

Abstract
Filamentous (F)-actin is a known regulator of the synaptic vesicle (SV) cycle, with roles in SV mobilization, fusion, and endocytosis. However, the molecular pathways that regulate its dynamic assembly within presynaptic boutons remain unclear. In this study, we have used shRNA-mediated knockdown to demonstrate that Piccolo, a multidomain protein of the active zone cytomatrix, is a key regulator of presynaptic F-actin assembly. Boutons lacking Piccolo exhibit enhanced activity-dependent Synapsin1a dispersion and SV exocytosis, and reduced F-actin polymerization and CaMKII recruitment. These phenotypes are rescued by stabilizing F-actin filaments and mimicked by knocking down Profilin2, another regulator of presynaptic F-actin assembly. Importantly, we find that mice with a targeted deletion of exon 14 from the Pclo gene, reported to lack >95% of Piccolo, continue to express multiple Piccolo isoforms. Furthermore, neurons cultured from these mice exhibit no defects in presynaptic F-actin assembly due to the expression of these isoforms at presynaptic boutons. These data reveal that Piccolo regulates neurotransmitter release by facilitating activity-dependent F-actin assembly and the dynamic recruitment of key signaling molecules into presynaptic boutons, and highlight the need for new genetic models with which to study Piccolo loss of function.
Introduction
Synaptic transmission depends on the regulated release of neurotransmitter from specialized domains of the axonal plasma membrane called active zones (AZs). This process involves synaptic vesicle (SV) exocytosis and endocytosis, as well as mobilization from the reserve (RP) to readily releasable pool (RRP) during periods of sustained neuronal activity (Südhof, 2004). Although the specialized activities of many proteins are required for these processes, only one molecule, actin, modulates each of these steps. For instance, filamentous (F)-actin negatively regulates SV release probability (Pvr) by creating a barrier to restrain SV fusion at the AZ (Morales et al., 2000; Cingolani and Goda, 2008), maintains the RP and mediates SV translocation to the RRP through interactions with synapsins (Greengard et al., 1994; Hilfiker et al., 1999; Jensen et al., 2007; Cingolani and Goda, 2008), and can regulate SV endocytosis together with dynamin, Abp1, and synapsin (Kessels et al., 2001; Shupliakov et al., 2002; Bloom et al., 2003; Engqvist-Goldstein and Drubin, 2003; Dillon and Goda, 2005; Evergren et al., 2007). Despite its many roles, there is currently little known about where and how presynaptic F-actin assembly is coordinated.
One likely site of presynaptic F-actin regulation is the AZ. Ultrastructural and imaging studies indicate that actin is a component of the active zone cytomatrix (CAZ) (Hirokawa et al., 1989; Morales et al., 2000; Phillips et al., 2001; Bloom et al., 2003; Li et al., 2010), an electron-dense structure associated with SV release sites. Moreover, F-actin depolymerization has been shown to transiently increase Pvr (Morales et al., 2000), indicating that it negatively regulates SV fusion at the AZ (Morales et al., 2000; Cingolani and Goda, 2008). It remains unclear how F-actin is linked to the CAZ and SV fusion machinery, or how its assembly is regulated in response to synaptic activity, although several CAZ-associated proteins have been suggested to have roles in these processes, including Piccolo, Rab3a-interacting molecules (RIMs), and neurexins (Morales et al., 2000). Of these candidates, Piccolo, the largest CAZ protein (∼560 kDa), is uniquely capable of spanning multiple presynaptic subdomains and scaffolding a series of actin regulatory molecules, including Abp1, GIT1, and profilin (Wang et al., 1999; Fenster et al., 2003; Kim et al., 2003). In addition, Piccolo knockdown enhances both activity-dependent SV exocytosis and Synapsin1a dispersion out of presynaptic boutons (Leal-Ortiz et al., 2008), phenotypes similar to those observed following actin depolymerization (Sankaranarayanan et al., 2003).
