared to PPA patients with FTLD-tau or FTLD-TDP pathology(3, 14). Despite the value of this approach at a group level, follow-up observations have shown that quantitative neuropsychological measures are unreliable at identifying histopathology in individual patients(15).
Structural and functional imaging studies associate the distinct features of a clinical FTLD syndrome with partially selective interruptions of large-scale networks that are associated with PPA or bvFTD. These anatomic distributions of disease also may reflect a particular histopathologic abnormality.
An example of the anatomic distribution of cortical thinning in the three major PPA syndromes is illustrated in Figure 1(8). naPPA is related largely to anterior peri-Sylvian atrophy involving inferior, opercular, insular and dorsolateral portions of the left frontal lobe(16, 17). Longitudinal imaging studies are rare, but personal observations suggest that atrophy associated with PNFA often extends medially into orbital frontal and anterior cingulate regions, inferiorly into the anterior superior temporal lobe, and posteriorly along the Sylvian fissure into the parietal lobe. svPPA has a different anatomic distribution, causing left anterior temporal atrophy that affects lateral and ventral surfaces as well as the anterior hippocampus and the amygdala(18, 19). Longitudinal studies show atrophy extending in a posterior and superior direction in the ipsilateral temporal lobe, and often involve the adjacent frontal lobe and anterior temporal regions of the right hemisphere(20–22). lvPPA is associated with left posterior-peri-Sylvian atrophy in the area of temporal-parietal junction(16). Longitudinal imaging tends to extend into more inferior regions of the temporal lobe, and anteriorly along the Sylvian fissure into the frontal lobe.
FIGURE 1. CORTICAL ATROPHY CORRESPONDING TO PRIMARY PROGRESSIVE APHASIA SYNDROMES.
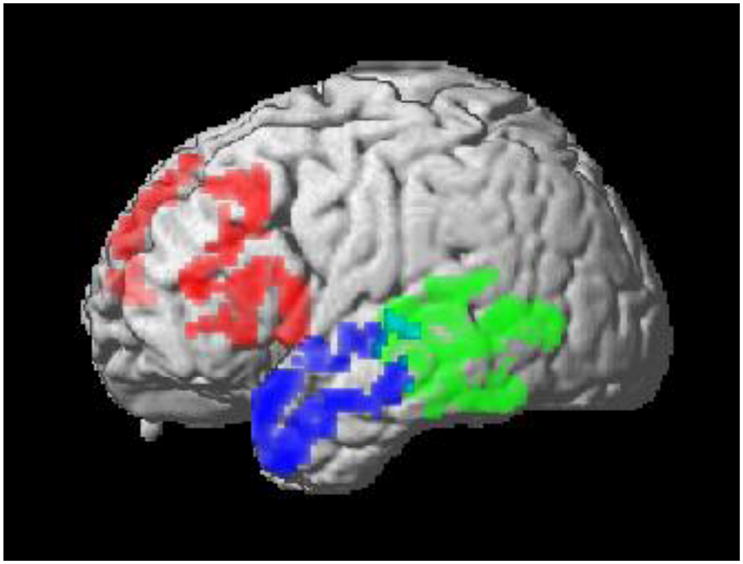
NOTE: 1. Frontal = non-fluent/agrammatic variant PPA; anterior temporal = semantic variant; posterior temporal-parietal = logopenic variant.
Comparative imaging studies in patients with pathologically-confirmed disease are crucial for validating an association between an anatomic distribution of disease and an underlying histopathologic abnormality. However, these studies have been rare. MRI gray matter thinning has been assessed in small groups of naPPA patients with tau-positive disease and svPPA patients with FTLD-U pathology presumably due to TDP-43(23). The naPPA patients showed superior temporal and inferior frontal thinning that was more prominent in the left hemisphere than the right hemisphere, and the svPPA patients showed prominent left anterior temporal thinning.
