

Abstract
The Notch pathway is crucial for a wide variety of developmental processes including the formation of tissue boundaries. That it may function in calvarial suture development and figure in the pathophysiology of craniosynostosis was suggested by the demonstration that heterozygous loss of function of JAGGED1 in humans can cause Alagille syndrome, which has craniosynostosis as a feature. We used conditional gene targeting to examine the role of Jagged1 in the development of the skull vault. We demonstrate that Jagged1 is expressed in a layer of mesoderm-derived sutural cells that lie along the osteogenic-non-osteogenic boundary. We show that inactivation of Jagged1 in the mesodermal compartment of the coronal suture, but not in the neural crest compartment, results in craniosynostosis. Mesodermal inactivation of Jagged1 also results in changes in the identity of sutural cells prior to overt osteogenic differentiation, as well as defects in the boundary between osteogenic and non-osteogenic compartments at the coronal suture. These changes, surprisingly, are associated with increased expression of Notch2 and the Notch effector, Hes1, in the sutural mesenchyme. They are also associated with an increase in nuclear β-catenin. In Twist1 mutants, Jagged1 expression in the suture is reduced substantially, suggesting an epistatic relationship between Twist1 and Jagged1. Consistent with such a relationship, Twist1-Jagged1 double heterozygotes exhibit a substantial increase in the severity of craniosynostosis over individual heterozygotes. Our results thus suggest that Jagged1 is an effector of Twist1 in coronal suture development.
Introduction
Establishing and maintaining tissue boundaries is essential for morphogenesis and patterning (Dahmann and Basler, 1999; Irvine and Rauskolb, 2001; Tepass et al., 2002). Boundaries not only provide physical separation between tissues, but also function as signaling interfaces, influencing the behavior of cells at the boundary and in flanking tissues. In developmental systems as diverse as the Drosophila wing disc (Bray, 1998; Buceta et al, 2007; Major and Irvine, 2005) and the mammalian hindbrain (Kiecker and Lumsden, 2005), loss of boundary integrity results in abnormal development. Our recent work implicates a boundary between osteogenic and non-osteogenic compartments in the development of the skull vault and in the pathological condition of craniosynostosis, the premature fusion of the calvarial bones (Merrill et al., 2006; Ting et al., 2009).
The skull vault consists of the paired frontal and parietal bones and the single interparietal bone. The frontal bones are derived from neural crest, the parietal bones from mesoderm (Chai and Maxson, 2006; Jiang et al., 2002). The interparietal bone is a composite, its medial portion originating from neural crest and its lateral portion from mesoderm. Interposed between the bones of the skull vault are sutures, fibrous joints that maintain attachments between the bones while accommodating the growth of the brain and providing flexibility to the overall head structure (Opperman, 2000; Rice, 2008; Slater et al., 2008).
Craniosynostosis is a common birth defect (1/2500 live births) resulting in abnormalities in skull shape and, in some instances, in neurological deficiencies (Wilkie, 1997). In humans, it is caused by mutations in a number of genes (Cohen, 2006; Morriss-Kay and Wilkie, 2005), including FGFRs 1-3 (Hajihosseini, 2008), MSX2 (Jabs et al., 1993), TWIST1 (Howard et al., 1997), FIBRILLIN-1 (FBN1) (Sood et al., 1996), TGFBR1, TGFBR2 (Loeys et al., 2005), EPHRINA4 (EFNA4) (Merrill et al., 2006), EFNB1 (Twigg et al., 2004), RAB23 (Jenkins et al., 2007), and the Notch ligand, JAGGED1 (Kamath et al., 2002). Studies in mice have implicated, in addition, EphA4 (Ting et al., 2009), Axin2, (Yu et al., 2005), Dusp6 (Li et al., 2007), Gdf6 (Settle et al., 2003), Pdgfr alpha (Moenning et al., 2009) and Nell1 (Zhang et al., 2002). These genes are components of several signaling pathways: Tgfbr1, Msx2, and Gdf6 function in the Bmp pathway, Jagged1 in the Notch pathway, EphrinA4, EphrinB1, EphA4, Dusp6, Pdgfr alpha, and Fgfr1-3 in the RTK pathway. RAB23 is a component of the Hedgehog pathway, and Axin2 of the Wnt pathway. Twist1 functions to coordinate the activities of the Bmp and RTK pathways (Connerney et al., 2008; Rice et al., 2000; Ting et al., 2009). Clearly a diverse set of genes and pathways can cause craniosynostosis, potentially implicating a wide range of processes in its pathophysiology.
The mechanisms underlying craniosynostosis are still largely unknown (Boyadjiev, 2007). We investigated the developmental basis of Saethre-Chotzen syndrome, caused by heterozygous loss of function of Twist1 (Merrill et al., 2006; Ting et al., 2009). We made use of Twist1+/- mutant mice, which exhibit a suture fusion phenotype that resembles that seen in humans (Carver et al., 2002; el Ghouzzi et al., 1997). We showed that Twist1 mutants have a defect in the boundary between neural crest and mesoderm in the coronal suture (Merrill et al., 2006). This boundary coincides with the boundary between osteogenic and non-osteogenic compartments within the suture. We showed further that ephrin-Eph signaling, controlled by Twist1, has a role in the maintenance of this boundary: Reduced dosage of Twist1 and EphA4 results in inappropriate targeting of migratory osteogenic cells to the coronal suture (Ting et al., 2009). This pathfinding defect, we proposed, is a key cause of craniosynostosis in Twist1 and EphA4 mutants. We noted that the synostosis phenotype caused by loss of EphA4 function is less severe than that of Twist1+/- mutants, leading us to postulate that there are additional genes downstream of Twist1 functioning in the maintenance of the osteogenic-non-osteogenic boundary and in the pathophysiology of craniosynostosis. Here we provide evidence that the Notch ligand, Jagged1 is such a gene.
Constituents of the Notch pathway in vertebrates include the membrane receptors, Notch1-3, and membrane bound ligands, Jagged1, Jagged2 and Dll1, Dll3 and Dll4 (Kopan and Ilagan, 2009). After ligand activation, the intracellular domain (NICD) of Notch receptor is released by proteolysis and translocated to the nucleus. Within the nucleus, NICD replaces repressors from the DNA binding protein, CSL, and recruits a coactivator to form a transcription complex that modulates the expression of downstream genes such as Hes and Hey family genes (Bray, 2006). Notch signaling is critical for a variety of developmental processes, including developmental boundary formation and cell type specification (Bolós et al., 2007; Lai, 2004).
In humans, heterozygous loss of function of the Notch ligand, JAGGED1, results in Alagille syndrome, a multi-organ disorder characterized by impaired development of intrahepatic bile ducts, as well as defects in the heart, eye, kidney, face, and skull (Alagille, 1987; Emerick et al., 1999; Kamath et al., 2004; Krantz et al., 1998; Oda et al., 1997). Synostosis of the coronal suture occurs at low frequency (Kamath et al., 2002). Mutations in NOTCH2, a receptor for JAGGED1, also cause many of the defects of Alagille syndrome, though craniosynostosis has not been reported to be among them (McDaniell et al, 2006).
In contrast to humans with JAGGED1 mutations, Jagged1+/- mice develop largely normally. However, homozygous mutants die at E10.5 with vascular defects (Xue et al., 1999). Jagged1/Notch2 compound mutants exhibit a set of deficiencies similar to those of humans affected with Alagille syndrome, although these mutants do not have craniosynostosis or other craniofacial defects (McCright et al., 2002). Recently, conditional Jagged1 alleles were created, and several studies have documented conditional phenotypes in different tissues, including the inner ear (Brooker et al., 2006; Kiernan et al., 2006), the anterior chamber of the eye (Le et al., 2009), and ducts of the pancreas (Golson et al., 2009a). The consequences of inactivation of Jagged1 in tissues of the skull vault have not yet been investigated.
That elements of the Notch pathway have roles in Alagille syndrome, as well in developmental boundary formation, prompted us to examine the role of Jagged1 in the development of the skull vault. We demonstrate here that Jagged1 is expressed in a layer of mesoderm-derived sutural cells that lie along the osteogenic-non-osteogenic boundary between the frontal and parietal bones. We show that conditional inactivation of Jagged1 in the mesodermal compartment of the coronal suture results in craniosynostosis. Jagged1 expression in the coronal suture is reduced substantially in Twist1+/- mutants, suggesting an epistatic relationship between Twist1 and Jagged1. Consistent with such a relationship, Twist1+/-;Jagged1+/- double mutants exhibit a substantial increase in the severity of craniosynostosis over individual mutants, as well as an expansion of Notch signaling activity prior to the appearance of synostosis. We conclude that Jagged1 is an effector of Twist1 in coronal suture development.
Materials and Methods
Mouse mutants and genotyping
All genetically modified mice used in this study, as well as methods for determining their genotypes, were as described previously: Jagged1 (Xue et al., 1999.), Twist1 (Chen and Behringer, 1995), Jagged1 flox (Brooker et al., 2006.), Wnt1-Cre (Danielian et al., 1998), Mesp1-Cre (Saga et al., 1999), Dermo1-Cre (Yu et al., 2003) and R26R (Soriano, 1999).
Skull preparation
The heads of postnatal day 21 mice were skinned and stained with Alizarin Red S (80mg/l in 1% KOH) to reveal mineralized bone. The skulls were then cleared and stored in 100% glycerol.
Histochemical staining for alkaline phosphatase (ALP) and ®-galactosidase activity
The ALP staining procedures for whole-mount and tissue sections were performed as described previously (Ishii et al., 2003), with minor modifications. Briefly, E14.5 embryo heads were fixed in 70% EtOH for two days and bisected sagittally for whole-mount staining. Brains and skins were removed for clear illustration. The specimens were then stained with 0.01% BCIP and 0.025% NBT in NTMT solution. Cre-activated ®-galactosidase activity in Mesp1-Cre;R26R, Mesp1-Cre;R26R;Jagged1cko/cko, Dermo1-Cre;R262, Wnt1-Cre;R26R embryos were detected by X-gal staining on 10-μm cryostat sections as described previously (Ishii et al., 2003). Cryostat sections of E14.5 embryos derived from a cross between Wnt1-Cre;Twist1+/-;Jagged1+/- males and R26R females were stained with X-gal followed by immunostaining for Jagged1 as described below.