In the current study, we show that Piccolo indeed regulates presynaptic F-actin assembly. Piccolo knockdown phenotypes are rescued by F-actin stabilization and phenocopied by knockdown of Profilin2, another presynaptic actin regulator. Intriguingly, Piccolo-mediated F-actin assembly regulates not only Synapsin1a dynamics and SV exocytosis, but also the activity-dependent recruitment of CaMKII into presynaptic boutons. Since CaMKII is implicated in presynaptic plasticity (Ninan and Arancio, 2004; Wang, 2008), these data suggest that Piccolo may have roles in both basal neurotransmission and plasticity mechanisms. Finally, we find that mice reported to lack >95% of Piccolo (Mukherjee et al., 2010) do not exhibit defects in presynaptic F-actin assembly, due to the continued expression of multiple synaptically localized Piccolo isoforms. These findings demonstrate that Piccolo regulates neurotransmitter release by facilitating F-actin assembly, and highlight the need for new genetic models with which to study the synaptic, circuit, and behavioral consequences of Piccolo loss.
Materials and Methods
Reagents
Antibodies against Piccolo (rabbit) and MAP2 (rabbit and mouse) were used as previously described (Zhai et al., 2000). Tubulin (mouse) antibodies were from Sigma-Aldrich, Profilin2 (mouse) and synaptophysin (rabbit) antibodies were from Santa Cruz, GFP (mouse) antibody was from Roche, and Homer1 (rabbit) and synapsin (mouse) antibodies were from Synaptic Systems. FM4-64 was purchased from Invitrogen; 4-[(2S)-2-[(5-isoquinolinylsulfonyl)methylamino]-3-oxo-3-(4-phenyl-1-piperazinyl)propyl]phenyl isoquinolinesulfonic acid ester (KN62), (9R,10S,12S)-2,3,9,10,11,12-hexahydro-10-hydroxy-9-methyl-1-oxo-9,12-epoxy-1H-diindolo[1,2,3-fg:3′,2′,1′-kl]pyrrolo[3,4-i][1,6]benzodiazocine-10-carboxylic acid, hexyl ester (KT5720), and 2-(2-amino-3-methoxyphenyl)-4H-1-benzopyran-4-one (PD98) from Tocris; latrunculin A from Calbiochem; and jasplakinolide from Calbiochem, Invitrogen, and Axxora. Unless otherwise indicated, all other chemicals are from Sigma-Aldrich.
Design of short hairpin RNAs and FUGW lentiviral vectors
The short hairpin RNA (shRNA) against Profilin2 was designed as described previously (Leal-Ortiz et al., 2008). The target sequence (Pfn380; AGGCATACTCAATGGCAAA; from Rattus norvegicus Profilin2; GenBank accession no. NM_030873) was subcloned into pZOff 2.0 (modified from pZOff 1.0 to have a U6 instead of H1 promoter) at the BglII and HindIII sites using the following primers (5′ to 3′: GATCTCAGGCATACTCAATGGCAAAttcaagagaTTTGCCATTGAGTATGCCTTTTTTGGAA (forward) and AGCTTTTCCAAAAAAGGCATACTCAATGGCAAAtctcttgaaTTTGCCATTGAGT ATGCCTGA (reverse). From pZOff 2.0, the sequence containing the U6 promoter and Pfn380, flanked by Acc1 and EcoR1, was subcloned into the FUGW H1+ vector [described by Leal-Ortiz et al. (2008)] at the Bsiw1 and Pac1 sites to create the FUGW vector for Pfn380 expression. The combined Pclo28/Pfn380 double knockdown vector was created by modifying the FUGW H1+ vector in two steps. First, a 750 nt stuffer sequence containing 5′ EcoR1 and BstB1 sites was inserted at the Bsiw1 site. Second, the sequence containing the U6 promoter and Pfn380 shRNA, again flanked by EcoR1 and Acc1 sites, was subcloned in at the EcoR1 and BstB1 sites. The resulting FUGW vector drives expression of the Pclo28 shRNA via the H1 promoter and, separated by 750 nt, the Pfn380 shRNA via the U6 promoter, an arrangement that allows for efficient lentivirus production and simultaneous knockdown of both Piccolo and Profilin2. EGFP-tagged Synapsin1a wild type, S/A phosphomutants, EGFP-actin, YFP-CaMKII, or EGFP-Profilin2 were subcloned into these FUGW vectors in place of soluble EGFP.