Another study assessed the validity of MRI structural imaging in 34 non-fluent PPA patients with autopsy- or CSF-based evidence consistent with underlying FTLD or AD pathology(15). Statistically significant differences in the anatomic distribution of cortical atrophy were described in the FTLD and AD subgroups, but a receiver operating characteristic (ROC) curve analysis of cortical atrophy revealed an area under the curve of only 0.64, with 72.7% sensitivity and 66.7% specificity.
Recent work also has demonstrated the utility of diffusion tensor imaging (DTI) to distinguish clinical syndromes on the basis of white matter abnormalities, although DTI studies in patients with autopsy-confirmed disease are rare. One preliminary report evaluated clinically-diagnosed FTLD patients who had autopsy- or CSF- based biomarkers consistent with FTLD or AD pathology(24). Significantly reduced fractional anisotropy (FA) was seen in FTLD compared to AD in the anterior corpus callosum and anterior portions of the inferior longitudinal fasciculus.
Another study combined cortical atrophy and FA changes to classify 50 individual cases of clinically diagnosed FTLD who had autopsy- or CSF-based findings consistent with FTLD or AD pathology(25). A support vector machine technique selected the optimal voxels from T1 and DTI imaging, and applied the resulting algorithm to categorize individual participants. In FTLD, changes were most prominent in frontal and anterior temporal regions, while changes in temporal-parietal brain areas were more evident in AD. The algorithm combining T1 and DTI accurately categorized 49 (98%) of the 50 participants.
Arterial spin labeling (ASL) perfusion MRI is a functional neuroimaging technique that has the potential to be more sensitive than structural MRI imaging because differences do not depend on a late change that involves death in a large population of neurons. Using ASL, abnormal hypoperfusion in frontal regions and hyperperfusion in parietal regions was seen in patients with autopsy- and CSF- defined FTLD(26). Moreover, in contrast to patients with autopsy- and CSF-defined AD, perfusion in FTLD was significantly lower in frontal regions and significantly elevated in temporal-parietal regions.
PET and SPECT functional imaging technologies incorporate tracers such as Pittsburgh Compound B (PiB) that radiolabel beta-amyloid (A)(27). This protein accumulates in AD but not FTLD, and thus may help distinguish between cases of PPA or a social disorder that may be caused by AD rather than FTLD spectrum pathology. One report described reduced PiB uptake in eight of 12 clinically diagnosed FTLD patients(28). A follow-up study noted PiB uptake in all four of the lvPPA patients who were evaluated, and signal was strongest in a left temporal-parietal distribution(29). By comparison, PiB uptake was less common in other PPA syndromes, where left frontal uptake was seen in only one of six naPPA patients and left temporal uptake was seen in only one of five svPPA patients. Unfortunately there was no autopsy confirmation of disease in the handful of patients that have been studied.
BIOFLUID MARKERS OF DISEASE
While clinical phenotype, quantitative neuropsychology and brain imaging provide useful screening information suggesting FTLD spectrum pathology, additional information is needed to define the histopathologic abnormality in an individual patient with PPA or bvFTD. Biomarkers thus must be developed to supplement the clinical examination. A biomarker is a validated measure that reliably reflects the underlying pathology of a disease. This is crucial in FTLD, where a clinical phenotype can be associated with several different pathologies. Some biomarkers are categorical and provide diagnostic information. This class of biomarkers must distinguish between healthy controls and patients. A panel of biomarkers may be needed to identify each of several neurodegenerative diseases. This would confer specificity to support distinguishing between different age-associated neurodegenerative conditions like FTLD-TDP, FTLD-tau and AD. Other biomarkers may reflect disease severity and the course of the condition. This class of biomarkers would be useful in treatment trials to determine whether the disease is responding to the treatment. Several biomarker modalities are potentially available to help determine the basis for FTLD, including blood and serum studies, and studies of analytes in the cerebrospinal fluid (CSF).