Immunostaining and in situ hybridization
The heads of E12.5, E13.5 and E14.5 embryos were fixed with 4% paraformaldehyde and embedded in HistoPrep (Fisher Scientific). Transverse frozen sections were cut in a cryostat at 10-μm thickness (or 6-μm for β-catenin antibody staining). Immunoperoxidase staining was performed by using streptavidin-biotin-peroxidase method (Histostain-SP Kit, Zymed), followed by Diaminobenzidin tetrahydrochloride (DAB) substrate (Zymed) with or without hematoxylin counterstain according to the manufacturer's instructions. Primary antibodies were purchased from the following companies and used at the indicated dilutions: goat anti-Jagged1 (Santa Cruz, sc-6011, 1:800), rabbit anti-Notch2 (sc-5545, 1:500), goat anti-Dll1 (sc-12531, 1:500), goat anti-Hes1 (sc-13842, 1:500), rabbit anti-P-Smad1/5/8 antibody (Cell Signaling, #9511, 1:100), rabbit anti-β-catenin (Sigma, C2206, 1:500) and rabbit anti-P-Erk1/2 antibody (Cell Signaling, #4376, 1:400). Indirect immunofluorescence staining was performed by using Rhodamin Red-X goat anti-rabbit IgG secondary antibody (Invitrogen, R6394, 1:100). The cell nuclei were revealed by co-staining with DAPI. Section in situ hybridization was carried out as described previously (Chen et al., 2007). Digoxigenin-labeled Twist1 antisense RNA probe was generated as reported by Ishii et al. (Ishii et al., 2003).
Results
Jagged1 is required in mesoderm and not in neural crest for the development of the coronal suture
We examined the expression of Jagged1 at E14.5, when the boundary between osteogenic and non-osteogenic cells becomes evident (Merrill et al., 2006; Ting et al., 2009). Immunostaining with a Jagged1-specific antibody revealed Jagged1 protein in a specific group of cells within the prospective coronal suture, which is derived from mesoderm (Yoshida et al., 2008) (Fig. 1A, arrow). These cells are located below a line extending outward from the parietal bone and above a line extending from the frontal bone. From previous work, most or all of the cells in this area contribute to the coronal suture (Yoshida et al., 2008). Whether the Jagged1 positive cells are a subset of cells in the suture or all of the cells is not clear. We note that cells medial to the frontal bone osteogenic front, which are derived from neural crest, do not contribute to the suture but instead appear to form bone as the leading edge extends (our unpublished observations). Cells medial to the parietal bone are derived from mesoderm and thus cannot be distinguished from sutural cells with mesoderm Cres. However, by analogy with the corresponding cells adjacent to the frontal bone, we believe these cells are ultimately incorporated into the parietal bone.
Fig. 1. Jagged1 is expressed in the prospective coronal suture.
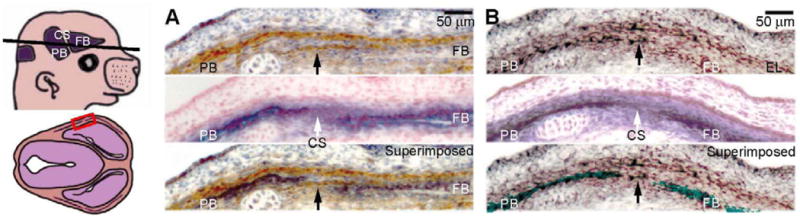
(A) We used an antibody against Jagged1 to stain transverse sections (as depicted) of E14.5 embryos. An adjacent section was stained for the early osteoblast marker, alkaline phosphatase (ALP) with nuclear fast red counterstain. A superimposition of the Jagged1 immunostaining and ALP is shown for clarity. (B) We also show the expression of the suture marker and craniosynostosis gene, Twist1 by in situ hybridization (upper panel) with an adjacent ALP-stained section (middle panel) and their superimposed image (lower panel). Note that Jagged1 and Twist1 are both expressed in the prospective coronal suture (arrows), as well in an ectocranial cell layer (EL). CS, prospective coronal suture; EL, ectocranial layer; FB, prospective frontal bone; PB, prospective parietal bone. Scale bars: 50 μm.
Jagged1 staining was also apparent in a layer of cells outside the prospective bone layer. This layer, which is also derived from mesoderm, is of interest because osteogenic precursor cells migrate within it (Ting et al., 2009). There was little or no expression in the neural crest-derived frontal bone, indicating that Twist1 expression overlapped significantly with that of Jagged1, both in the suture and in the layer of cells outside the prospective bone (Fig. 1B).
This pattern of Jagged1 expression suggested that it might function in the specification of sutural cells or in the development of osteogenic precursor cells. We used a genetic approach to examine these possibilities. Mice homozygous for a conventional Jagged1 knockout allele die at E10.5, precluding an examination of Jagged1 function in the development of the skull vault and sutures (Xue et al., 1999). Jagged1+/- mice have no detectible defects in skull vault development (Xue et al., 1999; our unpublished observations). We therefore used a conditional Jagged1 allele (Brooker et al., 2006) to address the function of Jagged1 in the skull.
We used two different Cre alleles, Mesp1-Cre (Saga et al., 1999), and Dermo1-Cre (Yu et al., 2003), to assess Jagged1 function in mesoderm-derived tissues of the developing coronal suture (Fig. 2). We used Wnt1-Cre to assess function in the adjacent neural crest-derived cells of frontal bone and dura. The Dermo1-Cre allele was generated by knocking the Cre gene into the Twist2 locus. Because Twist2 is related to Twist1, we were concerned that the Dermo1-Cre allele might interact with Twist1 or Jagged1 in coronal suture development. Three sets of findings argue against these possibilities. First, loss of Dermo1 function does not result in a defect in coronal suture development (Bialek et al., 2004). Second, a test cross of Dermo1-Cre with Twist1 mutants revealed that mice with the genotype Dermo1-Cre;Twist1+/- did not exhibit a more severe coronal suture defect than Twist1+/- mice in a comparison of 3 littermate pairs (data not shown). Third, mice with the genotype Dermo1-Cre;Jagged1cko/+ (n =10), like Jagged1+/- mice did not exhibit a coronal suture defect (data not shown). These results suggest that Dermo1 does not interact with either Twist1 or Jagged1 in the coronal suture and thus support the use of Dermo1-Cre for investigating Jagged1 function in the coronal suture.
Fig. 2. Conditional inactivation of Jagged1 in mesoderm and not in neural crest results in craniosynostosis and defects in the boundary between the coronal suture and prospective frontal and parietal bones.

(A-C) The R26R allele served as an indicator of either Mesp1-Cre, Dermo1-Cre or Wnt1-Cre activation in E14.5 embryos. Staining for ALP (below lacZ-stained sections) revealed osteogenic cells and the coronal suture. Punctate lacZ staining is apparent in the prospective coronal suture, parietal bone, and dermis of Mesp1-Cre;R26R and Dermo1-Cre;R26R embryos. Wnt1-Cre resulted in robust lacZ staining for the frontal bone (FB) and meninges (M). (D-G) Immunostains for Jagged1 to assess the influence of Mesp1-Cre, Dermo1-Cre or Wnt1-Cre on Jagged1 protein. Note Jagged1 expression in ectocranial layer (above ALP) and in prospective sutural cells (arrow). In Mesp1-Cre mutant, Jagged1 is selectively lost in sutural cells. In the Dermo1-Cre mutant, Jagged1 is reduced in both the ectocranial layer and suture. In the Wnt1-Cre mutant, Jagged1 is unaffected. (H-L) We produced mice with the floxed Jagged1 allele in combination with the indicated Cre lines. Mesp1-Cre;Jagged1cko/cko embryos died around E13.5-E15.5; consequently we examined the morphology of the developing coronal suture at E14.5 by means of whole mount ALP staining (H, I). Mesp1-Cre;Jagged1cko/cko mutants exhibited a narrowing of the prospective coronal suture (CS) (I, arrow). We examined the skulls of Dermo1-Cre;Jagged1cko/cko and Wnt1-Cre;Jagged1cko/cko mice at P21 by staining with Alizarin Red S (J-L). Note that coronal synostosis in the Dermo1-Cre;Jagged1cko/cko mutant (K, arrows). Sutures of Wnt1-Cre;Jagged1cko/cko mice were normal, but an ossification defect was apparent in the frontal bone (L, arrow). (M, N) To assess the boundary between the coronal suture and adjacent bone territories we produced mice with the R26R allele with Mesp1-Cre;Jagged1cko/cko (N). Note substantial numbers of lacZ positive cells in the frontal bone territories (N, arrowheads). Heads were sectioned as in Fig. 1. CS, coronal suture; FB, frontal bone; M, meninges; PB, parietal bone; SS, sagittal suture. Scale bars: 50 μm in A-G, M, N; 500 μm in H, I; 1mm in J-L.
To test the ability of Mesp1-Cre and Dermo1-Cre to cause recombination in the mesoderm-derived portion of the coronal suture, we crossed mice carrying each of these alleles with mice carrying R26R. Staining of sections through the coronal suture for lacZ revealed that both Dermo1-Cre and Mesp1-Cre drove recombination in areas of the developing suture known to be derived from mesoderm, including cells of the suture itself, the parietal bone, and the ectocranial layer (Fig. 2A, B). Staining was punctate, indicating that recombination did not occur in all mesoderm-derived cells. Wnt1-Cre, as expected, drove recombination in cells of the frontal bone and underlying dura (Fig. 2C).
We next produced mice with the genotypes Mesp1-Cre;Jagged1cko/cko, Dermo1-Cre;Jagged1cko/cko, and Wnt1-Cre;Jagged1cko/cko, and used immunostaining with a Jagged1 antibody to assess the expression of Jagged1 in mutant sutures at E14.5. As is evident in Fig. 2, D-G, Mesp1-Cre selectively reduced Jagged1 expression in sutural cells (Fig. 2E, arrowhead), and had a smaller effect on expression in cells of the ectocranial layer. Dermo1-Cre caused loss of Jagged1 expression in both sutural and ectocranial cells (Fig. 2F). Wnt1-Cre had no discernible effect on Jagged1 expression at either location, confirming that in the area of the coronal suture, Jagged1 was expressed selectively in mesoderm-derived cells (Fig. 2G).
We did not obtain any pups with the genotype Mesp1-Cre;Jaggedcko/cko (of 90 examined), suggesting that homozygous inactivation of Jagged1 in mesoderm causes lethality during embryogenesis. We examined Mesp1-Cre;Jagged1cko/cko embryos at E14.5 for evidence of craniosynostosis. In both sections and whole mounts, staining for the osteoblast marker, alkaline phosphatase (ALP), revealed a narrowing of the prospective coronal suture, consistent with early craniosynostosis (Fig. 2H, I, arrows) (Ting et al., 2009). Dermo1-Cre;Jagged1cko/cko mutants were viable. Alizarin Red stains of P21 mutant skulls revealed synostosis of the coronal sutures with a penetrance of 86% (n=14) (Fig. 2J, K). Mice with the genotype Wnt1-Cre;Jagged1cko/cko were viable at least through P21. Their skulls showed no evidence of synostosis of the coronal suture (Fig. 2L). However, an ossification defect was apparent in the frontal bone (Fig. 2L, arrow). The developmental mechanism underlying this defect is not clear; one reasonable explanation is that Jagged1 is expressed in a subset of neural crest that contribute to the medial portion of the frontal bone, and that inactivation of Jagged1 in these cells results in the defect.
