

Abstract
Backbone N-methylation is common among peptide natural products and has a significant impact on both the physical properties and the conformational states of cyclic peptides. However, the specific impact of N-methylation on passive membrane diffusion in cyclic peptides has not been investigated systematically. Here we report a method for the selective, on-resin N-methylation of cyclic peptides to generate compounds with drug-like membrane permeability and oral bioavailability. The selectivity and degree of N-methylation of the cyclic peptide was determined by backbone stereochemistry, suggesting that conformation dictates the regiochemistry of the N-methylation reaction. The permeabilities of the N-methyl variants were corroborated by computational studies on a 1024-member virtual library of N-methyl cyclic peptides. One of the most permeable compounds, a cyclic hexapeptide (MW = 755) with three N-methyl groups, showed an oral bioavailability of 28% in rat.
Introduction
Macrocyclic compounds have gained increasing attention as an untapped structural class in drug lead discovery programs1,2. Because of their size and relative complexity, macrocycles can show greater specificity and potency for biological targets compared to smaller acyclic compounds3 and may provide useful scaffolds for modulating more challenging biological targets such as protein-protein interactions or biomolecules that lack well-defined small molecule binding sites. Although there are over 100 macrocyclic drugs in clinical use today (e.g., cyclosporine A (CSA), romidepsin, rapamycin, vancomycin, and rifampacin), macrocycles as a class are underrepresented in commercial screening libraries. Many of these drugs are based on natural and synthetic cyclic peptide scaffolds1, which are generally larger and more complex than traditional synthetic small molecule drugs. Cyclic peptides tend to have molecular weights and polar group counts that place them outside most classical definitions of “drug-likeness”, challenging us to devise new rules for predicting pharmacokinetic properties (e.g., oral absorption) for this class of compounds.
Backbone N-methylation is a hallmark of cyclic peptide natural products, and seems to be correlated with increased membrane permeability in this class of compounds. Indeed, N-methylation has been shown to improve oral bioavailability while maintaining or even enhancing the bioactivity and selectivity of the modified scaffold4–6. In a recent study of a series of cyclic hexapeptides, Ovadia et al.7 demonstrate that both the number of N-methyl groups and, more importantly, their relative positions along the backbone, can have a dramatic effect on cell permeability, although they did not explicitly investigate the role of conformation in determining permeability.
The effect of N-methylation on the biological activity of cyclic peptides has also been examined, notably in the cases of somatostatin4, Arg-Gly-Asp (RGD)8, and the melanocortin5 family of peptides. The observation that most bioactive cyclic peptide natural products are N-methylated suggests that N-methylation may facilitate membrane permeability in these compounds; however, the relationship between N-methylation, conformation, and passive membrane diffusion in cyclic peptides has not been investigated systematically.
Crystal structures of N-methylated cyclic peptides (CSA9, ABA10, and the destruxins11) suggest that N-methylation may serve to increase membrane permeability by allowing (or possibly stabilizing) intramolecular hydrogen bonding in the membrane-associated state (Figure 1a). This hypothesis is consistent with previous reports studying the effect of conformation on membrane permeability in cyclic peptides12–16 and non-peptides17 in which intramolecular hydrogen bonding is a key determinant of passive membrane diffusion in flexible molecules containing hydrogen bond donors and acceptors. Based on this conformational hypothesis, we investigated whether intramolecular hydrogen bonding could be used to direct the N-methylation of a cyclic peptide scaffold on the solid phase (Figure 1b), and whether selective N-methylation could be used to improve the passive membrane permeability of the parent scaffolds. We reasoned that the same conformational determinants that influence permeability in non-methylated cyclic peptides could be employed to impart selectivity by directing N-methylation to the most exposed N-H groups. Moreover, we postulated that the resulting N-methyl derivatives would exhibit improved membrane permeability relative to the non-methylated parent compounds.
Figure 1.

a) Schematic representation of the conformational hypothesis of membrane permeability applied to N-methyl cyclic peptides. b) General strategy for on-resin N-methylation of cyclic peptides to generate scaffolds with improved permeability.
Here we report the use of cyclic hexapeptide scaffolds to investigate the interplay between N-methylation, conformation, and permeability, and we report a novel synthetic method for obtaining cyclic peptides with pharmacokinetic properties that rival those of typical small molecule drugs. We found that certain partially N-methylated cyclic peptides were more permeable than the corresponding fully N-methylated species, and that, consistent with detailed computational predictions, the permeabilities of cyclic peptides with the same number of N-methyl groups varied widely. In general, the relative permeabilities predicted by our model are borne out in in vitro and in vivo pharmacokinetics studies.
Results
We selected the cyclic hexapeptide sequence cyclo[Leu, Leu, Leu, Leu, Pro, Tyr] as a test scaffold to screen for conditions that might afford regioselective N-methylation on the solid phase. A single Pro residue is known to promote efficient cyclization, even in all-L hexapeptides18, and similar cyclic hexapeptides are known to adopt different conformations depending on the arrangement of D- and L-residues in the sequence12. We synthesized the linear peptides by linking the Tyr side chain to the resin with C-terminal protection as the allyl ester, followed by deprotection and cyclization on the solid phase13. After testing a variety of diastereomers under a range of N-methylation conditions, we identified a sequence, cyclo[Leu, D-Leu, Leu, Leu, D-Pro, Tyr] (1), that gave excellent selectivity in the on-resin N-methylation reaction. Thus, when we treated the resin-bound cyclic peptide with excess LiOtBu in THF, followed by excess CH3I in DMSO, we observed nearly complete conversion to a single trimethylated species (Table S4 entry 9). These conditions had been reported previously for the on-resin peralkylation of linear peptides19, 20, suggesting that the selectivity we observed was due to the cyclic nature of the starting peptide. A more detailed evaluation of reaction conditions for 1 is described in the Supplemental Information.
N-methylation regiochemistry
Since α-carbon stereochemistry can have a significant impact on conformation in cyclic peptides21, we examined the effect of stereochemistry on the selectivity of N-methylation by performing the methylation reaction on 32 stereoisomers based on the sequence cyclo[D/LLeu-D/LLeu-D/LLeu-D/LLeu-D/LPro-LTyr] (Figure 2). Among the 29 diastereomers that cyclized efficiently on the solid phase, 6 sequences yielded specific N-methyl adducts with >95% selectivity when treated with the same on-resin N-methylation conditions described above for 1 (Figure 2, arrows). Three of these sequences yielded a trimethyl product (scaffolds 3, 19, and 27), and three yielded a tetramethyl product (scaffolds 2, 18, and 26). The remaining sequences yielded permethylated product (2 sequences), returned starting material (4 sequences), or gave mixtures of varying complexity (17 sequences).