Familial disorders are frequent in FTLD. Thus, one important source of information about the pathology causing a FTLD syndrome is DNA found in blood samples. Up to 45% of index cases can have a strongly positive family history(30, 31), and about 15% of FTLD patients have an identifiable mutation. , naPPA and bvFTD may be associated with a mutation of the GRN gene coding progranulin (PGRN) on chromosome 17(32, 33). GRN mutations are reliably associated with FTLD-TDP pathology(34–37). Less common mutations associated with FTLD-TDP pathology include VCP on chromosome 9, CHMP2B on chromosome 3, and TARDBP on chromosome 1. Other familial patients with an FTLD syndrome have a mutation of the microtubule-associated protein tau (MAPT) gene coding tau on chromosome 17. This is associated with tau histopathology(38–40). An important point of caution is that a clinical FTLD syndrome can be highly variable in patients with the identical GRN mutation(41) or members of a family with the same mutation of GRN(42, 43). Likewise, the phenotype associated with a specific MAPT mutation may be highly variable(44).
These genetic factors may be used to help define the risk associated with the pathologic basis for FTLD in sporadic cases. Tau haplotypes are derived from the region of chromosome 17 where the MAPT gene is coded. The H1 haplotype is over-represented in conditions associated with tau pathology such as corticobasal degeneration, progressive supranuclear palsy, and sporadic FTLD(45–47). However, a tau haplotype has been associated directly with tau pathology only rarely(48). A recent genome-wide association study (GWAS) of over 1,000 patients with autopsy-confirmed CBD and PSP has identified several highly significant single nucleotide polymorphisms (SNPs) that are risk factors for FTLD-tau. Some are associated with the MAPT region coding tau at 17q21, including rs8070723 and rs242557(49). Other highly significant SNPs include rs1411478 at chromosome 1q25.3 coding for STX6, rs1768208 coding for MOBP at chromosome 3p22.1, and rs7299371 coding for NELL2 at chromosome 12q12. Each of these SNPs is associated with the biology of these tauopathies: For example, MOBP appears to be strongly associated with brainstem white matter pathology in PSP.
Similar work has been pursued in identifying markers of TDP-43 pathology. Homozygosity for the T allele at rs5848 in the region coding GRN on chromosome 17 appears to be an important risk marker for FTLD-TDP pathology(50). A risk biomarker associated with TDP-43 pathology was derived from a GWAS of >500 autopsy-proven cases with FTLD-TDP pathology. An increased risk for the presence of TDP-43 pathology was associated with TMEM106B, coded on chromosome 7p21. The proportions of FTLD patients (46%) and AD patients (22%) with elevated plasma levels of TDP-43 correspond roughly to the percentages of these groups with TDP-43 detected in the brains at autopsy(51). Plasma levels of phosphorylated TDP-43 are correlated with the density of FTLD-TDP brain pathology, but plasma levels of TDP-43 do not distinguish between patients with FTLD and AD(52).
Cerebrospinal fluid (CSF) is another potential source of biomarker information. About 20% of individual patients with FTLD had significantly low levels of CSF tau relative to healthy controls, although this was never seen in patients with AD(53). In patients with known pathology, the tau:Aβ1–42 ratio was significantly lower in FTLD than AD. A ROC curve analysis showed that a ratio of 1.06 had excellent sensitivity and specificity for distinguishing FTLD from AD(54), although this was based on an ELISA analysis of CSF that has considerably greater variability than recent measurement techniques involving a Luminex platform(55).
TDP-43 levels have been assayed in the CSF of patients with a clinical diagnosis of FTLD. Analyses also have been performed in ALS, where over 95% of patients with sporadic disease have TDP-43 pathology(56, 57). Significantly elevated CSF levels of TDP-43 have been found. However, the participants in these studies did not have autopsy-proven disease, and TDP-43 levels of patients overlapped with those of controls.