Our previous findings showed that the boundary between osteogenic and non-osteogenic cells in the coronal suture is deficient in craniosynostosis (Merrill et al., 2006; Ting et al., 2009). Together with the well-documented role of Notch signaling in boundary formation, these findings prompted us to test for boundary defects within the coronal sutures of Jagged1 conditional mutants. The osteogenic-non-osteogenic boundary coincides with the neural crest-mesoderm boundary in the coronal suture. To examine the osteogenic-non-osteogenic boundary, we crossed R26R into Mesp1-Cre;Jagged1cko/cko mice and examined the distribution of lacZ-expressing cells in the coronal suture. As is evident in Fig. 2N, lacZ positive cells crossed from the mesoderm compartment to the neural crest compartment in substantial numbers (arrowheads). Jagged1 is thus required in the mesoderm to maintain the integrity of the boundary between osteogenic and non-osteogenic cells in the coronal suture.
NOTCH2 is mutated in some cases of Alagille syndrome (McDaniell et al, 2006). It is also known to function cooperatively with Jagged1 in the development of bile ducts (Lozier et al., 2008) and lens fiber cells (Saravanamuthu et al., 2009), and can be negatively regulated by Jagged1 (Yuan et al., 2006). This potential functional relationship between Jagged1 and Notch2 prompted us to assess the influence of Jagged1 on the expression of Notch2 in developing coronal sutures.
In wild type embryos at E14.5, Notch2 was expressed in ALP-positive cells of the prospective frontal and parietal bones and was excluded from the coronal suture (Fig. 3A). In Mesp1-Cre;Jagged1cko/cko and Dermo1-Cre;Jagged1cko/cko mutants, the domain of Notch2 expression expanded into the suture (Fig. 3B, D). This expansion did not occur in Wnt1-Cre;Jagged1cko/cko mutants (Fig. 3C). These results suggest that Jagged1 functions in mesoderm to exclude Notch2 expression or Notch2-expressing cells from the coronal suture.
Fig. 3. Inactivation of Jagged1 in mesoderm causes an expansion of Notch2 and Hes1 expression domains in the prospective coronal suture.
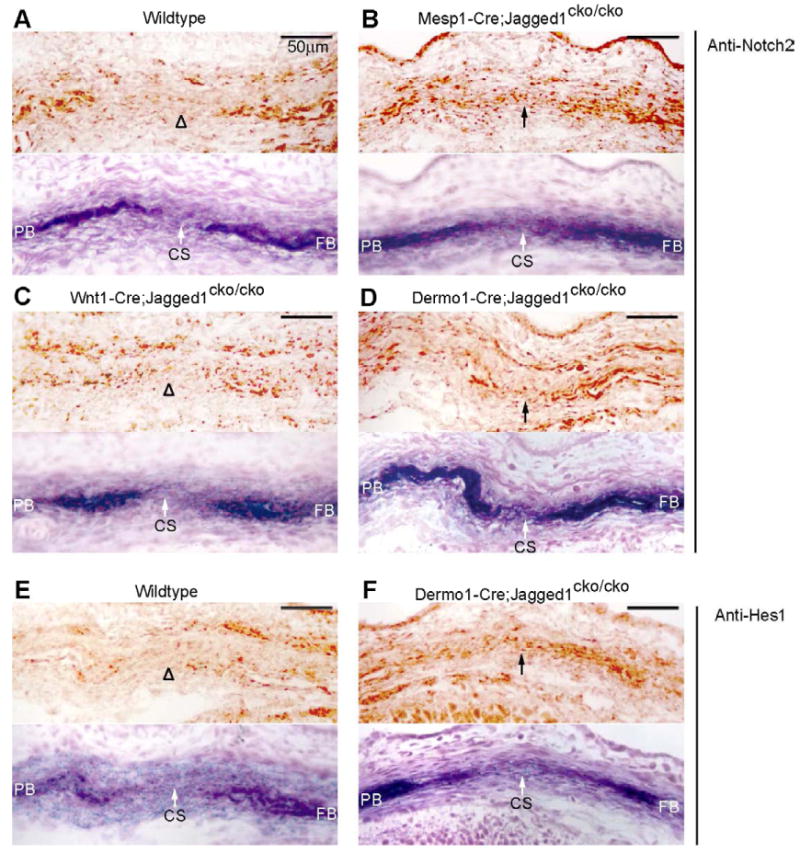
We used immunostaining to examine the influence of lineage-specific inactivation of Jagged1 on the distribution of the Jagged1 receptor, Notch2 (A-D) and the Notch target, Hes1 (E, F). Embryos at E14.5 of the indicated genotypes were sectioned as shown in Fig. 1 and subjected to immunostaining. Alternate sections were stained for ALP to reveal prospective bone. Note that in wildtype embryos (A), Notch2 was expressed in the prospective bone territories and was excluded from the coronal suture. Hes1 was expressed similarly (E). In both Mesp1-Cre- and Dermo1-Cre-mediated knockouts of Jagged1, Notch2 and Hes1 expression expanded into the prospective suture (B, D, F). No change in Notch2 expression was evident in a Wnt1-Cre-mediated Jagged1 knockout (C). CS, coronal suture; FB, frontal bone; PB, parietal bone. Scale bars: 50 μm.
We next examined the expression of Hes1, a downstream effector of Notch signaling (Jarriault et al., 1995), as an indicator of Notch signaling activity. Hes1 was not expressed in sutural mesenchyme of wild type mice, but was readily detectible in mutant sutures (Fig. 3E, F), suggesting that inactivation of Jagged1 resulted not only in an expansion of the Notch2 expression domain but also in an increase in Notch signaling in the sutural mesenchyme. These data suggest that loss of Jagged1 function results in an activation of Notch signaling in the suture by a mechanism that involves either an increased expression of the Notch2 protein in sutural cells or a movement of Notch2-expressing cells into the suture.
Interactions between Jagged1 and P-Erk1/2, P-Smad1/5/8, Wnt-β-catenin, and ephrinA-EphA pathways in the coronal suture
We asked whether loss of Jagged1 function influenced the activity of the RTK, Bmp, and Wnt/β-catenin pathways in the developing suture. Each is known to function during osteogenic differentiation and has been implicated in craniosynostosis (Morriss-Kay and Wilkie, 2005). Moreover, in previous work we noted a reduction in RTK signaling, as assessed by P-Erk1/2, in EphA4 mutants, which exhibit craniosynostosis and defects in the osteogenic-non-osteogenic boundary (Ting et al., 2009). Consistent with our previous results, in control embryos, P-Erk1/2 was expressed in cells of prospective bone, osteogenic fronts, and an ectocranial cell layer, but not in the suture. In Dermo1-Cre;Jagged1cko/cko mutants, P-Erk1/2 expression expanded, coincident with the expanded ALP expression at the leading edges of the frontal and parietal bones (Fig. 4A, B). The expression of ephrinA2 in the ectocranial layer was unaffected, suggesting that loss of Jagged1 function in mesoderm did not affect ephrinA2 signaling at the ligand level (Fig. 4G, H).
Fig. 4. Interactions between Jagged1 and P-Erk1/2, P-Smad1/5/8, Wnt-β-catenin, and ephrinA-EphA pathways in the coronal suture.
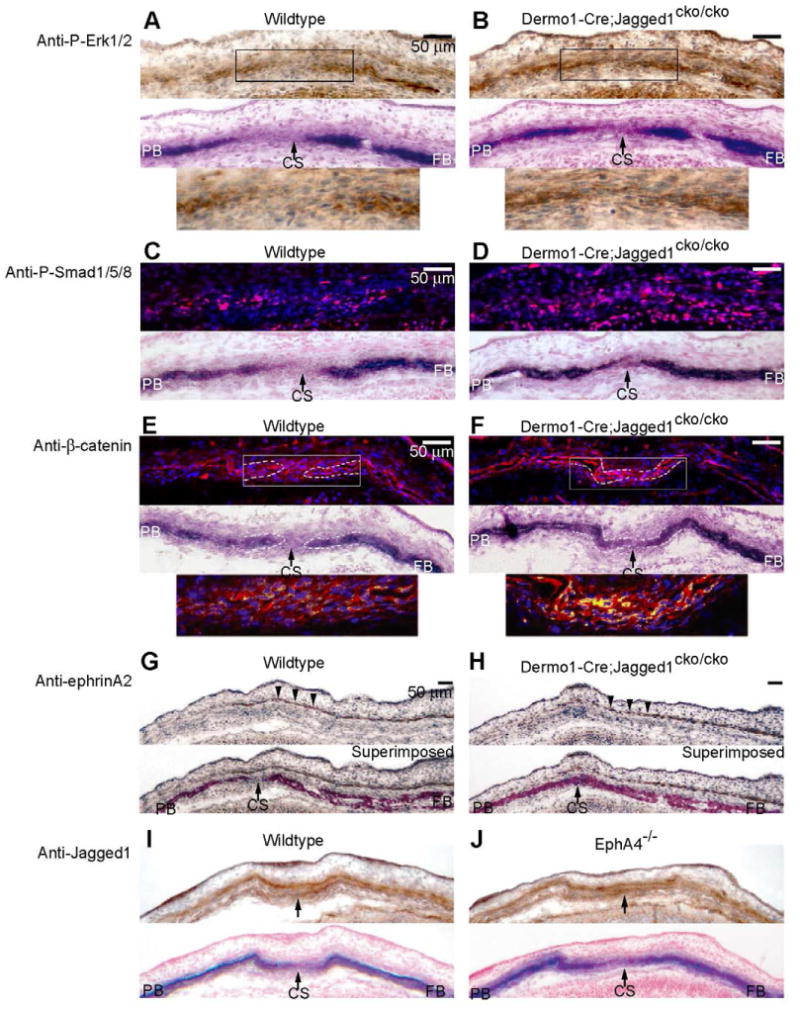
We examined the influence of mesoderm-specific inactivation of Jagged1 on the distribution of P-Erk1/2 (A, B), P-Smad1/5/8 (C, D), β-catenin (E, F) and ephrinA2 (G, H) in the coronal suture. (I, J) We also assessed the influence of loss of EphA4 function on Jagged1 expression. Wildtype and mutant embryos at E14.5 were prepared as in Fig. 1 and sections were stained with indicated antibodies. For P-Erk1/2, ephrinA2 and Jagged1, the antibodies were visualized with DAB (brown color) and sections were counterstained with hematoxylin. For P-Smad1/5/8 and β-catenin, the antibodies were visualized with Rhodamine Red-X-conjugated secondary antibody (red color). Nuclei were stained with DAPI (blue). In the case of β-catenin, the pink color of DAPI, β-catenin double positive nuclei was transformed to yellow to make it more visible. ALP stains of adjacent sections are shown below images of immunostains. The lower panels in A, B and E, F are enlargements of the boxed areas in the upper panels. Note that in wildtype embryos, P-Erk1/2, P-Smad1/5/8 were expressed in prospective bone and osteogenic fronts but not in the suture. β-catenin was expressed in the suture but was largely cytoplasmic. In Dermo1-Cre;Jagged1cko/cko mutants, P-Erk1/2 expression was coincident with ALP expression in the frontal and parietal bones. In contrast, P-Smad1/5/8 expression expanded into the sutural mesenchyme, similar to Notch2 and Hes1 expression (Fig. 3D, F), and ephrinA2 expression was unaltered (G, H). Jagged1 expression did not change in the EphA4-/- mutant coronal suture (I, J). CS, coronal suture; FB, frontal bone; PB, parietal bone. Scale bars: 50 μm.