Figure 2.

a) Diastereomer library screened for N-methylation selectivity. Arrows represent sequences that showed >95% selectivity for a single partially methylated species; all other sequences showed no reaction, permethylation, or mixtures of N-methyl products of varying complexity. Scaffolds 18 and 19 correspond to compounds 2 and 1, respectively. b) Consensus sequences for trimethyl- and tetramethyl-selective compounds.
A pattern emerged in the relationship between stereochemistry and selectivity among the 6 highly selective scaffolds. The three trimethylated scaffolds were identical in four contiguous residues (Tyr6-Leu1-Leu2-Leu3) with the consensus sequence cyclo[LLeu-DLeu-LLeu-D/LLeu-D/LPro-LTyr] (Figure 2b). The three tetramethylated scaffolds were identical at the same four contiguous residues, but shared a different consensus sequence: cyclo[DLeu-LLeu-LLeu-D/LLeu-D/LPro-LTyr]. We chose two of the selective, partially N-methylated products for further study: the trimethyl variant of scaffold 19 (3) and the tetramethyl variant of scaffold 18 (4).
Using 2D NMR techniques (HMBC, HMQC, TOCSY, COSY and NOESY), we determined the pattern of N-methylation for 1 and 2 (Figure 3a). Compound 1 yielded exclusively the trimethyl adduct shown in Figure 3a (3), in which residues D-Leu2, Leu3 and Tyr6 were N-methylated. Diastereomer 2 yielded a single tetramethyl adduct (4), in which residues D-Leu1, Leu2, Leu4, and Tyr6 were N-methylated, (Figure 3a). Thus, the transposition of only two stereocenters between 1 and 2 (Leu1 and Leu2) led to a complete shift in both the pattern and degree of N-methylation, highlighting the impact that small stereochemical differences can have on the global conformations of small cyclic peptides.
Figure 3.

a) Reaction sequence in the on-resin N-methylation of 1 and 2, and the resulting pattern of alkylation to generate 3 and 4, respectively. H/D exchange studies on 1 and 3 (b) along with 2 and 4 (c). Amide N-H resonances were monitored as a function of time after addition of 5% MeOH-d4/0.5% HOAc-d4.
H/D exchange studies
To test the hypothesis that intramolecular hydrogen bonding exerts regiochemical control in the N-methylation reaction by protecting certain amide N-H groups from alkylation (Figure 1), we performed H/D exchange studies on 1, 2, and their N-methylated derivatives, 3 and 4 (Figures 3b and 3c). H/D exchange experiments showed that, while all of the amide protons in 1 exchanged within minutes or hours, the two nonmethylated amides in 3, Leu1 and Leu4, exchanged much more slowly (over a period of several days; Figure 3b). Likewise with 2, the nonmethylated precursor showed an H/D exchange profile similar to that of 1, in which most of the amide N-H groups exchanged with solvent over a few hours. By contrast, the remaining amide N-H in 4 showed negligible exchange over 24 h (Figure 3c). Thus, the free amide N-H groups in both 3 and 4 were either protected sterically from solvent or were involved in highly stable intramolecular hydrogen bonds in CDCl3. In addition, N-methylation appeared to stabilize these hydrogen bonds relative to the nonmethylated precursors, since the amides that were protected from exchange in the methylated species were among ones that exchanged rapidly in the nonmethylated precursors.
Passive membrane permeability studies
We tested the passive membrane diffusion rates of 1 and 2, their selectively N-methylation products, 3 and 4, and their permethylated derivatives, 5 and 6, in a parallel artificial membrane permeability assay (PAMPA)22. PAMPA distinguishes passive membrane diffusion behavior from active transport mechanisms23, and the resulting permeability values correlate well with in vivo absorption rates24 for drugs that diffuse by transcellular mechanisms.
For both scaffolds, the partially methylated derivatives 3 and 4 were significantly more permeable than their nonmethylated precursors (Figure 4a). Indeed, the PAMPA permeabilities of 3 and 4 were comparable to that of the orally bioavailable drug propranolol. Interestingly, both permethylated derivatives, 5 and 6, were significantly less permeable by PAMPA than their partially methylated derivatives, with permeabilitiesas as low as those of the nonmethylated precursors. This result suggested that each scaffold has an optimal number of N-methyl groups with respect to membrane permeability, and that, at least for some small, conformationally constrained cyclic peptides, membrane permeability is not simply proportional to the number of N-methyl groups in the backbone.
Figure 4.

(a) Passive membrane permeabilities of compounds based on diastereomer 1 (white bars), diastereomer 2 (hashed bars), and the orally bioavailable positive control propranolol (grey bar) determined using the parallel artificial membrane permeability assay (PAMPA). %T calculated as the percentage of analyte entering the acceptor compartment after 16 h, where 100% T corresponds to equilibrium between donor and acceptor compartments. (b) Schematic structure of 7, an isomer of 3 with a different pattern of N-methylation, and a stereoview of its predicted membrane-associated conformation. (c) Table showing H-D exchange data for compounds 3 and 7.
To further test the hypothesis that conformation is the main determinant of membrane permeability in 3 and 4, we synthesized43 a regioisomer of 3 to generate 7 (Figure 4b), and investigated its passive membrane diffusion rate by PAMPA. Although 7 differs from 3 by the placement of a single N-methyl group, computational studies (see below) predict that 7 is significantly less permeable due to its inability to form internal hydrogen bonds in a low dielectric environment. In contrast to the compact β-turn structure of 3, the predicted membrane-associated conformation of 7 has an irregular geometry in which one of the three N-methyl groups is in the cis amide configuration and both free amide N-H groups are exposed to solvent (Figure 4b).
Solution NMR studies
As predicted from H/D exchange data, the NMR solution structures of 3 and 4 in CDCl3 showed intramolecular hydrogen bonds between the nonmethylated amide N-H groups and backbone carbonyls (Figures 3a and S3a). For 3, the Leu1 and Leu4 residues each form two transannular hydrogen bonds with each other and are flanked by two β–turns. For 4, a single hydrogen bond between the backbone carbonyl of D-Pro5 and Leu2 generates two opposing nonclassical turns, resulting in a twisted geometry (Figures 3a and S3b). For both 3 and 4, all of the N-methyl amides are in the trans configuration, and the N-methyl groups point away from the ring into solution, consistent with the hypothesis proposed in Figure 1a.