Hippocampal culture and lentiviral infection
Primary hippocampal cultures were prepared using a modified Banker culture protocol, as previously described (Waites et al., 2009). Neurons were infected with lentivirus containing EGFP-tagged proteins in the absence or presence of shRNAs on DIV 0, prepared as previously described (Leal-Ortiz et al., 2008; Waites et al., 2009).
Mouse hippocampal cultures were similarly prepared from P0–P1 pups of either sex (strain name, B6;129S6-Pclotm1Sud/J; The Jackson Laboratory), except that dissociated neurons were plated onto poly-l-lysine-coated coverslips precoated with a glial feeder layer (one hippocampus/18-mm-diameter coverslip), and maintained in Neurobasal A medium supplemented with B27 and GlutaMAX. Neurons were infected with lentivirus (5 μl/coverslip) on the day of plating and used for experiments on DIV 10. Genotyping was performed according to the protocol on The Jackson Laboratory website (www.jax.org) for strain B6;129S6-Pclotm1Sud/J.
Immunocytochemistry
Neurons were fixed and processed for immunofluorescence as previously described (Leal-Ortiz et al., 2008), using primary antibodies against synaptophysin, Homer, MAP2, Piccolo, and GFP [for stimulated emission depletion (STED) microscopy]. Alexa 568 and 647 (anti-mouse and anti-rabbit; Invitrogen) were used as secondary antibodies, and Alexa 568-labeled phalloidin (Invitrogen) was also used to label dendritic spines. For STED microscopy, anti-mouse Atto 647 (Sigma-Aldrich) was used to detect EGFP-actin. For conventional immunostaining, images were acquired on a spinning disc confocal microscope (Zeiss Axiovert 200M with PerkinElmer spinning disc and Melles Griot 43 series ion laser), using a 63× Plan-Apochromat objective (NA 1.4), photometrics Cascade 512B digital camera (Roper Scientific), and MetaMorph software (Molecular Devices).
STED microscopy
A Leica TCS STED setup with a 100×, 1.4 NA oil objective (Leica) was used to acquire STED images. The dye (goat anti-mouse Atto 647N; Sigma-Aldrich; 1:200) was excited with a pulsed laser at 635 nm and depleted at 760 nm (Mai Tai Ti:Sapphire; Newport/Spectra Physics). Avalanche photodiodes were used to detect wavelengths between 650 and 710 nm. Images were acquired using the Leica Application Suite Advanced Fluorescence software. STED images were processed using a linear deconvolution algorithm integrated into the ImSpector Data Acquisition and Analysis Environment (Max Planck Innovation). Regularization parameters ranged from 2e-11 to 1e-12. The point spread function was generated by using a 2D Lorentzian function with its half-width and half-length fitted to the half-width and half-length obtained by images of 25 nm crimson beads conjugated to Atto 647N.
Western blot
Immunoblots of cellular lysates were prepared from lentivirally infected hippocampal neurons as described previously (Leal-Ortiz et al., 2008). Protein levels were standardized by loading equal amounts of α-tubulin in all lanes. For mouse experiments, postnuclear supernatant from total brain homogenates were prepared as previously described (Zhai et al., 2000), and protein levels were standardized using the Bradford assay.
Live imaging
All live-imaging experiments were performed on a custom-built (by C. C. Garner) scanning confocal microscope (Zeiss Axiovert 200M) equipped with a 40× objective (1.3 NA; Zeiss Plan Neofluar), 488–514 nm laser (Spectra Physics), and using OpenView software (written by Dr. Noam Ziv, Haifa, Israel). Neuronal coverslips were mounted in a custom-built chamber designed for perfusion and electrical stimulation, heated to 37°C by forced-air blower, and perfused with Tyrode's saline solution (25 mm HEPES, 119 mm NaCl, 2.5 mm KCl, 30 mm glucose, 2 mm CaCl, 2 mm MgCl2, 50 μm CNQX, 10 μm APV, pH 7.4).