The distribution of P-Smad1/5/8 expressing cells was altered in Dermo1-Cre;Jagged1cko/cko mutants in a manner similar to the changes we described previously in Twist1-EphA4 mutants (Ting et al., 2009). Whereas in wild type sutures, P-Smad1/5/8 positive cells were concentrated in the osteogenic fronts, in mutants, they were distributed throughout the suture (Fig. 4C, D).
The expression of β-catenin was also changed in Dermo1-Cre;Jagged1cko/cko mutant sutures compared with controls (Fig. 4E, F). In control embryos, β-catenin was expressed throughout the suture, and was distributed largely in the cytoplasm of sutural cells. (Fig. 4E). In mutants, there was a substantial increase in the overall level of β-catenin staining (Fig. 4F). Moreover, the proportion of stained nuclei increased sharply in the area of the suture.
Together, these results suggest that Jagged1 influences RTK, Bmp, and Wnt/β-catenin signaling, but not ephrinA-EphA signaling, in the coronal suture. Finally, to determine whether ephrinA-EphA signaling influenced Jagged1 activity, we examined Jagged1 expression in EphA4 mutants. As is evident in Fig 4I, J, there was no change in Jagged1 expression, either in the prospective suture or in the ectocranial mesenchyme. These results suggest that there is no discernible interaction between Jagged1 and ephrinA-EphA signaling in the coronal suture. Thus, in aggregate, our results suggest that the Notch and ephrinA-EphA pathways function in parallel, downstream of Twist1.
A genetic interaction between Jagged1 and Twist1
The overlap in Twist1 and Jagged1 expression in the coronal suture (Fig. 1), together with the similarity in Jagged1 and Twist1 mutant phenotypes suggested that Twist1 and Jagged1 might interact genetically. As a first test of this possibility, we asked whether heterozygous loss of Jagged1 function exacerbated the synostosis defect in Twist1 mutant mice. Twist1-/- mice die at E11.5, Jagged1-/- mice at E10.5. Therefore we focused our analysis on compound heterozygotes, which are viable. We crossed Twist1+/- and Jagged1+/- conventional mutant mice and examined the coronal sutures in compound mutant offspring (Fig. 5). Coronal synostosis in the Twist1 substrain used in this series of experiments had a penetrance of 44%, lower than that of Twist1 mutant strains examined previously (Merrill et al., 1986; Ting et al., 2009). At P21, Twist1+/-;Jagged1+/mice exhibited a substantial increase in the penetrance of synostosis of the coronal suture in comparison to Twist1+/- mice. Increases in penetrance were also apparent in the squamosal, lambdoid, occipitointerparietal (OIP), and interfrontal sutures (Table 1).
Fig. 5. Increased severity of craniosynostosis in Twist1-Jagged1 combination mutants.
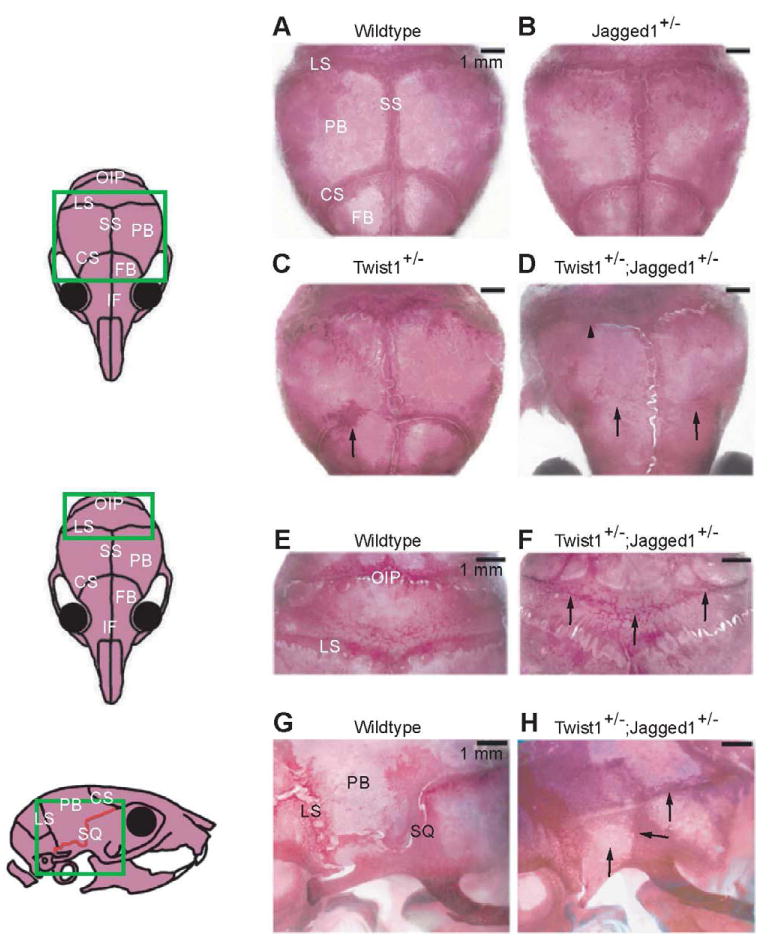
To test for a genetic interaction between Twist1 and Jagged1, we compared skull morphologies of individual mutants with those of compound heterozygotes. Pups of the indicated genotypes were taken at P21 and the skulls were stained with Alizarin Red S. (A-D) Note bilateral coronal synostosis in Twist1+/-;Jagged1+/- skull (D, arrows) compared with partial unilateral synostosis in Twist1+/- skull (C, arrow) and normal suture morphology in wildtype and Jagged1+/- skulls (A, B). The lambdoid (LS), squamosal (SQ) and occipitointerparietal (OIP) sutures also exhibited more severe synostosis in the compound heterozygotes compared with individual mutants (D-H). CS, coronal suture; FB, frontal bone; IF, interfrontal suture; LS, lambdoid suture; OIP, occipitointerparietal suture; PB, parietal bone; SQ, squamosal suture; SS, sagittal suture. Scale bars: 1 mm. See Tables 1 and 2.
Table 1. Penetrance of craniosynostosis in Jagged1+/-; Twist1+/- and Twist1+/-;Jagged1+/- mice*.
| Genotype | n | Coronal L+R** (%) | Squamosal L+R (%) | Lambdoid L+R (%) | OIP (%) | Interfrontal (%) | Penetrance in eight sutures (%) |
|---|---|---|---|---|---|---|---|
| Wildtype | 21 | 0 | 0 | 0 | 0 | 0 | 0 |
| Jagged1+/- | 14 | 0 | 0 | 0 | 0 | 0 | 0 |
| Twist1+/- | 18 | 44 | 28 | 56 | 72 | 0 | 94 |
| Twist1+/-;Jagged1+/- | 22 | 77 | 100 | 95 | 100 | 32 | 100 |
The percentage of postnatal day 21 mice with suture fusion phenotypes was examined. The synostosis phenotypes of five different sutures (totally eight sutures), including left coronal, right coronal, left squamosal, right squamosal, left lambdoid, right lambdoid, occipitointerparietal (OIP) and interfrontal suture, were examined under microscope on alizarin red S-stained skulls.
We used a quantitative measure of synostosis, the craniosynostosis index (CI) (Oram and Gridley, 2005) to assess the severity of the craniosynostosis phenotype (Table 2). The CI includes the coronal, lambdoid, squamosal, OIP and interfrontal sutures. The CI for each individual suture was elevated significantly in Twist1+/-;Jagged1+/- mutants compared with other genotypes. Whereas the sum score was 3.8±2.3 for Twist1+/- mice at P21, it was 10.1±2.6 for Twist1+/-;Jagged1+/- compound heterozygous mutants. These results are consistent with a strong genetic interaction between Twist1 and Jagged1 in the developing skull vault.
Table 2. Craniosynostosis index (CI) of Jagged1+/-; Twist1+/- and Twist1+/-;Jagged1+/- mice*.
| Genotype | n | Coronal L+R** | Squamosal L+R | Lambdoid L+R | OIP | Interfrontal | Sum score of eight sutures |
|---|---|---|---|---|---|---|---|
| Wildtype | 21 | 0.0±0.0 | 0.0±0.0 | 0.0±0.0 | 0.0±0.0 | 0.0±0.0 | 0.0±0.0 |
| Jagged1+/- | 14 | 0.0±0.0 | 0.0±0.0 | 0.0±0.0 | 0.0±0.0 | 0.0±0.0 | 0.0±0.0 |
| Twist1+/- | 18 | 0.6±0.8§ | 0.8±1.3§ | 1.7±1.8§ | 0.7±0.5§ | 0.0±0.0 | 3.8±2.3§§ |
| Twist1+/-;Jagged1+/- | 22 | 1.5±1.2§ | 3.5±1.6§ | 3.5±1.8§ | 1.2±0.5§ | 0.4±0.7§ | 10.1±2.6§§ |
The Craniosynostosis index (CI) is a scoring method adopted from Oram and Gridley, 2005. Eight sutures as indicated in Table 1 were scored for each skull. The degree of fusion was assessed microscopically and scored (0: unfused; 1: <50% fused; 2: ≥50% fused and 3: 100% fused.) Scores for left and right sutures of the same type were added. For example, the maximum score given to two coronal sutures in each skull is six. The mean score±SD for each type of sutures were calculated separately. The sum score of the eight sutures represents the severity of craniosynostosis in each genotype. Pairwise comparisons among four genotypes were analyzed by two-tailed Student's t-test and significant differences are marked with (§) for P<0.05 or (§§) for P<0.000001.