A targeted proteomic search has identified a panel of novel CSF biomarkers that distinguishes between autopsy-confirmed cases of AD and a group of non-AD patients where 92% had FTLD spectrum pathology(58). Phosphorylated-tau181, Aβ1–42, and agouti-related peptide (AgRP) were found to be useful classifying analytes. A follow-up study examined the CSF of autopsy-confirmed cases with FTLD-TDP and FTLD-tau pathology(59). In addition to AgRP, four analytes were identified as distinguishing biomarkers, including adrenocorticotropic hormone (ACTH), eotaxin-3, Fas, and interleukin 17 (IL-17). Together, these analytes distinguished between FTLD-tau and FTLD-TDP with reasonable sensitivity and specificity.
Once screened by clinical and imaging markers of disease, recent advances suggest that biofluid biomarkers are likely to be useful in the identification of the specific histopathologic abnormality causing a FTLD clinical syndrome. A two-stage diagnostic approach thus may help identify the histopathology abnormality in FTLD. First, imaging studies appear to be useful at distinguishing FTLD from patients with AD pathology. Based on a family history, biomarkers in blood then can be obtained to identify patients with a genetic mutation that is associated with FTLD-tau or FTLD-TDP. Unfortunately, this represents only about 15% of patients with a FTLD clinical syndrome. Studies of CSF and plasma are becoming increasingly useful at identifying a specific histopathologic abnormality in the remaining patients with a sporadic FTLD syndrome. Although not yet definitive, these findings suggest that will be able to identify patients who are eligible for trials targeted a specific histopathologic abnormality causing FTLD.
Acknowledgments
This report was supported in part by funding from NIH (AG15116, AG17586, NS44266, NS53488, AG32953)
References
- 1.Mackenzie IR, Neumann M, Bigio E, Cairns NJ, Alafuzoff I, Kril JJ, et al. Nomenclature for neuropathologic subtypes of frontotemporal lobar degeneration: consensus recommendations. Acta Neuropathologica. 2009;117(1):15–8. doi: 10.1007/s00401-008-0460-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neumann M, Sampathu DM, Kwong LK, Truax AC, Micseny MC, Chou TT, et al. Ubiquinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclereosis. Science. 2006;314:130–3. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- 3.Forman MS, Farmer J, Johnson JK, Clark CM, Arnold SE, Coslett HB, et al. Frontotemporal dementia: Clinicopathological correlations. Annals of Neurology. 2006;59:952–62. doi: 10.1002/ana.20873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Neumann M, Rademakers R, Roeber S, Baker M, Kretzschmar HA, Mackenzie IRA. Frontotemporal lobar degeneration with FUS pathology. Brain. 2009 August 11;:2009:awp214. doi: 10.1093/brain/awp214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Snowden J, Neary D, Mann D. Frontotemporal lobar degeneration: clinical and pathological relationships. Acta Neuropathologica. 2007;114(1):31–8. doi: 10.1007/s00401-007-0236-3. [DOI] [PubMed] [Google Scholar]