Synapsin dispersion.
Dispersion of EGFP-Synapsin1a was induced by electrical stimulation (10 Hz, 90 s), as previously described (Chi et al., 2001). Images were acquired before stimulation and every 5 s during stimulation. For experiments with latrunculin A (10 μm), KN62 (10 μm), or jasplakinolide (5 μm), neurons were preincubated for 5 or 20 min (for KN62), with the drugs diluted 1:1000 in Tyrode's solution [from 10 mm (latrunculin, KN62) or 5 mm (jasplakinolide) stocks in DMSO]. Images were acquired before and after drug treatment (before stimulation) to insure that the drugs themselves had no effect on EGFP-Synapsin1a dispersion.
FM loading/destaining.
Presynaptic boutons were labeled with FM4-64 by 45 s incubation in high-potassium Tyrode's solution (90 mm KCl, 31.5 mm NaCl) containing ∼1 μg/ml FM dye, followed by 30 s incubation in normal Tyrode's with 1 μg/ml FM dye. Neurons were then washed in Tyrode's solution for ∼2 min before imaging. Destaining was performed by 10 Hz, 90 s electrical stimulation. Simultaneous images of EGFP-synapsin and FM4-64 were acquired before stimulation and every 5 s during stimulation. For experiments with latrunculin A or jasplakinolide, FM loading was performed before drug treatment (5 min for latrunculin A, 10 min for jasplakinolide), and images were acquired before and after treatment to assess whether the drugs affected basal FM levels.
Actin/CaMKII/Profilin2 clustering.
EGFP-actin, YFP-CaMKII, or EGFP-Profilin2 clustering was induced by 60 s incubation in high-potassium Tyrode's solution while simultaneously labeling boutons with FM4-64. Three images were acquired before stimulation and again following the FM4-64 labeling/washing procedure. For experiments with jasplakinolide and latrunculin A, images were acquired before drug treatment, after treatment (5 min for latrunculin A, 10 min for jasplakinolide), and following high-K+ stimulation.
Quantification of synapsin dispersion/FM destaining
Image analysis and quantification were performed with OpenView software and Microsoft Excel. GraphPad Prism was used for curve fitting, graph plotting, and statistical analyses.
As described previously (Chi et al., 2001), dispersion or destaining curves for EGFP-Syn or FM4-64 puncta were obtained using the following equation: (Fo − Ft)/Fo, where Fo represents initial fluorescence intensity before stimulation (obtained by averaging puncta intensities from two images taken 5 s apart), and Ft represents fluorescence intensity at each of the 18 time points t during stimulation, from t = 5 to t = 90 s. Curves for each puncta in a field of view were pooled and averaged to give a single destaining curve/coverslip. Curves for individual puncta containing values >0 for time points >20 s were eliminated from the analysis (to correct for imperfections in the puncta-tracking software).
To calculate the average extent of EGFP-Syn dispersion or FM4-64 destaining for each coverslip, the ΔF/Fo intensity values for the last five time points (t = 70–90 s) were averaged to give avg Ft = 70–90 (Fig. 1A). To express percentage increase in EGFP-Syn dispersion or FM destaining for condition B (i.e., Pclo28 knockdown) versus condition A (i.e., wild type), both imaged on a given day, the following equation was used: [(avg Ft = 70–90 (B)/avg Ft = 70–90 (A)) − 1] × 100 (Fig. 1A). Resulting data points were then plotted on a column graph, enabling comparisons between experiments performed in different batches of neurons (Fig. 1B).
Figure 1.