L+R: Left+Right sutures
We next used the conditional Jagged1 allele, together with Wnt1-Cre or Mesp1-Cre, and the conventional Twist1 allele, to compare the strength of the interaction between Jagged1 and Twist1 in neural crest and mesoderm. From data presented in Table 3, mice with the genotype Mesp1-Cre;Jagged1cko/+;Twist1+/- exhibited a significantly greater craniosynostosis index (4.0±1.8) than mice with the Twist1 heterozygous knockout alone (2.0±1.9). In contrast, when Wnt1-Cre was used in place of Mesp1-Cre, there was no significant difference in the craniosynostosis index. These data suggest that Jagged1 is required in mesoderm for its interaction with Twist1.
Table 3. Jagged1 is required in the mesoderm for its interaction with Twist1 in the coronal suture*.
| Genotype | n | CI |
|---|---|---|
| Mesp1-Cre | 15 | 0.1±0.3 |
| Mesp1-Cre;Jagged1cko/+ | 12 | 0.0±0.0 |
| Mesp1-Cre;Twist1+/- | 10 | 2.0±1.9 |
| Mesp1-Cre;Jagged1cko/+;Twist1+/- | 14 | 4.0±1.8 |
| Genotype | n | CI |
|
| ||
| Wnt1-Cre | 19 | 0.0±0.0 |
| Wnt1-Cre;Jagged1cko/+ | 16 | 0.0±0.0 |
| Wnt1-Cre;Twist1+/- | 13 | 1.4±1.7 |
| Wnt1-Cre;Jagged1cko/+;Twist1+/- | 16 | 2.1±1.5 |
Skulls were collected at postnatal day 21 and stained with Alizarin Red S. The Craniosynostosis index (CI) was calculated as in Table 2. All pairwise comparisons (two-tailed Student's t-test) were significantly different (P<0.05), except for the following: (1) Mesp1-Cre vs. Mesp1-Cre;Jagged1cko/+, (2) Wnt1-Cre vs. Wnt1-Cre;Jagged1cko/+, and (3) Wnt1-Cre;Twist1+/- vs. Wnt1-Cre;Jagged1cko/+;Twist1+/-. (P>0.1)
To investigate the basis of the interaction between Twist1 and Jagged1, we assessed the influence of reduced dosage of both genes individually and together on the expression domains of Jagged1, Notch2 and Hes1 (Figs 6, 7) at several stages of development, including E12.5 and E13.5 prior to any boundary defects in Jagged1 or Twist1 mutant embryos, as well as at E14.5 when such defects are evident.
Fig. 6. Influence of Twist1 and Jagged1 on Notch signaling in the prospective coronal suture.

We assessed the influence of Twist1 and Jagged1 on the expression of Notch2 and Hes1 in a developmental series of embryos. Sections of E12.5 (A-H) and E13.5 (I-T) embryos of the indicated genotypes were prepared as in Fig. 1 and stained with antibodies against Jagged1 (A-D, I-L), Notch2 (E-H, M-P), and Hes1 (Q-T). At E13.5, Jagged1 is expressed in two layers. The lower of these is reduced preferentially in Twist1+/- and Twist1+/-;Jagged1+/- mutants (K, L, arrowheads). This layer gives rise to the coronal suture (see Fig. 7A-D). Alternate sections were stained for ALP. A superimposition of the Jagged1 immunostaining and ALP, as well as a schematic diagram, are shown below the E13.5 sections for clarity (I-L). Note expansion of Notch2 into the prospective suture in both Twist1+/- and Twist1+/-;Jagged1+/- mutants at E12.5 (G, H, arrows) and E13.5 (O, P, arrows). Also note expansion of Hes1, indicating an increase in Notch signaling in the suture (S, T, arrows). CS, coronal suture; FB, frontal bone; PB, parietal bone. Scale bars: 50 μm.
Fig. 7. Twist1 and Jagged1 function together to maintain a boundary between the coronal suture and adjacent osteogenic territories.

(A-D) We produced mice with the genotype Wnt1-Cre;R26R together with the allelic combinations Jagged1+/-, Twist1+/- and Twist1+/-;Jagged1+/-. We examined the distribution of lacZ positive (neural crest-derived) cells in combination with Jagged1 immunostaining in such mutants at E14.5 (A-D, upper panels). We also examined the expression of ALP by histochemistry (A-D, middle panels). The lower panels in A-D show a superimposition of the two images above. Note that Jagged1 expression is selectively reduced in the sutural mesenchyme in mutants (compare A, arrow, with B, C, D). Also note lacZ positive cells crossing into sutural mesenchyme (C,D, arrowheads). (E-L) The expansion of Notch2 (compare G, H, arrows with E, F, arrowheads) and Hes1 (compare K, L arrows with I, J, arrowheads) into sutural mesenchyme of Twist1+/- and Twist1+/-;Jagged1+/- mutants. CS, coronal suture; FB, frontal bone; PB, parietal bone. Scale bars: 50 μm. (M) Twenty serial sections (forty coronal sutures) from a similar level were selected from each of two embryos and the total number of lacZ-positive cells in the coronal mid-suture mesenchyme (as indicated by arrowheads in C, D) were counted. Error bars represent one standard deviation based on two biological replicates.
At E12.5, Jagged1 protein was located in the frontal and parietal bone territories, which were identified by the expression of ALP. In the parietal bone territory, Jagged1 expression extended beyond the ALP domain into an area of prospective osteogenic tissue and further into the coronal suture (Fig. 6A). Notch2 expression overlapped with ALP activity and was detectable but reduced in the prospective coronal suture (Fig. 6E).
In Jagged1+/- embryos, Jagged1 expression was downregulated slightly (Fig. 6B), as expected. In Twist1+/- mutant embryos at this stage Jagged1 expression changed little if at all (Fig. 6C), but the Notch2 expression domain expanded into the sutural mesenchyme (Fig. 6G). This suggests that Twist1 is required to exclude Notch2 from the prospective suture. Combination Twist1+/-;Jagged1+/- mutants exhibited a reduction in Jagged1 expression and an expansion of Notch2 expression similar to that of Twist1+/- embryos (Fig. 6D, H). This expansion of Notch2 at E12.5 presages changes in ALP activity that accompany craniosynostosis.
In control embryos at E13.5, Jagged1 expression in the parietal bone territory resolved into two bands of cells flanking the prospective parietal bone (Fig. 6I, arrows). The upper band was continuous across the coronal suture ectocranial to the developing frontal bone. Notch2 was expressed in the prospective bone territories endocranial to the outer band of Jagged1 expression, and was excluded from the prospective coronal suture (Fig. 6M). Hes1 was expressed at low levels in both the frontal and parietal bone territories and was also excluded from the prospective coronal suture (Fig. 6Q).
These patterns were virtually identical in Jagged1+/- mutants (Fig. 6J, N, R), but were significantly different in Twist1+/- and Twist1+/-;Jagged1+/- mutants. Jagged1 expression in the endocranial band was reduced in Twist1+/- mutants and was largely lost in Twist1+/-;Jagged1+/- mutants (Fig. 6K, L). In Twist1+/- and Twist1+/-;Jagged1+/- mutants, both Notch2 and Hes1 expanded into the coronal suture (Fig. 6O, P, S, T).
Similarly, at E14.5, in wildtype embryos, Jagged1 continued to be expressed in sutural cells (Fig 7A, arrow) and in the ectocranial layer. Expression in sutural cells was selectively lost in Twist1 mutants. Notch2 and Hes1 continued to be expressed in the osteogenic layer (Fig. 7E, I), and in Twist1+/- and Twist1+/-;Jagged1+/- combination mutants, Notch2 and Hes1 expanded into the Jagged1 domain in the midsutural mesenchyme (Fig. 7G, H, K, L). We note finally that Twist1 expression was not influenced by Jagged1 gene dosage (data not shown).
Jagged1 and Twist1 cooperatively control the boundary between osteogenic and sutural cells in the coronal suture
We also assessed the status of the neural crest-mesoderm boundary in Twist1+/-;Jagged1+/- mutants. We used Wnt1-Cre;R26R to mark the neural crest territory, which coincides with the osteogenic cells of the frontal bone (Ting et al., 2009). As is evident in Fig. 7C and 7D, lacZ positive cells crossed into the Jagged1 sutural expression domain in both Twist1+/- and Twist1+/-;Jagged1+/- mutants. The total number of boundary-crossing LacZ-positive cells, summed over the whole length of the coronal suture, increased significantly in the Twist1+/-;Jagged1+/- mutants compared with Twist1+/- mutants (Fig 7M). The average number of boundary-crossing cells per section was approximately the same (not shown). Thus the genetic combination of Twist1 and Jagged1 resulted in an increase in the severity of the boundary defect, defined by the length of the coronal suture over which boundary-crossing events occurred.
Discussion
We sought to test the role of the Notch pathway in craniosynostosis and in the formation of a compartment boundary in the coronal suture. Our focus was the Notch ligand, Jagged1, because of its function in boundary formation and Alagille syndrome, which has craniosynostosis as a feature (Kamath et al., 2002). Here, by means of conditional targeting, we show that inactivation of Jagged1 in the coronal suture results in craniosynostosis. It also results in changes in the identity of sutural cells as early as E12.5 prior to overt osteogenic differentiation, as well as defects in the boundary between osteogenic and non-osteogenic cells in the coronal suture. Surprisingly these changes are associated with increased Notch2 expression and Notch signaling activity in the sutural mesenchyme. Finally, we found that Twist1 controls Jagged1 and Notch2 expression in the prospective coronal suture and Twist1 and Jagged1 exhibit a strong genetic interaction. Thus in addition to its role in the regulation of ephrin-Eph signaling in the coronal suture (Merrill et al., 2006; Ting et al., 2009), Twist1 also regulates the Notch pathway, linking Alagille and Saethre-Chozen syndromes at the pathophysiological level.
It is becoming increasingly clear that at least some forms of craniosynostosis have their beginnings during embryonic development. Our earlier work showed that heterozygous loss of Twist1 function causes defects in the boundary between osteogenic and non-osteogenic cells in the coronal suture as early as E14.5 (Merrill et al., 2006). Recently, we demonstrated that these changes are mediated in part by ephrin-Eph signaling (Ting et al., 2009). Mice carrying the Apert Fgfr2S252W mutant allele exhibit osteogenic differentiation of the sutural mesenchyme at E13.5 (Holmes et al., 2009). The present study pushes back the detection of a change in sutural cells in a craniosynostosis disorder to E12.5 with our finding of expansion of the Notch2 expression domain into the prospective suture and a corresponding increase in the activity of the Notch effector, Hes1. The expansion of Notch2 presages changes in ALP activity that precede craniosynostosis. Notch2 is normally expressed in osteogenic cells of the frontal and parietal bones. Increased Notch signaling can promote osteogenic differentiation in MC3T3-E1 osteoblastic cells and primary human bone marrow mesenchymal stem cells (hMSCs) (Nobta et al., 2005; Tezuka et al., 2002). Also, Hes1 can serve as a coactivator for Runx2 in rat ROS17/2.8 osteoblastic cells (McLarren et al., 2000) and MC3T3-E1 cells (Suh et al., 2008). Therefore upregulation of Notch signaling in the coronal sutures of Jagged1 mutants is consistent with the subsequent change in sutural cells to an osteogenic identity. Conversely, exclusion of Notch activity from sutural cells may serve to maintain such cells in a non-osteogenic state.