- 6.Josephs KA, Duffy JR, Strand EA, Whitwell JL, Layton KF, Parisi JE, et al. Clinicopathological and imaging correlates of progressive aphasia and apraxia of speech. Brain. 2006 Jun 01;129:1385–98. doi: 10.1093/brain/awl078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mesulam M, Wicklund A, Johnson N, Rogalski E, Leger GC, Rademaker A, et al. Alzheimer and frontotemporal pathology in subsets of primary progressive aphasia. Annals of Neurology. 2008;63(6):709–19. doi: 10.1002/ana.21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grossman M. Primary progressive aphasia: Clinical-pathological correlations. Nature Reviews Neurology. 2010;6:88–97. doi: 10.1038/nrneurol.2009.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hu WT, Mandrekar JN, Parisi JE, Knopman DS, Boeve BF, Petersen RC, et al. Clinical Features of Pathologic Subtypes of Behavioral-Variant Frontotemporal Dementia. Arch Neurol. 2007 November 1;64(11):1611–6. doi: 10.1001/archneur.64.11.1611. [DOI] [PubMed] [Google Scholar]
- 10.Johnson JK, Head E, Kim R, Starr A, Cotman CW. Clinical and pathological evidence for a frontal variant of Alzheimer disease. Archives of Neurology. 1999;56:1233–9. doi: 10.1001/archneur.56.10.1233. [DOI] [PubMed] [Google Scholar]
- 11.Josephs KA, Whitwell JL, Duffy JR, Vanvoorst WA, Strand EA, Hu WT, et al. Progressive aphasia secondary to Alzheimer disease vs FTLD pathology. Neurology. 2008 January 1;70(1):25–34. doi: 10.1212/01.wnl.0000287073.12737.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grossman M, Xie SX, Libon DJ, Wang X, Massimo L, Moore P, et al. Longitudinal decline in autopsy-defined frontotemporal lobar degeneration. Neurology. 2008 May 27;70(22):2036–45. doi: 10.1212/01.wnl.0000303816.25065.bc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Grossman M, Libon DJ, Forman MS, Massimo L, Wood E, Moore P, et al. Distinct antemortem profiles in patients With pathologically defined frontotemporal dementia. Arch Neurol. 2007 November 1;64(11):1601–9. doi: 10.1001/archneur.64.11.1601. [DOI] [PubMed] [Google Scholar]
- 14.Rascovsky K, Salmon DP, Ho GJ, Galasko D, Peavy GM, Hansen LA, et al. Cognitive profiles differ in autopsy-confirmed frontotemporal dementia and AD. Neurology. 2002;58:1801–8. doi: 10.1212/wnl.58.12.1801. [DOI] [PubMed] [Google Scholar]
- 15.Hu WT, McMillan C, Libon DJ, Leight S, Forman MS, Lee VM-Y, et al. Multi-modal predictors for Alzheimer’s disease in non-fluent primary progressive aphasia. Neurology. 2010 doi: 10.1212/WNL.0b013e3181ed9c52. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gorno-Tempini M, Dronkers NF, Rankin KP, Ogar JM, Phengrasamy L, Rosen HJ, et al. Cognition and anatomy in three variants of primary progressive aphasia. Annals of Neurology. 2004;55:335–46. doi: 10.1002/ana.10825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Peelle JE, Troiani V, Gee JC, Moore P, McMillan C, Vesely L, et al. Sentence comprehension and voxel-based morphometry in progressive nonfluent aphasia, semantic dementia, and nonaphasic frontotemporal dementia. Journal of Neurolinguistics. 2008;21:418–32. doi: 10.1016/j.jneuroling.2008.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mummery CJ, Patterson K, Price CJ, Hodges JR. A voxel-based morphometry study of semantic dementia: Relationship between temporal lobe atrophy and semantic memory. Annals of Neurology. 2000;47:36–45. [PubMed] [Google Scholar]
- 19.Bonner MF, Vesely L, Price C, Powers C, Richmond L, Farag C, et al. Reversal of the concreteness effect in semantic dementia. Cognitive Neuropsychology. 2009;26:568–79. doi: 10.1080/02643290903512305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Avants B, Anderson C, Grossman M, Gee JC. Spatiotemporal normalization for longitudinal analysis of gray matter atrophy in frontotemporal dementia. Medical Image Computing and Computer-Assisted Intervention: MICCAI. 2007;10:309–10. doi: 10.1007/978-3-540-75759-7_37. [DOI] [PubMed] [Google Scholar]