Illustration of how percentage increase in EGFP-Synapsin1a dispersion was calculated. A, Graph depicting time course of EGFP-Syn dispersion for two sets of experiments from 2 different days, #1 (black) and #2 (red). Each curve represents the averaged values from >100 puncta. Note the pronounced week-to-week differences in absolute extent of EGFP-Syn dispersion observed (black curves vs red curves). Note also that wild-type curves (squares) always exhibit less dispersion relative to Pclo28 curves (triangles). The first step, indicated by gray “1),” for calculating percentage increase in EGFP-Syn dispersion (Pclo28 vs wild type) was to average the last five time points of each curve from a given day (values in gray boxes), producing the values shown. These values were then put into the equation [((avg Ft = 70–90 (Pclo28)/avg Ft = 70–90 (wt)) − 1)] × 100 (see Materials and Methods). B, Column graph depicting the final values for percentage increase in EGFP-Syn dispersion (Pclo28 vs wt) and their average (black line). Note that these ratioed values are reasonably similar when compared across different weeks (red circle vs black circle). C, Graph depicting time course of EGFP-Syn dispersion for one set of experiments from a single day, demonstrating how percentage increase in dispersion was calculated when there were multiple coverslips for each condition (wt 1, 2, 3 and Pclo28 1, 2, 3). The last five time points for all wild-type curves (black; in gray box) were averaged together to give a single value (−0.28). The last five time points for each Pclo28 curve (blue) were averaged to give a separate value for each coverslip. These values were each put into the above equation and plotted individually on the column graph (D).
To express percentage decrease in dispersion or destaining for a coverslip of condition A (i.e., wild-type neurons) versus condition B (i.e., Pclo28-expressing neurons), the same general strategy was used, with the following equation: [1 − (avg Ft = 70–90 (A)/avg Ft = 70–90 (B))] × 100. This measurement was used to assess rescue of Synapsin1a dispersion or FM destaining in the Pclo28 background by Synapsin1a S/A phosphomutants or jasplakinolide (as in Figs. 3, 4).
Figure 3.
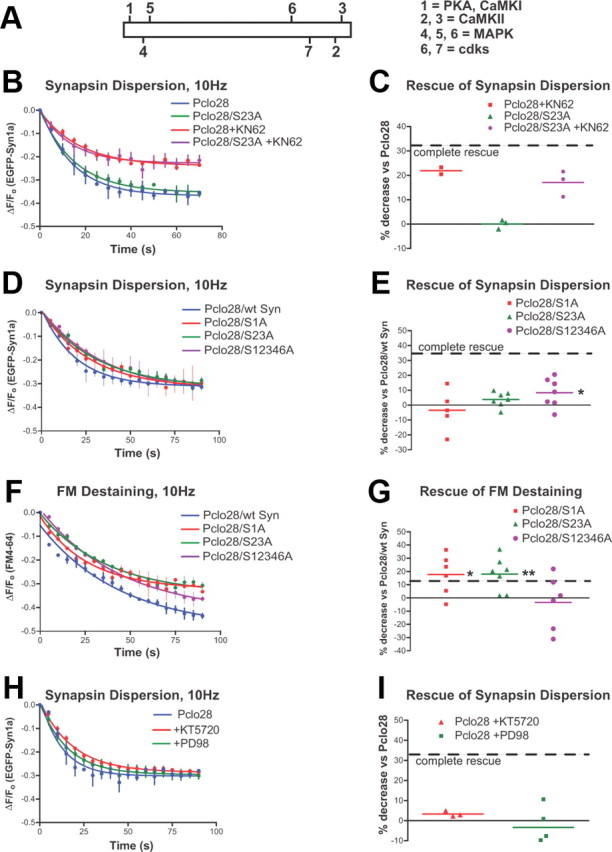