In addition to a potential role in controlling the specification of sutural cells, Jagged1 may also be part of a mechanism that maintains a boundary between the osteogenic and non-osteogenic compartments of the coronal suture. The expression of Jagged1 in the sutural mesenchyme and not in the adjacent osteogenic territory supports such as role. Moreover, our finding that mesoderm-derived cells cross into the non-osteogenic territory of the coronal suture demonstrates such role directly. Whether the change in sutural cell identity or the mixing of osteogenic cells with sutural cells is the primary cause of the inappropriate osteogenic differentiation of sutural cells remains unclear. We did not detect cell mixing until E14.5, after the expansion of Notch2 and Hes1 expression at E12.5 and E13.5. Ectopic ALP expression was evident at E14.5. Given this temporal sequence, we propose that an early event predisposes sutural cells to osteogenic differentiation—but does not cause overt differentiation—and subsequent mixing of osteogenic cells with sutural cells provides a final stimulus for such differentiation.
Work performed largely in Drosophila has shown that a major function of compartment boundaries is to establish specialized border cells between the two compartments (Blair, 2003; Irvine and Rauskolb, 2001). These cells are sources of morphogens that influence the fate of cells flanking the boundary. The Notch pathway has a prominent role in the development of such cells in several developmental settings, including the dorsal-ventral boundary of the wing imaginal disc (Irvine and Rauskolb, 2001; Micchelli et al., 1997). In this tissue, Notch is first expressed broadly on both sides of the boundary. Subsequently, through the actions of the dorsally expressed selector gene, Apterous, the Notch ligand Serrate is stimulated dorsally, the Notch ligand Delta is repressed, and the Fringe glycosyl transferase is induced dorsally. Fringe alters the glycosylation of Notch in dorsal cells, making it more sensitive to Delta but insensitive to Serrate. The end result is activation of Notch in a narrow band of cells at the D/V boundary.
We suggest that in the coronal suture, the Notch pathway also functions in the specification of a group of border cells—sutural cells crucial for the development of the suture. At E12.5, Notch2 is expressed in the frontal and parietal territories and at a lower level in the area of the prospective coronal suture (Fig. 6). This reduced expression in the prospective suture requires Twist1. By E14.5, Notch2 expression, and Notch pathway activity as assessed by Hes1 expression, are excluded from the prospective suture. Jagged1 expression begins broadly, overlapping Notch2, but becomes localized to two narrow bands of cells, one ectocranial to the prospective bone, the other located at the boundary between the osteogenic compartments of the frontal and parietal bones. This latter band of cells exhibits reduced expression of Notch2 and Hes1. These cells, we propose, serve as border cells. Loss of Twist1 or Jagged1 function results in expansion of Notch2 and Hes1 into the area of these cells. The result is an increase in cells expressing P-Smad1/5/8, nuclear β-catenin, and P-Erk1/2, as well as a loss of integrity of the osteogenic-non-osteogenic boundary.
Golson and colleagues (2009b) made the interesting observation that during pancreas development, Jagged1 loss of function phenocopies an increase in Notch signaling, consistent with our demonstration of an inhibitory role for Jagged1. These authors suggest that the Notch glycosyltransferase, Mfng, may figure in this process, by causing Jagged1 to act as a competitive inhibitor of signaling mediated by the Notch ligand, Dll1. In a survey of Notch ligands expressed in the coronal suture, we found that Dll1 was expressed in a very limited domain in the central portion of the suture, and that its expression did not change in Jagged1 mutants (data not shown). Thus we believe that a competition between Jagged1 and Dll1 is not likely to be part of the mechanism by which Jagged1 inhibits Notch signaling in the suture. The nature of this mechanism remains obscure.
We note that NOTCH2 is mutated in some cases of Alagille syndrome (McDaniell et al., 2006). Affected individuals, similar to Notch2 mutant mice, have a spectrum of defects that overlap with those present in individuals with Jagged1 mutations. However, these defects do not include craniosynostosis. Our results may provide an explanation for this lack of craniosynostosis in individuals carrying Notch2 mutation: That loss of Jagged1 function in mice results in an increase in Notch signaling in the suture—and craniosynostosis— predicts that gain of function rather than loss of function mutations in NOTCH2 in humans would cause craniosynostosis. Thus far germline gain of function mutations in NOTCH2 have not been reported.
Molecular epistasis experiments did not identify cross regulatory interactions between Jagged1 and ephrinA-EphA signaling in the coronal suture (Fig. 4). Thus Jagged1 is not likely to be upstream of the ephrin-Eph or Fgf pathways. Whether Erk1/2 signaling controls Jagged1 remains to be determined. We did detect an increase in nuclear β-catenin-expressing cells in sutures of Dermo1-Cre;Jagged1cko/cko mutants. Liu and colleagues showed that inactivation of the Wnt pathway inhibitor, Axin2, results in craniosynostosis, and that a deficiency of β-catenin rescues this phenotype (Liu et al., 2007). These findings, together with our demonstration that β-catenin is upregulated in Jagged1 mutant sutures raises the possibility that a key function of Jagged1 is to exclude β-catenin/Wnt activity from sutural cells and thus maintain such cells in a non-osteogenic state. We also noted a change in the distribution of P-Smad1/5/8 expressing cells in Jagged1 mutants. This change was similar to a change in Twist1-EphA4 mutants that we described previously (Ting et al., 2009). In wildtype sutures, P-Smad1/5/8 positive cells were concentrated in the osteogenic fronts; in mutants, they were distributed throughout the suture, consistent with sutural cells assuming an osteogenic identity.
Our present findings, taken together with our previous results on an interaction between Twist1 and EphA4 (Ting et al., 2009), provide an outline of regulatory interactions in coronal suture development (Fig. 8). These results place Twist1 at the top of a regulatory hierarchy, controlling two independent pathways, ephrin-Eph and Jagged1/Notch. Ephrin-Eph functions in the guidance of osteogenic cells along the ectocranial mesenchyme to their destinations in the developing frontal and parietal bones. A failure of this process results in mis-migration of osteogenic precursor cells into the coronal suture. Jagged1/Notch functions in the initial specification of sutural cells and in the boundary between the osteogenic and non-osteogenic compartments in the coronal suture. Jagged1 is also required to exclude β-catenin from sutural cells. Heterozygous loss of Twist1 function results in downregulation of both EphA4 and Jagged1 and consequent changes in osteogenic precursor migration and the osteogenic-non-osteogenic boundary in the coronal suture. Genetic combinations of EphA4-/- or Jagged1+/- with Twist1+/- exacerbate these defects, as expected.
Fig. 8. Schematic model of regulatory interactions during coronal suture development and craniosynostosis.
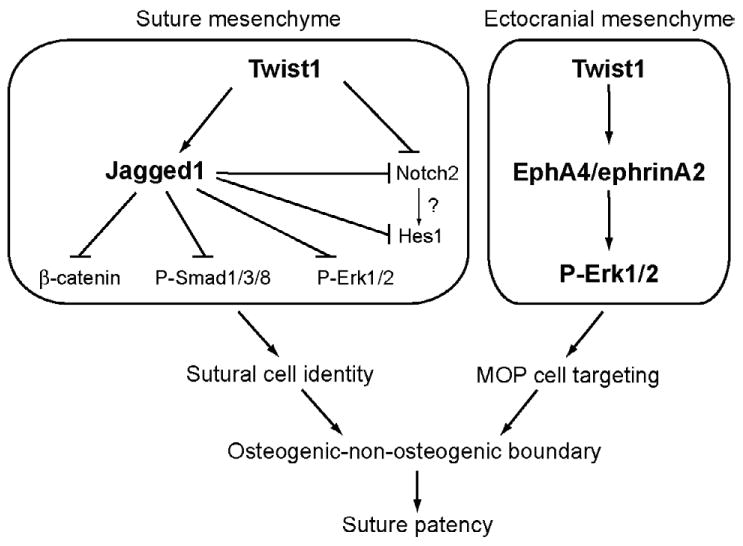
The present findings, together with our previous results on an interaction between Twist1 and EphA4 (Ting et al., 2009), place Twist1 at the top of a regulatory hierarchy, controlling two independent pathways, Jagged1/Notch and ephrinA-EphA. Twist1 positively regulates Jagged1 in sutural mesenchyme. Jagged1 represses Notch2. Jagged1/Notch functions in the initial specification of sutural cells and in the boundary between the osteogenic and non-osteogenic compartments in the coronal suture. Jagged1 is also required to exclude β-catenin, P-Smad1/5/8, and P-Erk1/2 from sutural cells. Twist1 is required for the expression of EphA4 and ephrinA2 in the ectocranial mesenchyme (Ting et al., 2009). EphrinA-EphA signaling, functioning through the RTK pathway, controls the guidance of osteogenic cells along the ectocranial mesenchyme to their destinations in the developing frontal and parietal bones. A failure of this process results in mis-migration of osteogenic precursor cells into the coronal suture. Heterozygous loss of Twist1 function results in downregulation of both EphA4 and Jagged1 and consequent changes in osteogenic precursor migration and the osteogenic-non-osteogenic boundary in the coronal suture. Genetic combinations of EphA4-/- or Jagged1+/- with Twist1+/- exacerbate these defects, as expected. Jagged1 and EphA4 do not influence each other's activity. Thus the Notch and ephrinA-EphA pathways act in parallel downstream of Twist1.
Our results show that in Twist1-Jagged1 mutants, synostosis occurs in the squamosal, lambdoid, occipitointerparietal, and interfrontal sutures in addition to the coronal, implying that mechanisms similar to those we have identified in the coronal suture may operate in other sutures. These sutures differ from the coronal in that the boundary between non-osteogenic cells of the suture and osteogenic cells of the adjacent bone is not coincident with known lineage boundaries. We point out, however, that the absence of a lineage boundary does not prove that developmental boundary does not exist. A lineage restriction is a consequence of heritable gene regulatory mechanisms operating in two compartments (Irvine and Rauskolb, 2001). If the expression of cell-cell interaction genes crucial for boundary formation is not regulated by heritable mechanisms, but instead by local signaling, then a precise lineage boundary need not be present in order for a developmental boundary to be established. Thus we predict that a set of mechanisms similar to those we have documented in the coronal suture, involving ephrin-Eph and Notch signaling under the control of Twist1, also functions in other sutures to establish and maintain boundaries between osteogenic and non osteogenic compartments.