Effect of side chain substitution on N-methylation
A series of cyclic peptides based on compound 1 were synthesized in which Leu1, Leu3 and Leu4 were replaced with various residues of differing size, functionality, and side chain protection (Table S5). The selectivity observed for 1 in the N-methylation reaction was also observed for Leu-to-X substitutions, where X = Ala, Asp, Glu, Phe, Ser, Thr, and Tyr. No other N-methylation products were observed for these compounds. Not surprisingly, compounds that bore a carbamate or amide NH in the protected side chain (e.g., Asn, Gln, or Lys) became tetramethylated (data not shown). We performed a full 2D NMR analysis of the Leu4-to-Ser substitution (1-L4S), and found that the N-methylation pattern was the same as that of 3, indicating that the regiochemistry was preserved (see Supplemental Information). For other amino acids such as histidine and methionine, the on-resin N-methylation resulted in product mixtures of varying complexity. However, for side chains that were protected with appropriate groups such as esters and ethers, the N-methylation selectivity was preserved, indicating that the backbone was the driving force behind the N-methylation selectivity.
Computational studies
We used computational simulations to further examine the relationship between N-methylation and permeability in the cyclic peptide series cyclo[Leu-Leu-Leu-Leu-Pro-Tyr]. In previous studies, we successfully predicted the solution structures of two cyclic peptides in CDCl312 using a computational method originally developed for predicting protein loop conformations25. For N-methyl cyclic peptides, we modified our model to take into account the additional steric constraints and changes in solvation energy imposed by the N-methyl group (Figure S1).
We then applied this method to calculate the free energy cost of desolvation (i.e., the energy cost of transferring from water into the membrane) for 1, 2, and the 32 possible N-methyl variants of each diastereomer (Figure 5a, b). Of the 32 N-methyl variants based on scaffold 1, the most permeable variant predicted using the computational model was 3, the major product of the on-resin methylation chemistry. For scaffold 2, the major N-methylation product, 4, was the second-most permeable N-methyl variant predicted computationally, and the most permeable among its 4 tetramethylated regioisomers. The computations predict that the free energy cost of desolvation for 5 is greater than that for 3, in accord with the experimental results from the PAMPA studies, while 6 and 4 are predicted to have roughly the same desolvation energies. Upon closer inspection of the membrane-associated conformations predicted for the partially and fully N-methylated version of 1, the solvent exposure of backbone carbonyls in the permethylated compound likely accounts for its higher desolvation energy compared with 3. Like amide N-H groups, carbonyl oxygens also form favorable interactions with water that must be overcome upon membrane insertion. Since these carbonyls cannot participate in internal hydrogen bonds, they are consequently more hydrophilic than their hydrogen-bonded analogs. Our calculations suggest that the experimental results may be generalized: partially N-methylated cyclic peptides are more permeable than their permethylated analogs as long as the pattern of N-methylation favors a geometry in which the remaining free NH groups can either participate in internal hydrogen bonds or otherwise be sterically protected from solvent. We postulate that the selective on-resin N-methylation conditions discussed above, in particular the use of a low-dielectric solvent (THF) in the deprotonation step, tend to favor the formation of these highly permeable products.
Figure 5.

Predicted free energies of desolvation of all N-methyl variants for scaffolds 1 (a) and 2 (b). Compounds 3 and 4 are indicated with red circles. Compound 7 is indicated with a blue circle. c) Predicted free energies of desolvation of all N-methyl variants for all diastereomers based on the sequence cyclo[D/L-Leu; D/L-Leu; D/L-Leu; D/L-Leu; D/L-Pro-LTyr]. d) Boxplot showing the PAMPA permeabilities (%T) as a function of the number of N-methyl groups for starting materials (0 N-methyls) and products (1–5 N-methyls) derived from the on-resin N-methylation of scaffolds 12, 19, 21, and 23 (Figure 2a) under three different solvent conditions: 1) LiOtBu (THF)/CH3I (THF), 2) LiOtBu (DCM)/CH3I (DCM), 3) LiOtBu (THF)/CH3I (DMSO).
To explore this model further, we performed computational predictions on a comprehensive virtual library of N-methyl cyclic peptide diastereomers based on the sequence cyclo[D/L-Leu; D/L-Leu; D/L-Leu; D/L-Leu; D/L-Pro; L-Tyr], where all possible stereoisomers (32) and their N-methyl variants (32 per isomer) were sampled (1024 compounds; Figure 5c). The results of these calculations are consistent with our experimental observations for 1 and 2, predicting that the free energy cost of desolvation is highly dependent on both stereochemistry and backbone N-methylation, and that even compounds with the same degree of N-methylation are expected to have widely varying permeabilities.
As predicted, the passive membrane diffusion rate of 7 was significantly lower than its congener 3. Indeed, this N-methyl variant was less permeable (%T = 5.7 ± 0.9) than the non-methylated parent compound 1 (%T = 9.5 ± 1.9), suggesting that patterns of N-methylation that disfavor passive membrane diffusion can expose polar groups and decrease permeability relative to the non-methylated forms. H-D exchange studies showed that, in contrast to the long exchange half lives of the two free amides of 3 (each >24 h), one of the amide N-H groups of 7 exchanged completely within 3 min and the other amide exchanged with a half life of ~2 h. These results are consistent with the observed difference in membrane permeability between 3 and 7, and suggest that H-D exchange rates can provide a sensitive means of predicting relative membrane permeability in congeneric cyclic peptides.