Aberrant Synapsin1a phosphorylation does not cause the Pclo28 phenotypes. A, Schematic of Synapsin1a showing its seven phospho-sites and the kinases responsible for phosphorylation. B, Time course of dispersion for wild-type and S23A mutant Synapsin1a in the Piccolo background, in the presence or absence of KN62. KN62 attenuates the dispersion of both wild-type and S23A Synapsin1a. SEM values are shown (n = 2 experiments/condition). C, Average extent of EGFP-Syn dispersion in the absence or presence of KN62, expressed as percentage decrease in EGFP-Syn dispersion for S23A versus wild-type EGFP-Syn in the Pclo28 background (n = 2 coverslips for EGFP-Syn plus KN62, 3 for S23A, 3 for S23A plus KN62; 2 batches of neurons). The dashed line denotes “complete rescue” of Pclo28 phenotype, defined as the percentage decrease in EGFP-Syn dispersion observed in wild-type neurons versus Pclo28 neurons (=33%). D, Time course of dispersion for wild-type, S1A, S23A, and S12346A EGFP-Syn constructs in the Pclo28 background. S12346A partially rescues the EGFP-Syn dispersion phenotype, with 8.4% less dispersion than wt EGFP-Syn in Pclo28 boutons (*p < 0.05, t test). E, Average extent of EGFP-Syn dispersion, expressed as percentage decrease in EGFP-Syn dispersion for phosphomutants versus wild-type EGFP-Syn in the Pclo28 background. The dashed line denotes complete rescue of the Pclo28 phenotype as in C. F, Time course of FM destaining at boutons expressing wild-type, S1A, S23A, and S12346A EGFP-Syn constructs in the Pclo28 background. SEM values are shown (n > 2 experiments/condition). G, Average extent of FM destaining, expressed as percentage decrease in FM destaining for phosphomutants versus wild-type EGFP-Syn in the Pclo28 background (n = 5 coverslips for S1A, 7 for S23A and S12346A; 3 batches of neurons). The dashed line denotes complete rescue of Pclo28 phenotype, defined as the percentage decrease in FM destaining observed in wild-type neurons versus Pclo28 neurons (=11.6%). S1A and S23A both rescue the enhanced FM destaining (by 17.6 and 17.9%, respectively), but S12346A does not (*p < 0.05; **p < 0.005, t test). H, Time course of EGFP-Syn dispersion in the Pclo28 background, in the presence of PKA (KT5720) and MAP kinase (PD98) blockers. SEM values are shown (n > 3 experiments/condition). I, Average extent of synapsin dispersion in the presence of PKA and MAPK inhibitors, expressed as in C and E. Neither has a significant effect on EGFP-Syn dispersion.
Figure 4.
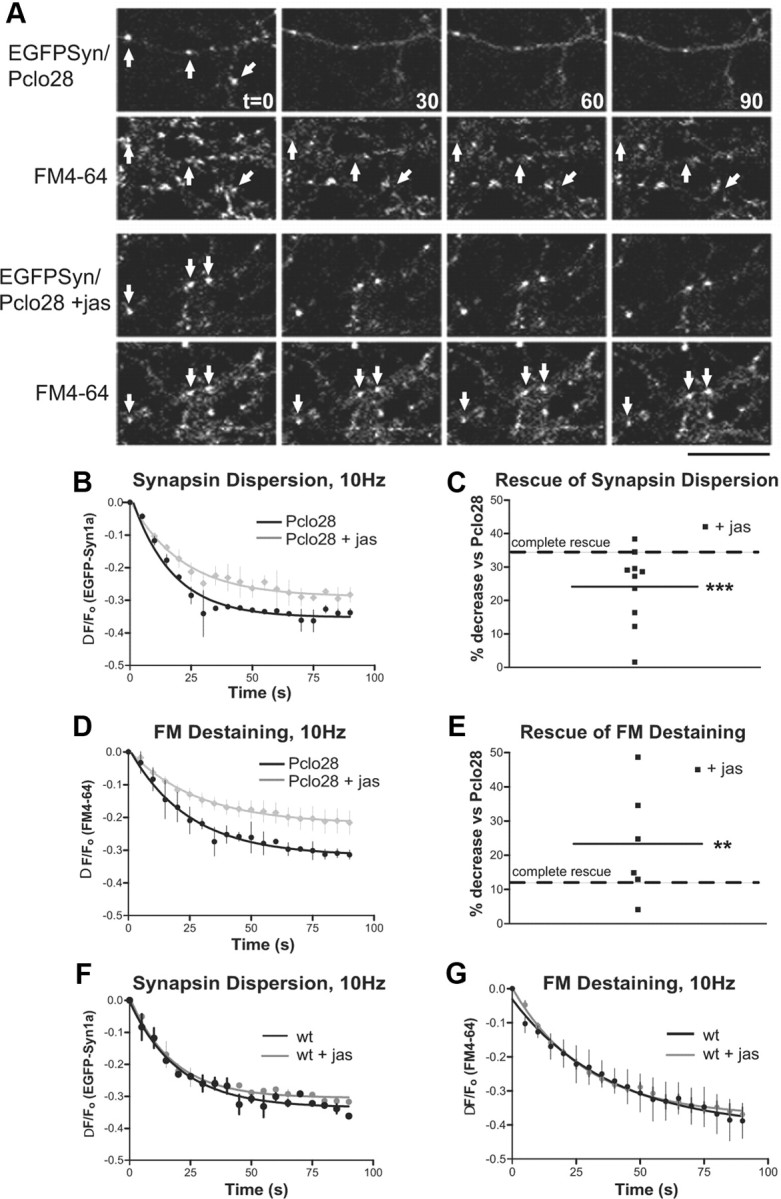