Acknowledgments
We thank Nancy Wu, Mamoru Ishii, Jingjing Sun, Youzhen Yen, Paul Roybal and Christopher Schafer for helpful discussions. This work was supported by NIH Grants DE19650 and DE16320 from the NIDCR to REM.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Alagille D, Estrada A, Hadchouel M, Gautier M, Odièvre M, Dommergues JP. Syndromic paucity of interlobular bile ducts (Alagille syndrome or arteriohepatic dysplasia): review of 80 cases. J Pediatr. 1987;110:195–200. doi: 10.1016/s0022-3476(87)80153-1. [DOI] [PubMed] [Google Scholar]
- Bialek P, Kern B, Yang X, Schrock M, Sosic D, Hong N, Wu H, Yu K, Ornitz DM, Olson EN, Justice MJ, Karsenty G. A twist code determines the onset of osteoblast differentiation. Dev Cell. 2004;6:423–435. doi: 10.1016/s1534-5807(04)00058-9. [DOI] [PubMed] [Google Scholar]
- Blair SS. Lineage compartments in Drosophila. Curr Biol. 2003;13:R548–551. doi: 10.1016/s0960-9822(03)00469-x. [DOI] [PubMed] [Google Scholar]
- Bolós V, Grego-Bessa J, de la Pompa JL. Notch signaling in development and cancer. Endocr Rev. 2007;28:339–363. doi: 10.1210/er.2006-0046. [DOI] [PubMed] [Google Scholar]
- Boyadjiev SA. International Craniosynostosis Consortium. Genetic analysis of non-syndromic craniosynostosis. Orthod Craniofac Res. 2007;10:129–137. doi: 10.1111/j.1601-6343.2007.00393.x. [DOI] [PubMed] [Google Scholar]
- Bray S. Notch signalling in Drosophila: three ways to use a pathway. Semin Cell Dev Biol. 1998;9:591–597. doi: 10.1006/scdb.1998.0262. [DOI] [PubMed] [Google Scholar]
- Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol. 2006;7:678–689. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- Brooker R, Hozumi K, Lewis J. Notch ligands with contrasting functions: Jagged1 and Delta1 in the mouse inner ear. Development. 2006;133:1277–1286. doi: 10.1242/dev.02284. [DOI] [PubMed] [Google Scholar]
- Buceta J, Herranz H, Canela-Xandri O, Reigada R, Sagués F, Milán M. Robustness and stability of the gene regulatory network involved in DV boundary formation in the Drosophila wing. PLoS One. 2007;2:e602. doi: 10.1371/journal.pone.0000602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carver EA, Oram KF, Gridley T. Craniosynostosis in Twist heterozygous mice: a model for Saethre-Chotzen syndrome. Anat Rec. 2002;268:90–92. doi: 10.1002/ar.10124. [DOI] [PubMed] [Google Scholar]
- Chai Y, Maxson RE., Jr Recent advances in craniofacial morphogenesis. Dev Dyn. 2006;235:2353–2375. doi: 10.1002/dvdy.20833. [DOI] [PubMed] [Google Scholar]
- Chen YH, Ishii M, Sun J, Sucov HM, Maxson RE., Jr Msx1 and Msx2 regulate survival of secondary heart field precursors and post-migratory proliferation of cardiac neural crest in the outflow tract. Dev Biol. 2007;308:421–437. doi: 10.1016/j.ydbio.2007.05.037. [DOI] [PubMed] [Google Scholar]
- Chen ZF, Behringer RR. twist is required in head mesenchyme for cranial neural tube morphogenesis. Genes Dev. 1995;9:686–699. doi: 10.1101/gad.9.6.686. [DOI] [PubMed] [Google Scholar]
- Cohen MM., Jr The new bone biology: pathologic, molecular, and clinical correlates. Am J Med Genet A. 2006;140:2646–2706. doi: 10.1002/ajmg.a.31368. [DOI] [PubMed] [Google Scholar]
- Connerney J, Andreeva V, Leshem Y, Mercado MA, Dowell K, Yang X, Lindner V, Friesel RE, Spicer DB. Twist1 homodimers enhance FGF responsiveness of the cranial sutures and promote suture closure. Dev Biol. 2008;318:323–334. doi: 10.1016/j.ydbio.2008.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahmann C, Basler K. Compartment boundaries: at the edge of development. Trends Genet. 1999;15:320–326. doi: 10.1016/s0168-9525(99)01774-6. [DOI] [PubMed] [Google Scholar]
- Danielian PS, Muccino D, Rowitch DH, Michael SK, McMahon AP. Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr Biol. 1998;8:1323–1326. doi: 10.1016/s0960-9822(07)00562-3. [DOI] [PubMed] [Google Scholar]
- el Ghouzzi V, Le Merrer M, Perrin-Schmitt F, Lajeunie E, Benit P, Renier D, Bourgeois P, Bolcato-Bellemin AL, Munnich A, Bonaventure J. Mutations of the TWIST gene in the Saethre-Chotzen syndrome. Nat Genet. 1997;15:42–46. doi: 10.1038/ng0197-42. [DOI] [PubMed] [Google Scholar]
- Emerick KM, Rand EB, Goldmuntz E, Krantz ID, Spinner NB, Piccoli DA. Features of Alagille syndrome in 92 patients: frequency and relation to prognosis. Hepatology. 1999;29:822–829. doi: 10.1002/hep.510290331. [DOI] [PubMed] [Google Scholar]
- Golson ML, Le Lay J, Gao N, Brämswig N, Loomes KM, Oakey R, May CL, White P, Kaestner KH. Jagged1 is a competitive inhibitor of Notch signaling in the embryonic pancreas. Mech Dev. 2009b;126:687–699. doi: 10.1016/j.mod.2009.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golson ML, Loomes KM, Oakey R, Kaestner KH. Ductal malformation and pancreatitis in mice caused by conditional Jag1 deletion. Gastroenterology. 2009a;136:1761–1771.e1. doi: 10.1053/j.gastro.2009.01.040. [DOI] [PubMed] [Google Scholar]
- Hajihosseini MK. Fibroblast growth factor signaling in cranial suture development and pathogenesis. Front Oral Biol. 2008;12:160–177. doi: 10.1159/000115037. [DOI] [PubMed] [Google Scholar]
- Holmes G, Rothschild G, Roy UB, Deng CX, Mansukhani A, Basilico C. Early onset of craniosynostosis in an Apert mouse model reveals critical features of this pathology. Dev Biol. 2009;328:273–84. doi: 10.1016/j.ydbio.2009.01.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard TD, Paznekas WA, Green ED, Chiang LC, Ma N, Ortiz de Luna RI, Garcia Delgado C, Gonzalez-Ramos M, Kline AD, Jabs EW. Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre-Chotzen syndrome. Nat Genet. 1997;15:36–41. doi: 10.1038/ng0197-36. [DOI] [PubMed] [Google Scholar]
- Irvine KD, Rauskolb C. Boundaries in development: formation and function. Annu Rev Cell Dev Biol. 2001;17:189–214. doi: 10.1146/annurev.cellbio.17.1.189. [DOI] [PubMed] [Google Scholar]
- Ishii M, Merrill AE, Chan YS, Gitelman I, Rice DP, Sucov HM, Maxson RE., Jr Msx2 and Twist cooperatively control the development of the neural crest-derived skeletogenic mesenchyme of the murine skull vault. Development. 2003;130:6131–6142. doi: 10.1242/dev.00793. [DOI] [PubMed] [Google Scholar]
- Jabs EW, Müller U, Li X, Ma L, Luo W, Haworth IS, Klisak I, Sparkes R, Warman ML, Mulliken JB, et al. A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell. 1993;75:443–450. doi: 10.1016/0092-8674(93)90379-5. [DOI] [PubMed] [Google Scholar]
- Jarriault S, Brou C, Logeat F, Schroeter EH, Kopan R, Israel A. Signalling downstream of activated mammalian Notch. Nature. 1995;377:355–358. doi: 10.1038/377355a0. [DOI] [PubMed] [Google Scholar]
- Jenkins D, Seelow D, Jehee FS, Perlyn CA, Alonso LG, Bueno DF, Donnai D, Josifova D, Mathijssen IM, Morton JE, Orstavik KH, Sweeney E, Wall SA, Marsh JL, Nurnberg P, Passos-Bueno MR, Wilkie AO. RAB23 mutations in Carpenter syndrome imply an unexpected role for hedgehog signaling in cranial-suture development and obesity. Am J Hum Genet. 2007;80:1162–1170. doi: 10.1086/518047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Iseki S, Maxson RE, Sucov HM, Morriss-Kay GM. Tissue origins and interactions in the mammalian skull vault. Dev Biol. 2002;241:106–116. doi: 10.1006/dbio.2001.0487. [DOI] [PubMed] [Google Scholar]
- Kamath BM, Spinner NB, Emerick KM, Chudley AE, Booth C, Piccoli DA, Krantz ID. Vascular anomalies in Alagille syndrome: a significant cause of morbidity and mortality. Circulation. 2004;109:1354–1358. doi: 10.1161/01.CIR.0000121361.01862.A4. [DOI] [PubMed] [Google Scholar]
- Kamath BM, Stolle C, Bason L, Colliton RP, Piccoli DA, Spinner NB, Krantz ID. Craniosynostosis in Alagille syndrome. Am J Med Genet. 2002;112:176–180. doi: 10.1002/ajmg.10608. [DOI] [PubMed] [Google Scholar]
- Kiecker C, Lumsden A. Compartments and their boundaries in vertebrate brain development. Nat Rev Neurosci. 2005;6:553–564. doi: 10.1038/nrn1702. [DOI] [PubMed] [Google Scholar]
- Kiernan AE, Xu J, Gridley T. The Notch ligand JAG1 is required for sensory progenitor development in the mammalian inner ear. PLoS Genet. 2006;2:e4. doi: 10.1371/journal.pgen.0020004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell. 2009;137:216–233. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krantz ID, Colliton RP, Genin A, Rand EB, Li L, Piccoli DA, Spinner NB. Spectrum and frequency of jagged1 (JAG1) mutations in Alagille syndrome patients and their families. Am J Hum Genet. 1998;62:1361–1369. doi: 10.1086/301875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai EC. Notch signaling: control of cell communication and cell fate. Development. 2004;131:965–973. doi: 10.1242/dev.01074. [DOI] [PubMed] [Google Scholar]