Permeability of expanded set of N-methylation products
While screening reaction conditions for compound 1, we found that the choice of solvent for both the base treatment and the CH3I addition impacted the extent of N-methylation along with the total number and identity of the products observed. For example, the use of CH2Cl2 for both reactions tended to yield products with fewer N-methyl groups, while the use of THF in both steps caused greater N-methyl incorporation but less selectivity than when DMSO was used for the CH3I addition. We therefore selected a set of diastereomers (Scaffolds 12, 19, 21, and 23, Figure 2a) that showed varying selectivity in the on-resin N-methylation reaction, and then further increased the number of N-methyl products observed for each scaffold by varying the reaction conditions. By attenuating the selectivity of the N-methylation chemistry and using different stereoisomeric scaffolds, we were able to observe 48 distinct N-methyl products ranging from non-methylated to permethylated peptides. PAMPA permeability measurements were made on the 4 N-methyl product mixtures from each scaffold plus the non-methylated precursors. Independent control studies were performed on a set of pure compounds to verify that %T values from the product mixtures were the same as the %T values determined for the pure compounds. The results, summarized in Figure 5d, show that even among compounds with the same number of N-methyl groups (i.e., regio- and stereoisomers), permeabilities vary greatly. The results also corroborate the computational predictions in that the most permeable trimethylated compounds were more permeable than any of the permethylated species. All four scaffolds produced at least one N-methyl product with a PAMPA permeability (%T > 25%) comparable to that of many orally bioavailable drugs26. Thus, even among scaffolds that showed poor selectivity in the on-resin N-methylation chemistry (scaffolds 12, 21, and 23), N-methylated derivatives exist that are highly permeable, indicating that while selectivity in the on-resin N-methylation reaction is associated with excellent permeability, it is not a precondition for it. These compounds are accessible, in principle, using the type of site-specific N-methylation chemistry used to synthesize 7.
In vitro and in vivo pharmacokinetics studies
The permeabilities of peptides 1, 2, and their N-methylated analogs were also examined in a cell-based RRCK permeability assay.1 This assay uses a stable population of MDCK cells that were selected by flow cytometry to have little or no functional Pgp, and therefore provide a good measure of passive, transcellular permeability with minimal interference from active transport mechanisms associated with Pgp expression (unpublished results). Compounds 1 and 2 showed relatively low cell permeability in RRCK cells, while their partially methylated derivatives, 3 and 4, were significantly (3 to 10-fold) more permeable than the corresponding non-methylated precursors (Table 1). As in the PAMPA results, compound 5 appears to be less permeable than the partially methylated species in the RRCK assay. However, 6 was the most permeable among the compound 2 series in the RRCK assay, a result that contrasted with the PAMPA data showing 6 to be less permeable than partially methylated 4. Despite this discrepancy between the PAMPA and cell-based permeability data for 2, the computational data on these compounds predicted 4 and 6 to have essentially the same permeability. All of the N-methylated compounds in this study had better cell permeability in the RRCK assay than the orally bioavailable drug CSA.
Table 1.
In vitro cell permeability, microsomal stability, and serum stability data for selected cyclic peptides.
| compound | RRCK pH 7.4(× 106cm/s) | HLM Clint (μg/min/mg) | RLM Clint (μg/min/mg) | Human plasma stab (t1/2, min) |
|---|---|---|---|---|
| CSA | 1.1 | 46.8 | 14.1 | > 360 |
| 1 | 1.8 | 225 | 243 | > 360 |
| 3 | 4.9 | 110 | 30.4 | > 360 |
| 5 | 3.5 | >320 | 317 | >360 |
| 2 | 0.5 | 109 | 47.1 | >360 |
| 4 | 4.1 | >320 | >491 | >360 |
| 6 | 6.1 | 191 | 62.6 | >360 |
In addition to the cell permeability studies, the compound series based on 1 and 2 was tested in vitro for stability in human plasma and in liver microsomes from rats and humans. These assays provide a general measure of the stability of compounds toward degradation by proteolytic and CYP enzymes, respectively. All of the cyclic peptides studied were stable in human plasma (t1/2 > 360 min), consistent with the stabilizing effect of cyclization (and possibly the presence of D-residues) toward proteolytic degradation. In human liver microsomal stability assessments, compounds 1, 2, and their methylated variants were less stable than CSA, as reflected in the higher CLint values (Table 1). Further, the partially methylated compound 3 had greater microsomal stability than either its non- or permethylated derivative, while partially methylated 4 was significantly less stable than either 2 or 6. Overall, for both series, stability was greater in rat than human liver microsomes, suggestive of species-specific differences in metabolism. Surprisingly, compounds 1 and 2 had very different microsomal clearance rates even though they have the same sequence and molecular weight, differing only by the relative configurations at two stereocenters. Thus, conformation not only plays a large role in determining passive cell permeability in cyclic peptides, but it also has a significant impact on their stability toward oxidative metabolism.
Effect of side chain substitutions
Most N-methylated cyclic peptide natural products contain a preponderance of aliphatic residues, although many also contain neutral, polar side chains such as Thr and Ser. We investigated the effect of polar side chain substitutions on the pharmacokinetic parameters of compound 3 by making Leu-to-Ser substitutions at positions 1, 3 and 4, and testing the resulting compounds in the PAMPA and cell-based permeability assays along with the in vitro serum and microsomal stability assays (Table 2). A single Leu-to-Ser substitution in 3 caused the RRCK cell permeability to drop by ~65% for Leu1 and Leu3, and by 85% for Leu4. The Leu1-to-Ser and the Leu3-to-Ser substitutions improved HLM stability slightly relative to the parent compound, while the Leu4-to-Ser substitution significantly decreased HLM stability. The same trend was observed in the RLM stability measurements. As expected, these hydrophobic-to-polar group substitutions decreased cell permeability, although from the standpoint of potential oral bioavailability, this decrease is partially offset by a concomitant increase in microsomal stability. There is also a relatively strong positional effect on both permeability and microsomal stability, in that the Leu4-to-Ser replacement had a greater impact on both permeability and stability than the Leu1 and Leu3 substitutions.
Table 2.
Permeability and microsomal stability data on Leu-to-Ser substitutions in compound 3.