- 21.Rohrer JD, McNaught E, Foster J, Clegg SL, Barnes J, Omar R, et al. Tracking progression in frontotemporal lobar degeneration: Serial MRI in semantic dementia. Neurology. 2008 October 28;71(18):1445–51. doi: 10.1212/01.wnl.0000327889.13734.cd. [DOI] [PubMed] [Google Scholar]
- 22.Brambati SM, Rankin KP, Narvid J, Seeley WW, Dean D, Rosen HJ, et al. Atrophy progression in semantic dementia with asymmetric temporal involvement: A tensor-based morphometry study. Neurobiology of Aging. 2009;30(1):103–11. doi: 10.1016/j.neurobiolaging.2007.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rohrer JD, Warren JD, Modat M, Ridgway GR, Douiri A, Rossor MN, et al. Patterns of cortical thinning in the language variants of frontotemporal lobar degeneration. Neurology. 2009 May 5;72(18):1562–9. doi: 10.1212/WNL.0b013e3181a4124e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hu WT, Zhang H, McMillan C, Lee VMY, Trojanowski JQ. Distinct white matter changes in frontotemporal dementia associated with FTLD and Alzheimer’s pathology or CSF biomarkers. Neurology. 2010;74:A569. [Google Scholar]
- 25.Avants BB, Cook PA, Ungar L, Gee JC, Grossman M. Dementia induces correlated reductions in white matter integrity and cortical thickness: A multivariate neuroimaging study with sparse canonical correlation analysis. Neuroimage. 2010;50(3):1004–16. doi: 10.1016/j.neuroimage.2010.01.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hu WT, Wang Z, Lee VMY, Trojanowski JQ, Detre J, Grossman M. Distinct cerebral perfusion patterns in FTLD and AD. Neurology. 2010 doi: 10.1212/WNL.0b013e3181f11e35. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Klunk W, Engler H, Nordberg A, Wang Y, Blomqvist G, Holt DP, et al. Imaging brain amyloid in Alzheimer’s disease with Pittsburgh Compound-B. Annals of Neurology. 2004;55:306–19. doi: 10.1002/ana.20009. [DOI] [PubMed] [Google Scholar]
- 28.Rabinovici GD, Furst AJ, O’Neil JP, Racine CA, Mormino EC, Baker SL, et al. 11C-PIB PET imaging in Alzheimer disease and frontotemporal lobar degeneration. Neurology. 2007 Apr 10;68:1205–12. doi: 10.1212/01.wnl.0000259035.98480.ed. [DOI] [PubMed] [Google Scholar]
- 29.Gil D. Rabinovici WJJAJFJMOCARECMJPONRALNF. Abeta amyloid and glucose metabolism in three variants of primary progressive aphasia. Annals of Neurology. 2008;64(4):388–401. doi: 10.1002/ana.21451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chow TW, Miller BL, Hayashi VN, Geschwind DH. Inheritance of Frontotemporal Dementia. Archives of Neurology. 1999 Jul 01;56:817–22. doi: 10.1001/archneur.56.7.817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seelaar H, Kamphorst W, Rosso SM, Azmani A, Masdjedi R, de Koning I, et al. Distinct genetic forms of frontotemporal dementia. Neurology. 2008;71:1220–6. doi: 10.1212/01.wnl.0000319702.37497.72. [DOI] [PubMed] [Google Scholar]
- 32.Snowden JS, Pickering-Brown SM, Mackenzie IR, Richardson AMT, Varma A, Neary D, et al. Progranulin gene mutations associated with frontotemporal dementia and progressive non-fluent aphasia. Brain. 2006 November 1;129(11):3091–102. doi: 10.1093/brain/awl267. [DOI] [PubMed] [Google Scholar]
- 33.Mesulam M, Johnson N, Krefft TA, Gass JM, Cannon AD, Adamson JL, et al. Progranulin mutations in primary progressive aphasia: The PPA1 and PPA3 families. Archives of Neurology. 2007 Jan 01;64:43–7. doi: 10.1001/archneur.64.1.43. [DOI] [PubMed] [Google Scholar]