- Le TT, Conley KW, Brown NL. Jagged 1 is necessary for normal mouse lens formation. Dev Biol. 2009;328:118–126. doi: 10.1016/j.ydbio.2009.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Scott DA, Hatch E, Tian X, Mansour SL. Dusp6 (Mkp3) is a negative feedback regulator of FGF-stimulated ERK signaling during mouse development. Development. 2007;134:167–176. doi: 10.1242/dev.02701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Yu HM, Hsu W. Craniosynostosis caused by Axin2 deficiency is mediated through distinct functions of beta-catenin in proliferation and differentiation. Dev Biol. 2007;301:298–308. doi: 10.1016/j.physletb.2003.10.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, Meyers J, Leitch CC, Katsanis N, Sharifi N, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37:275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- Lozier J, McCright B, Gridley T. Notch signaling regulates bile duct morphogenesis in mice. PLoS One. 2008;3:e1851. doi: 10.1371/journal.pone.0001851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Major RJ, Irvine KD. Influence of Notch on dorsoventral compartmentalization and actin organization in the Drosophila wing. Development. 2005;132:3823–3833. doi: 10.1242/dev.01957. [DOI] [PubMed] [Google Scholar]
- McCright B, Lozier J, Gridley T. A mouse model of Alagille syndrome: Notch2 as a genetic modifier of Jag1 haploinsufficiency. Development. 2002;129:1075–1082. doi: 10.1242/dev.129.4.1075. [DOI] [PubMed] [Google Scholar]
- McDaniell R, Warthen DM, Sanchez-Lara PA, Pai A, Krantz ID, Piccoli DA, Spinner NB. NOTCH2 mutations cause Alagille syndrome, a heterogeneous disorder of the notch signaling pathway. Am J Hum Genet. 2006;79:169–173. doi: 10.1086/505332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLarren KW, Lo R, Grbavec D, Thirunavukkarasu K, Karsenty G, Stifani S. The mammalian basic helix loop helix protein HES-1 binds to and modulates the transactivating function of the runt-related factor Cbfa1. J Biol Chem. 2000;275:530–538. doi: 10.1074/jbc.275.1.530. [DOI] [PubMed] [Google Scholar]
- Merrill AE, Bochukova EG, Brugger SM, Ishii M, Pilz DT, Wall SA, Lyons KM, Wilkie AO, Maxson RE., Jr Cell mixing at a neural crest-mesoderm boundary and deficient ephrin-Eph signaling in the pathogenesis of craniosynostosis. Hum Mol Genet. 2006;15:1319–1328. doi: 10.1093/hmg/ddl052. [DOI] [PubMed] [Google Scholar]
- Micchelli CA, Rulifson EJ, Blair SS. The function and regulation of cut expression on the wing margin of Drosophila: Notch, Wingless and a dominant negative role for Delta and Serrate. Development. 1997;124:1485–1495. doi: 10.1242/dev.124.8.1485. [DOI] [PubMed] [Google Scholar]
- Moenning A, Jäger R, Egert A, Kress W, Wardelmann E, Schorle H. Sustained platelet-derived growth factor receptor alpha signaling in osteoblasts results in craniosynostosis by overactivating the phospholipase C-gamma pathway. Mol Cell Biol. 2009;29:881–891. doi: 10.1128/MCB.00885-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morriss-Kay GM, Wilkie AO. Growth of the normal skull vault and its alteration in craniosynostosis: insights from human genetics and experimental studies. J Anat. 2005;207:637–653. doi: 10.1111/j.1469-7580.2005.00475.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobta M, Tsukazaki T, Shibata Y, Xin C, Moriishi T, Sakano S, Shindo H, Yamaguchi A. Critical regulation of bone morphogenetic protein-induced osteoblastic differentiation by Delta1/Jagged1-activated Notch1 signaling. J Biol Chem. 2005;280:15842–15848. doi: 10.1074/jbc.M412891200. [DOI] [PubMed] [Google Scholar]
- Oda T, Elkahloun AG, Pike BL, Okajima K, Krantz ID, Genin A, Piccoli DA, Meltzer PS, Spinner NB, Collins FS, Chandrasekharappa SC. Mutations in the human Jagged1 gene are responsible for Alagille syndrome. Nat Genet. 1997;16:235–242. doi: 10.1038/ng0797-235. [DOI] [PubMed] [Google Scholar]
- Opperman LA. Cranial sutures as intramembranous bone growth sites. Dev Dyn. 2000;219:472–485. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1073>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- Oram KF, Gridley T. Mutations in snail family genes enhance craniosynostosis of Twist1 haplo-insufficient mice: implications for Saethre-Chotzen Syndrome. Genetics. 2005;170:971–974. doi: 10.1534/genetics.105.041277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice DP. Developmental anatomy of craniofacial sutures. Front Oral Biol. 2008;12:1–21. doi: 10.1159/000115028. [DOI] [PubMed] [Google Scholar]
- Rice DP, Aberg T, Chan Y, Tang Z, Kettunen PJ, Pakarinen L, Maxson RE, Thesleff I. Integration of FGF and TWIST in calvarial bone and suture development. Development. 2000;127:1845–1855. doi: 10.1242/dev.127.9.1845. [DOI] [PubMed] [Google Scholar]
- Saga Y, Miyagawa-Tomita S, Takagi A, Kitajima S, Miyazaki J, Inoue T. MesP1 is expressed in the heart precursor cells and required for the formation of a single heart tube. Development. 1999;126:3437–3447. doi: 10.1242/dev.126.15.3437. [DOI] [PubMed] [Google Scholar]
- Saravanamuthu SS, Gao CY, Zelenka PS. Notch signaling is required for lateral induction of Jagged1 during FGF-induced lens fiber differentiation. Dev Biol. 2009;332:166–176. doi: 10.1016/j.ydbio.2009.05.566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Settle SH, Jr, Rountree RB, Sinha A, Thacker A, Higgins K, Kingsley DM. Multiple joint and skeletal patterning defects caused by single and double mutations in the mouse Gdf6 and Gdf5 genes. Dev Biol. 2003;254:116–130. doi: 10.1016/s0012-1606(02)00022-2. [DOI] [PubMed] [Google Scholar]
- Slater BJ, Lenton KA, Kwan MD, Gupta DM, Wan DC, Longaker MT. Cranial sutures: a brief review. Plast Reconstr Surg. 2008;121:170e–178e. doi: 10.1097/01.prs.0000304441.99483.97. [DOI] [PubMed] [Google Scholar]
- Sood S, Eldadah ZA, Krause WL, McIntosh I, Dietz HC. Mutation in fibrillin-1 and the Marfanoid-craniosynostosis (Shprintzen-Goldberg) syndrome. Nat Genet. 1996;12:209–211. doi: 10.1038/ng0296-209. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Suh JH, Lee HW, Lee JW, Kim JB. Hes1 stimulates transcriptional activity of Runx2 by increasing protein stabilization during osteoblast differentiation. Biochem Biophys Res Commun. 2008;367:97–102. doi: 10.1016/j.bbrc.2007.12.100. [DOI] [PubMed] [Google Scholar]
- Tepass U, Godt D, Winklbauer R. Cell sorting in animal development: signalling and adhesive mechanisms in the formation of tissue boundaries. Curr Opin Genet Dev. 2002;12:572–582. doi: 10.1016/s0959-437x(02)00342-8. [DOI] [PubMed] [Google Scholar]
- Tezuka K, Yasuda M, Watanabe N, Morimura N, Kuroda K, Miyatani S, Hozumi N. Stimulation of osteoblastic cell differentiation by Notch. J Bone Miner Res. 2002;17:231–239. doi: 10.1359/jbmr.2002.17.2.231. [DOI] [PubMed] [Google Scholar]
- Ting MC, Wu NL, Roybal PG, Sun J, Liu L, Yen Y, Maxson RE., Jr EphA4 as an effector of Twist1 in the guidance of osteogenic precursor cells during calvarial bone growth and in craniosynostosis. Development. 2009;136:855–864. doi: 10.1242/dev.028605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigg SR, Kan R, Babbs C, Bochukova EG, Robertson SP, Wall SA, Morriss-Kay GM, Wilkie AO. Mutations of ephrin-B1 (EFNB1), a marker of tissue boundary formation, cause craniofrontonasal syndrome. Proc Natl Acad Sci USA. 2004;101:8652–8657. doi: 10.1073/pnas.0402819101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkie AO. Craniosynostosis: genes and mechanisms. Hum Mol Genet. 1997;6:1647–1656. doi: 10.1093/hmg/6.10.1647. [DOI] [PubMed] [Google Scholar]
- Xue Y, Gao X, Lindsell CE, Norton CR, Chang B, Hicks C, Gendron-Maguire M, Rand EB, Weinmaster G, Gridley T. Embryonic lethality and vascular defects in mice lacking the Notch ligand Jagged1. Hum Mol Genet. 1999;8:723–730. doi: 10.1093/hmg/8.5.723. [DOI] [PubMed] [Google Scholar]
- Yoshida T, Vivatbutsiri P, Morriss-Kay G, Saga Y, Iseki S. Cell lineage in mammalian craniofacial mesenchyme. Mech Dev. 2008;125:797–808. doi: 10.1016/j.mod.2008.06.007. [DOI] [PubMed] [Google Scholar]
- Yu HM, Jerchow B, Sheu TJ, Liu B, Costantini F, Puzas JE, Birchmeier W, Hsu W. The role of Axin2 in calvarial morphogenesis and craniosynostosis. Development. 2005;132:1995–2005. doi: 10.1242/dev.01786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Xu J, Liu Z, Sosic D, Shao J, Olson EN, Towler DA, Ornitz DM. Conditional inactivation of FGF receptor 2 reveals an essential role for FGF signaling in the regulation of osteoblast function and bone growth. Development. 2003;130:3063–3074. doi: 10.1242/dev.00491. [DOI] [PubMed] [Google Scholar]
- Yuan ZR, Kobayashi N, Kohsaka T. Human Jagged 1 mutants cause liver defect in Alagille syndrome by overexpression of hepatocyte growth factor. J Mol Biol. 2006;356:559–568. doi: 10.1016/j.jmb.2005.11.097. [DOI] [PubMed] [Google Scholar]
- Zhang X, Kuroda S, Carpenter D, Nishimura I, Soo C, Moats R, Iida K, Wisner E, Hu FY, Miao S, et al. Craniosynostosis in transgenic mice overexpressing Nell-1. J Clin Invest. 2002;110:861–870. doi: 10.1172/JCI15375. [DOI] [PMC free article] [PubMed] [Google Scholar]