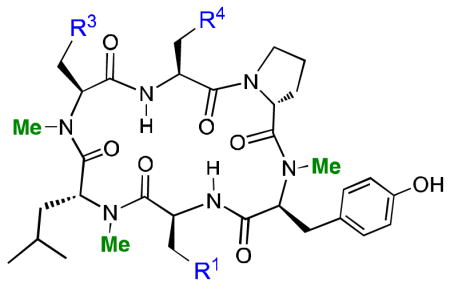
| ||||||||
|---|---|---|---|---|---|---|---|---|
| cpd | R1 | R3 | R4 | PAMPA (%T) | RRCK pH 7.4 (× 106cm/s) | HLM Clint (μg/min/mg) | RLM Clint (μg/min/mg) | Human plasma stab (t1/2, min) |
| 3 L1S | −OH | i-Pr | i-Pr | < 0.08 | 1.9 | 54 | 98 | > 360 |
| 3 L3S | i-Pr | −OH | i-Pr | < 0.08 | 1.6 | 85 | 53 | > 360 |
| 3 L4S | i-Pr | i-Pr | −OH | < 0.08 | 0.76 | 271 | 507 | >360 |
In vivo pharmacokinetics studies
The in vivo pharmacokinetics of 3 in male Wistar-Han rats are summarized in Table 3. The intravenous pharmacokinetics of 3 in rats are characterized by a low CLp (~ 4.5 mL/min/kg) and a moderate Vdss (1.1 L/kg), leading to a terminal elimination (t1/2) of 2.8 h. The absolute oral bioavailability (F) of 3 was determined to be ~ 28%, a value remarkably similar to that reported for CSA27–29. CSA and 3 also showed very similar AUC and Cmax values following the same oral dose of 10 mg/kg.
Table 3.
Rat Pharmacokinetics for compound 3 and CSA.
|
|
||||||
|---|---|---|---|---|---|---|
| i.v. administration | p.o. administration | |||||
|
| ||||||
| compound | CL (mL/min/kg) | Vdss (L/kg) | t1/2 (h) | AUC (ng.h/mL) | Cmax (ng/mL) | %F |
| 3 | 4.5 | 1.1 | 2.8 | 10.5 | 852 | 28% |
| CSA7 | 3.5 | 1.2 | 6.0 | 13.8 | 1440 | 29% |
Discussion
We have shown that cyclic peptides can be selectively N-methylated on the solid phase under conditions that for linear peptides are known to induce complete N-methylation19, 20. The fact that the observed selectivity was highly dependent on scaffold geometry points toward conformation as the key selectivity determinant. The H/D exchange results suggest that backbone N-methylation rigidifies the conformation of the peptide, since the free amide N-H groups exchanged much more slowly in 3 and 4 than in their nonmethylated precursors. This observation is consistent with the prediction that certain patterns of N-methylation stabilize hydrogen-bonded conformations by decreasing flexibility in the peptide backbone.
The H/D exchange results for the non-methylated peptides 1 and 2 suggested that they are in conformational flux among multiply hydrogen-bonded states on a time scale of seconds to minutes. However, the high degree of selectivity observed in the on-resin N-methylation for these compounds would seem to require restricted conformational freedom on this time scale. While this discrepancy may be due to solvent differences between the H/D exchange (chloroform) and N-methylation reactions (THF), we postulate that the amide deprotonation events are sequential and cooperative, such that deprotonation of the most exposed N-H limits the flexibility of the molecule and renders one or more of the remaining exposed amides more acidic. In other words, initial deprotonation of the most exposed N-H could shift the conformational equilibrium in the precursor toward the hydrogen-bonded conformation found in the product. This hypothesis is supported by a report showing that, while hydrogen bonding protects the involved amide N-H from H/D exchange, it increases the exchange rate of the amide whose carbonyl is participating as the H-bond acceptor30.
The permethylated versions of both 1 and 2 were significantly less permeable than their partially N-methylated counterparts, 3 and 4, in the PAMPA permeability measurements. For compound 1, the same trend was observed in the RRCK permeability assay, in which 3 was more permeable than either 1 or 5. This was likely due to the greater solvent exposure of hydrophilic carbonyl groups in the permethylated derivatives, which were sequestered in hydrogen bonds in the partially methylated compounds. Since none of the amide carbonyls in permethylated cyclic peptides can participate in intramolecular hydrogen bonding, we predict that these compounds will be less permeable, in general, than those partially methylated derivatives that can achieve optimal internal hydrogen bonding. Because the synthetic strategy outlined here afforded preferential N-methylation of exposed NH groups over those involved in internal hydrogen bonds, we postulate that these conditions can lead to methylated cyclic peptides with optimal or nearly optimal passive membrane permeability. In many cases, the most optimal conformation in terms of permeability may not be the predominant conformation selected by the N-methylation chemistry. However, given that hydrogen bonding is favorable to both permeability and selectivity, it is likely that the products of the on-resin N-methylation reaction will be among the most permeable of the many possible N-methyl variants.
For the compound 2 series, the cell-based permeability results are in partial conflict with the PAMPA data. In cells, 6 was more permeable than either 4 or the parent compound 2, while in PAMPA the 2 series followed the same trend as 1, with the partially methylated species showing the greater permeability. The discrepancy between the relative PAMPA and cell-based permeability values between 6 and 4 may lie in the differences between these two assays. The former is comprised of a relatively thick alkane layer impregnated with phospholipids, while the latter is a bona fide cell membrane. Where PAMPA and RRCK permeabilities disagree, the cell-based assay should be considered the more biologically relevant result with respect to passive membrane diffusion and is likely to be the better predictor of oral bioavailability.
We found that replacing a single Leu residue with Ser caused a significant loss in permeability. The magnitude of that loss, however, was dependent on the position of the substitution, indicating that the effect of polar side chains on permeability is dependent not only on the intrinsic polarity of the particular functional group, but also on its location along the backbone. This effect could result from a position-dependent interaction between the side chain hydroxyl and the backbone in a way that perturbs the backbone conformation, although preliminary calculations suggest that the beta structure of 3 is robust to side chain substitutions (data not shown).
While the cell-based permeabilities of the Leu-to-Ser substitutions were significantly lower than those of the all-Leu analogs, their PAMPA permeabilities were so low that they were undetectable. The threshold of detection for this assay put an upper limit on these values at 0.08% T, which, while consistent with the RRCK results in relative terms, predicted a more profound loss in permeability for these compounds than the cell-based results indicate. This difference in magnitude between the two results may also reflect the differences in the physical properties between the relatively thick hydrophobic spacer in the PAMPA vs. the true bilayer of a cell.