- 34.Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006 Aug 24;442:916–9. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- 35.Davidson Y, Kellley T, Mackenzie IR, Pickering-Brown S, Du Plessis D, Neary D, et al. Ubiquitinated pathological lesions in frontotemporal lobar degeneration contain the TAR DNA-binding protein, TDP-43. Acta Neuropathologica. 2007;113 doi: 10.1007/s00401-006-0189-y. [DOI] [PubMed] [Google Scholar]
- 36.Mackenzie I. The neuropathology and clinical phenotype of FTD with progranulin mutations. Acta Neuropathologica. 2007;114(1):49–54. doi: 10.1007/s00401-007-0223-8. [DOI] [PubMed] [Google Scholar]
- 37.Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Human Molecular Genetics. 2006 Oct 15;15:2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- 38.Hong M, Zhukareva V, Vogelsberg-Ragaglia V, zolek Z, ed LA, Miller BL, et al. Mutation-specific functional impairments in distinct tau isoforms of hereditary FTDP-17. Science. 1998;282:1914–7. doi: 10.1126/science.282.5395.1914. [DOI] [PubMed] [Google Scholar]
- 39.Lee VMY, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annual Review of Neuroscience. 2001;24:1121–59. doi: 10.1146/annurev.neuro.24.1.1121. [DOI] [PubMed] [Google Scholar]
- 40.Zhukareva V, Vogelsberg-Ragaglia V, Van Deerlin VMD, Bruce J, Shuck T, Grossman M, et al. Loss of brain tau defines novel sporadic and familial tauopathies with frontotemporal dementia. Annals of Neurology. 2001;49:165–75. doi: 10.1002/1531-8249(20010201)49:2<165::aid-ana36>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 41.Rademakers R, Baker M, Gass J, Adamson J, Huey ED, Momeni P, et al. Phenotypic variability associated with progranulin haploinsufficiency in patients with the common 1477COT (Arg493X) mutation: an international initiative. Lancet Neurology. 2007;6:857–68. doi: 10.1016/S1474-4422(07)70221-1. [DOI] [PubMed] [Google Scholar]
- 42.Le Ber I, Camuzat A, Hannequin D, Pasquier F, Guedj E, Rovelet-Lecrux A, et al. Phenotype variability in progranulin mutation carriers: a clinical, neuropsychological, imaging and genetic study. Brain. 2008 March 1;131(3):732–46. doi: 10.1093/brain/awn012. [DOI] [PubMed] [Google Scholar]
- 43.Leverenz JB, Yu CE, Montine TJ, Steinbart E, Bekris LM, Zabetian C, et al. A novel progranulin mutation associated with variable clinical presentation and tau, TDP43 and alpha-synuclein pathology. Brain. 2007 May 1;130(5):1360–74. doi: 10.1093/brain/awm069. [DOI] [PubMed] [Google Scholar]
- 44.Bird TD, Nochlin D, Poorkaj P, Cherrier M, Kaye J, Payami H, et al. A clinical pathological comparison of three families with frontotemporal dementia and identical mutations in the tau gene (P301L) Brain. 1999 April 1;122(4):741–56. doi: 10.1093/brain/122.4.741. [DOI] [PubMed] [Google Scholar]
- 45.Houlden H, Baker M, Morris HR, MacDonald N, Pickering-Brown S, Adamson J, et al. Corticobasal degeneration and progressive supranuclear palsy share a common tau haplotype. Neurology. 2001 Jun 26;56:1702–6. doi: 10.1212/wnl.56.12.1702. [DOI] [PubMed] [Google Scholar]
- 46.Hughes A, Mann DMA, Pickering-Brown S. Tau haplotype frequency in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Experimental Neurology. 2003;181:12–6. doi: 10.1016/s0014-4886(03)00024-4. [DOI] [PubMed] [Google Scholar]