It is worth noting that the scaffold and N-methylation pattern found in 3 is similar to that identified by Biron, et al.4 in their N-methyl scan of the Veber-Hirschmann somatostatin analog cyclo[Pro–Phe–D-Trp–Lys–Thr Phe]31. In their study, an extensive N-methyl scan led to 30 analogs, of which a subset cyclized efficiently to yield the desired N-methyl cyclic peptide. Of the 7 N-methyl variants whose affinity toward the somatostatin receptor was similar to that of the wild type sequence, only the parent sequence and a single trimethyl variant, cyclo[Pro–Phe–DTrp(NMe)–Lys(NMe)–Thr–Phe(NMe)], showed significant in vivo uptake. Although the relative placement of the conformation-determining D-Trp and Pro residues in this trimethylated version of the Veber-Hirschmann peptide differ from that of 3, both peptides adopt similar backbone structures in which the D-residue and the Pro form two opposing β-turns with the nonmethylated amides participating in transannular hydrogen bonds. This conformation, in which the positioning of the two D-residues templates the formation of two opposing β-turns, is likely the key determinant of both the N-methylation regiochemistry in compound 1 and the membrane permeability of its trimethyl adduct. The reported oral bioavailability of this peptide is 9%6, a value comparable to that of 28% for 3.
While the strategy reported here may help identify novel membrane permeable scaffolds, its ability to confer bioavailability to peptide sequences with known bioactivity is limited. In many cases, N-methylation is likely to diminish biological activity, either by directly blocking interactions of the backbone with its receptor or by driving the peptide into an inactive conformation. However, varying both stereochemistry and N-methyl placement for a given sequence may yield novel scaffolds that retain biological activity while showing improved permeability.
The observation that 3 is significantly more permeable than 5 may provide insight into the relative scarcity of permethylated cyclic peptide natural products. A survey of the online Dictionary of Natural Products revealed that, among ~353 cyclic peptide and depsipeptide natural products whose chemical structures have been deposited, 146 have partially N-methylated backbones whereas only two are completely N-methylated. While the biosynthesis of N-methyl cyclic peptides by non-ribosomal peptide synthetases (NRPS) may disfavor permethylated species simply due to steric strain in the cyclization step, it is tempting to speculate that the pattern of N-methylation observed in cyclic peptides of natural origin may be driven, at least in part, by selective pressure to maximize membrane permeability. Such selective pressure may exist for peptide natural products created by bacteria to interact with eukaryotic host organisms.
Large cyclic peptide natural products, and macrocycles in general, may occupy “islands of bioactivity”* in chemical space that become increasingly sparse with increasing molecular mass. We postulate that many of these islands are populated with bioactive natural products whose properties have been optimized by the action of natural selection, although it is likely that many other islands remain undiscovered. A combination of the synthetic and computational approaches described here may open these regions of chemical space up to the discovery of new macrocyclic scaffolds with drug-like oral bioavailability similar to 3, a 755-molecular weight compound that nonetheless achieves acceptable oral bioavailability.
Methods
Peptide synthesis and N-methylation
Cyclic peptides were synthesized starting with the allyl ester of Fmoc-protected tyrosine, Fmoc-Tyr(OH)-OAll, linked as a silyl ether via the phenolic OH to an all-alkyl silyl-tethered polystyrene resin according to published procedures12. For large-scale syntheses (>0.5 mmol, or 1 g resin), peptides were synthesized and cyclized using an automated peptide synthesizer (Prelude, Protein Technologies, Inc.). For the 32-diastereomer library, a 96-well deep-well filter plate was used (Arcticwhite, LLC) with a manifold for solvent removal and an 8-channel trigger dispenser to dispense wash solvents and the deprotection solution. In general, couplings were performed using 4 eq Fmoc-protected amino acid, 3.8 eq HBTU, and 6 eq DIPEA in DMF (0.1 M in amino acid) for 1.5–3 h. Fmoc deprotections were carried out with 2% DBU/DMF for 15 min. After each coupling and deprotection step, the resin was washed with DMF (3x), DCM (3x), and DMF (3x). After the addition of the final residue, deallylation and final Fmoc removal were performed simultaneously with a solution of 1 eq Pd(Ph3P)4 in THF containing 10% v/v piperidine for 3 h. A chelating wash was performed to remove traces of Pd using 5% v/v sodium diethyldithiocarbamate/5% v/v DIPEA in DMF, followed by the normal DMF/DCM/DMF resin wash sequence. Cyclization was performed with 3 eq HATU, 3.2 eq HOAT, and 5 eq DIPEA in DMF for 3 h, followed by resin washing with two final DCM washes to remove residual DMF. Peptides were either cleaved following cyclization, or N-methylated (see below) and then cleaved by treating the resin (100 mg) with 5% HF/pyridine in THF (5 mL) for 1 h. The filtrate was collected into a polypropylene tube, and the reaction was quenched with 500 μL ethoxytrimethyl silane. After evaporation, the peptide was taken up into ACN/H2O (3:1) and purified by reverse-phase preparative HPLC. For peptides that contained side chain protecting groups, a final deprotection was performed using a solution of 5% v/v trimethylsilane in TFA for 1 h. The solution was evaporated, and the peptide was taken up into a 1:1 solution of water/ACN and either purified immediately or lyophilized and stored at −20°C.
On-resin N-methylation
The following procedure was developed for on-resin N-methylation using 1 g of resin at a loading of ~0.13 mmol/g of peptide: A solution of LiOtBu (25 mL; 1.5 M) in dry THF was filtered through a 0.2 μ syringe filter and then added to the resin. After 30 min the base solution was drained and, without rinsing, a 10% (v/v) solution of methyl iodide in DMSO (25 mL) was added and the resin was agitated for 30 min. The resin was rinsed with water (1x), methanol (3x), DCM (3x), DMF (3x), and DCM (3x), and then dried under vacuum. The cyclic peptide was cleaved (see procedure above), and the crude peptide was purified by reversed-phase HPLC on a C-18 10 μ 250×20 mm column using a typical gradient of 40% H2O/ACN (with 0.1% TFA) to 100% ACN (0.1% TFA) over 1 h. The resulting peptide was lyophilized to yield 3 (26 mg) as a white solid (25% based on resin loading). LRMS (ESI): Calculated for C41H66N6O7 (MH+): 755.5; found: 755.6. The same procedure was followed for the N-methylation of all other N-methyl peptides. For 4, after purification and lyophilization, 15 mg of a white solid was obtained (15% based on resin loading). LRMS (ESI): Calculated for C42H66N6O7 (MH+): 769.0; found: 769.2.
On-resin N-methylation to generate permethylated compounds 5 and 6
Cyclic peptides were synthesized as above, and, without cleaving, the resin (0.1 g, 0.4 mmol/g) was dried in vacuo with phosphorus pentoxide for 24 h. A mixture of dry THF (3 mL) and dry DMSO (2.4 mL) was then added to the resin and agitated with dry N2. LiOtBu (32 mg, 10 eq) was added, and the mixture was agitated with N2 for 20 min. 600 μL of CH3I was added via syringe, and the reaction was agitated with N2 for 20 min. The resin was filtered and washed multiple times with THF and DCM. Cleavage of peptides from the resin and purification by preparative HPLC yielded 5 (5 mg) or 6 (3 mg) as white solids. LRMS (ESI) for both peptides: Calculated for C43H71N6O7 (MH+): 783.5; found: 783.6.
H/D exchange studies
All NMR spectra for the H/D exchange studies were recorded at 297 K on a 600 MHz Varian Inova spectrometer equipped with a 5 mm inverse detection probe. Spectra were referenced to residual solvent proton signals (1H 7.26 for CDCl3). Peptides were dissolved in 665 μL CDCl3 at 6 mg/mL. Prior to initiating the H/D exchange reaction, a spectrum was recorded (t = 0). The NMR tube was removed from the spectrometer, and a solution of 35 μL CD3OD containing 10% CD3CO2D was added by syringe to a final concentration of 5% v/v CD3OD and 0.5% CD3CO2D. Upon addition of the deuterated solvents and quick agitation, a stopwatch was started and the NMR tube was returned to the spectrometer. Spectra were recorded using 8 FID transients for each time point at increasing intervals over a period of 16 h. H/D exchange rates were measured by integrating each exchangeable amide resonance separately and recording the ratio of its peak area vs. the peak area for a downfield non-exchangeable C(α)H resonance.
Additional methods
The PAMPA assay, in vitro and in vivo pharmacokinetics studies, NMR spectroscopy and structure determination, and computational methods are described in detail in the Supplementary Methods.
Supplementary Material
Acknowledgments
The synthesis and analytical results were supported by NIH grant GM084530-06 (RSL). The computational results were supported by NIH grant R01-GM086602 to MPJ. MPJ is a consultant to Schrodinger LLC. Cell-based assays and in vivo pharmacokinetics were provided by Pfizer.
Footnotes
This term was coined by David Hepworth and Spiros Liras of Pfizer, Inc.
Author Contributions
T.R.W. synthesized diastereomer library, developed on-resin N-methylation chemistry, performed PAMPA studies, synthesized compounds 1-6, and the Leu-to-Ser analogs. C.M.R. performed NMR experiments and analyzed data to determine patterns of N-methylation. A.C.R. synthesized and performed H-D exchange studies on compound 7. T.R. contributed to the development of the on-resin N-methylation chemistry. C.M.M. coordinated experiments and contributed to manuscript preparation. V.M.G. assisted in analysis of 2D NMR data for 3 and 4. R.A.T. contributed to synthesis method development and analytical procedures for PAMPA and compound characterization. R.G.L. provided guidance in developing NMR methods to determine pattern of N-methylation for 3, 4, and 7. S.S.F.L. performed computational studies, generated virtual libraries, and predicted permeabilities of virtual compounds. A.S.K., J.N.B., Y.Z., S.L., D.A.P., A.M.M. designed pharmacokinetic studies, interpreted results of in vitro and in vivo ADME and PK data. M.P.J. developed computational methodology for predicting permeability, designed experiments and discussed results, R.S.L. conceived the on-resin N-methylation approach, designed experiments. A.S.K., J.N.B., Y.Z., S.L., D.A.P., A.M.M., M.P.J., R.S.L. discussed results, wrote the paper.
Competing financial interests
There are no competing financial interests on the part of any of the authors of this manuscript.
Supporting Information Available. HPLC traces of 1, 2, and their N-methylation products in pure form and crude reaction products as a function of base concentration. Tables of NMR assignments for 3 and 4. HPLC trace and NMR spectrum of 7. Ramachandran plots showing calculated energy landscape of N-Me-Ala-CONH(Me) and overlaid (φ, ψ) angles of known N-methyl cyclic peptides. Computational models of membrane-associated conformations of 3 and 4.
Contributor Information
Matthew P. Jacobson, Email: Matt.Jacobson@ucsf.edu.
R. Scott Lokey, Email: slokey@ucsc.edu.
References
- 1.Driggers EM, Hale SP, Lee J, Terrett NK. The exploration of macrocycles for drug discovery--an underexploited structural class. Nat Rev Drug Discov. 2008;7:608–624. doi: 10.1038/nrd2590. [DOI] [PubMed] [Google Scholar]
- 2.Bauer RA, Wurst JM, Tan DS. Expanding the range of ‘druggable’ targets with natural product-based libraries: an academic perspective. Curr Opin Chem Biol. 14:308–314. doi: 10.1016/j.cbpa.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kim YK, et al. Relationship of stereochemical and skeletal diversity of small molecules to cellular measurement space. J Am Chem Soc. 2004;126:14740–14745. doi: 10.1021/ja048170p. [DOI] [PubMed] [Google Scholar]
- 4.Biron E, et al. Improving oral bioavailability of peptides by multiple N-methylation: somatostatin analogues. Angew Chem Int Ed Engl. 2008;47:2595–2599. doi: 10.1002/anie.200705797. [DOI] [PubMed] [Google Scholar]
- 5.Doedens L, et al. Multiple N-methylation of MT-II backbone amide bonds leads to melanocortin receptor subtype hMC1R selectivity: pharmacological and conformational studies. J Am Chem Soc. 132:8115–8128. doi: 10.1021/ja101428m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chatterjee J, Gilon C, Hoffman A, Kessler H. N-methylation of peptides: a new perspective in medicinal chemistry. Acc Chem Res. 2008;41:1331–1342. doi: 10.1021/ar8000603. [DOI] [PubMed] [Google Scholar]