- 47.Pittman AM, Myers AJ, Duckworth J, Bryden L, Hanson M, Abou-Sleiman P, et al. The structure of the tau haplotype in controls and in progressive supranuclear palsy. Hum Mol Genet. 2004 June 15;13(12):1267–74. doi: 10.1093/hmg/ddh138. [DOI] [PubMed] [Google Scholar]
- 48.Morris HR, Baker M, Yasojima K, Houlden H, Khan MN, Wood NW, et al. Analysis of tau haplotypes in Pick’s disease. Neurology. 2002 August 13;59(3):443–5. doi: 10.1212/wnl.59.3.443. [DOI] [PubMed] [Google Scholar]
- 49.Schellenberg GD. A genome-wide association study of progressive supranuclear palsy and corticobasal degeneration: Genes that modify risk. Dementia and Geriatric Cognitive Disorders. 2010;30:18–9. [Google Scholar]
- 50.Rademakers R, Eriksen JL, Baker M, Robinson T, Ahmed Z, Lincoln SJ, et al. Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Human Molecular Genetics. 2008 December 1;17(23):3631–42. doi: 10.1093/hmg/ddn257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Foulds P, McAuley E, Gibbons L, Davidson Y, Pickering-Brown S, Neary D, et al. TDP-43 protein in plasma may index TDP-43 brain pathology in Alzheimer’s disease and frontotemporal lobar degeneration. Acta Neuropathologica. 2008;116(2):141–6. doi: 10.1007/s00401-008-0389-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Foulds P, Davidson Y, Mishra M, Hobson DJ, Humphreys KM, Taylor M, et al. Plasma phosphorylated-TDP-43 protein levels correlate with brain pathology in frontotemporal lobar degeneration. Acta Neuropathologica. 2009;118:647–58. doi: 10.1007/s00401-009-0594-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grossman M, Farmer J, Leight S, Work M, Moore P, Van Deerlin VMD, et al. Cerebrospinal fluid profile distinguishes frontotemporal dementia from Alzheimer’s disease. Annals of Neurology. 2005;57:721–9. doi: 10.1002/ana.20477. [DOI] [PubMed] [Google Scholar]
- 54.Bian H, van Sweiten JC, Leight S, Massimo L, Wood E, Forman MS, et al. Cerebrospinal fluid biomarkers in frontotemporal lobar degeneration with known pathology. Neurology. 2008;70:1827–35. doi: 10.1212/01.wnl.0000311445.21321.fc. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shaw LM, Korecka M, Clark CM, Lee VMY, Trojanowski JQ. Biomarkers of neurodegeneration for diagnosis and monitoring therapeutics. Nature Reviews: Drug Discovery. 2007;6:295–303. doi: 10.1038/nrd2176. [DOI] [PubMed] [Google Scholar]
- 56.Steinacker P, Hendrich C, Sperfeld AD, Jesse S, von Arnim CAF, Lehnert S, et al. TDP-43 in cerebrospinal fluid of patients With frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Archives of Neurology. 2008 November 1;65(11):1481–7. doi: 10.1001/archneur.65.11.1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kasai T, Tokuda T, Ishigami N, Sasayama H, Foulds P, Mitchell D, et al. Increased TDP-43 protein in cerebrospinal fluid of patients with amyotrophic lateral sclerosis. Acta Neuropathologica. 2009;117(1):55–62. doi: 10.1007/s00401-008-0456-1. [DOI] [PubMed] [Google Scholar]
- 58.Hu WT, Chen-Plotkin A, Arnold SE, Grossman M, Clark CM, Shaw LM, et al. Novel CSF biomarkers for Alzheimer’s disease and mild cognitive impairment. Acta neuropathologica. 2010 Mar 16; doi: 10.1007/s00401-010-0667-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hu WT, Chen-Plotkin A, Grossman M, Arnold SE, Clark CM, Shaw LM, et al. Novel CSF biomarkers for frontotemporal lobar degenerations. Neurology. 2010 doi: 10.1212/WNL.0b013e318200d78d. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]