- 7.Ovadia O, et al. The effect of multiple N-methylation on intestinal permeability of cyclic hexapeptides. Mol Pharm. 2011;8:479–487. doi: 10.1021/mp1003306. [DOI] [PubMed] [Google Scholar]
- 8.Dechantsreiter MA, et al. N-Methylated Cyclic RGD Peptides as Highly Active and Selective aVb3 Integrin Antagonists. J Med Chem. 1999;42:3033–3040. doi: 10.1021/jm970832g. [DOI] [PubMed] [Google Scholar]
- 9.Loosli HR, et al. Peptide conformations. Part 31. The conformation of cyclosporin A in the crystal and in solution. Helvetica Chimica Acta. 1985;68:682–704. [Google Scholar]
- 10.In Y, Ishida T, Takesako K. Unique molecular conformation of aureobasidin A, a highly amide N-methylated cyclic depsipeptide with potent antifungal activity: X-ray crystal structure and molecular modeling studies. Journal of Peptide Research. 1999;53:492–500. doi: 10.1034/j.1399-3011.1999.00046.x. [DOI] [PubMed] [Google Scholar]
- 11.Steiner JR, Barnes CL. Crystal and Molecular-Structure of Destruxin-B. International Journal of Peptide and Protein Research. 1988;31:212–219. doi: 10.1111/j.1399-3011.1988.tb00025.x. [DOI] [PubMed] [Google Scholar]
- 12.Rezai T, Yu B, Millhauser GL, Jacobson MP, Lokey RS. Testing the Conformational Hypothesis of Passive Membrane Permeability Using Synthetic Cyclic Peptide Diastereomers. J Am Chem Soc. 2006;128:2510–2511. doi: 10.1021/ja0563455. [DOI] [PubMed] [Google Scholar]
- 13.Rezai T, et al. Conformational flexibility, internal hydrogen bonding, and passive membrane permeability: successful in silico prediction of the relative permeabilities of cyclic peptides. J Am Chem Soc. 2006;128:14073–14080. doi: 10.1021/ja063076p. [DOI] [PubMed] [Google Scholar]
- 14.Boguslavsky V, Hruby VJ, Brien DFO, Misicka A, Lipkowski AW. Effect of peptide conformation on membrane permeability. The Journal of Peptide Research. 2003;61:287–297. doi: 10.1034/j.1399-3011.2003.00047.x. [DOI] [PubMed] [Google Scholar]
- 15.Gudmundsson OS, et al. The effect of conformation on the membrane permeation of coumarinic acid‐ and phenylpropionic acid‐based cyclic prodrugs of opioid peptides. The Journal of Peptide Research. 1999;53:383–392. doi: 10.1034/j.1399-3011.1999.00076.x. [DOI] [PubMed] [Google Scholar]
- 16.Knipp GT, Vander Velde DG, Siahaan TJ, Borchardt RT. The effect of beta-turn structure on the passive diffusion of peptides across Caco-2 cell monolayers. Pharm Res. 1997;14:1332–1340. doi: 10.1023/a:1012152117703. [DOI] [PubMed] [Google Scholar]
- 17.Atwood VA, et al. Utilization of an intramolecular hydrogen bond to increase the CNS penetration of an NK1 receptor antagonist. J Med Chem. 2001;44:2276–2285. doi: 10.1021/jm010825z. [DOI] [PubMed] [Google Scholar]
- 18.Davies JS. The cyclization of peptides and depsipeptides. J Pept Sci. 2003;9:471–501. doi: 10.1002/psc.491. [DOI] [PubMed] [Google Scholar]
- 19.Houghten RA, et al. Libraries from libraries: generation and comparison of screening profiles. Molecular diversity. 1996;2:41–45. doi: 10.1007/BF01718699. [DOI] [PubMed] [Google Scholar]
- 20.Dorner B, Ostresh J, Blondelle S, Dooley C, Houghten R. In: Peptidomimetic Synthetic Combinatorial Libraries. Abell A, editor. Vol. 1. JAI Press; London: 1997. [Google Scholar]
- 21.Gurrath M, Muller G, Kessler H, Aumailley M, Timpl R. Conformation/activity studies of rationally designed potent anti-adhesive RGD peptides. European journal of biochemistry/FEBS. 1992;210:911–921. doi: 10.1111/j.1432-1033.1992.tb17495.x. [DOI] [PubMed] [Google Scholar]
- 22.Kansy M, Senner F, Gubernator K. Physicochemical high throughput screening: Parallel artificial membrane permeation assay in the description of passive absorption processes. J Med Chem. 1998;41:1007–1010. doi: 10.1021/jm970530e. [DOI] [PubMed] [Google Scholar]
- 23.Fujikawa M, Ano R, Nakao K, Shimizu R, Akamatsu M. Relationships between structure and high-throughput screening permeability of diverse drugs with artificial membranes: application to prediction of Caco-2 cell permeability. Bioorg Med Chem. 2005;13:4721–4732. doi: 10.1016/j.bmc.2005.04.076. [DOI] [PubMed] [Google Scholar]
- 24.Bermejo M, et al. PAMPA--a drug absorption in vitro model 7. Comparing rat in situ, Caco-2, and PAMPA permeability of fluoroquinolones. Eur J Pharm Sci. 2004;21:429–441. doi: 10.1016/j.ejps.2003.10.009. [DOI] [PubMed] [Google Scholar]
- 25.Jacobson MP, et al. A hierarchical approach to all-atom protein loop prediction. Proteins. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- 26.Hwang KK, Martin NE, Jiang L, Zhu C. Permeation prediction of M100240 using the parallel artificial membrane permeability assay. J Pharm Pharm Sci. 2003;6:315–320. [PubMed] [Google Scholar]
- 27.Fukushima K, et al. Effect of serum lipids on the pharmacokinetics of atazanavir in hyperlipidemic rats. Biomed Pharmacother. 2009;63:635–642. doi: 10.1016/j.biopha.2009.01.008. [DOI] [PubMed] [Google Scholar]
- 28.Jin M, et al. Long-term levothyroxine treatment decreases the oral bioavailability of cyclosporin A by inducing P-glycoprotein in small intestine. Drug Metab Pharmacokinet. 2005;20:324–330. doi: 10.2133/dmpk.20.324. [DOI] [PubMed] [Google Scholar]
- 29.Fukushima K, et al. Long-term pharmacokinetic efficacy and safety of low-dose ritonavir as a booster and atazanavir pharmaceutical formulation based on solid dispersion system in rats. Biol Pharm Bull. 2008;31:1209–1214. doi: 10.1248/bpb.31.1209. [DOI] [PubMed] [Google Scholar]
- 30.Steffel LR, Cashman TJ, Reutershan MH, Linton BR. Deuterium exchange as an indicator of hydrogen bond donors and acceptors. Journal of the American Chemical Society. 2007;129:12956–12957. doi: 10.1021/ja076185s. [DOI] [PubMed] [Google Scholar]
- 31.Veber DF, et al. A potent cyclic hexapeptide analogue of somatostatin. Nature. 1981;292:55–58. doi: 10.1038/292055a0. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.