Jasplakinolide rescues the Pclo28 phenotypes. A, EGFP-Synapsin1a dispersion and FM4-64 destaining for EGFP-Syn/Pclo28 or EGFP-Syn/Pclo28 in the absence or presence of 5 μm jasplakinolide. The arrows denote colocalized EGFP-Syn and FM4-64 puncta. Scale bars, 15 μm. B, Time course of EGFP-Syn dispersion at boutons lacking Piccolo, in the absence (black) or presence (gray) of jasplakinolide. SEM bars are shown (n = 2 experiments/condition). C, Average extent of EGFP-Syn dispersion at Pclo28-expressing boutons treated with jasplakinolide (n = 10 coverslips). Complete rescue (dashed line) is defined as in Figure 2C. Jasplakinolide decreases the extent of dispersion by 24.1% (***p < 0.0001, t test). D, Time course of FM4-64 destaining at boutons lacking Piccolo, in the absence (black) or presence (gray) of jasplakinolide. SEM values are shown (n = 2 experiments/condition). E, Average extent of FM destaining at Pclo28 boutons treated with jas (n = 6). Complete rescue (dashed line) defined as in Figure 3G. Jasplakinolide decreases the extent of FM destaining by 23.3% (**p < 0.005, t test). F, Time course of EGFP-Syn dispersion in wild-type neurons in the absence (black) or presence (blue) of jasplakinolide. SEM bars are shown (n = 3 experiments/condition). G, Time course of FM destaining in the absence (black) or presence (blue) of jasplakinolide. SEM bars are shown (n = 3 experiments wt, 5 experiments plus jasplakinolide). Wild-type boutons treated with jasplakinolide exhibit no change in either EGFP-Syn dispersion or FM destaining.
Quantification of EGFP-actin, YFP-CaMKII, and EGFP-Profilin2 clustering
EGFP-actin, YFP-CaMKII, or EGFP-Profilin2 fluorescence intensity at presynaptic boutons (based on colocalization with FM4-64) was measured with OpenView software. Intensity values from each set of three prestimulation images were averaged to give avg Fo, and those from poststimulation to give avg Fpoststim. These results were then expressed as percentage increase versus initial fluorescence using the following equation: ((avg Fpoststim/avg Fo) − 1) * 100, and averaged for all EGFP-actin/Profilin2 clusters in a field of view using Microsoft Excel. GraphPad Prism was used for graph plotting and statistical analyses. ImageJ and Excel were used to measure and compute number of EGFP-actin/YFP-CaMKII/EGFP-Profilin2 puncta per unit axon length for single images acquired immediately before and after high-K+ stimulation, again for each condition (i.e., wild type, Pclo28). Both the fluorescence intensity and number of clusters per pixel values were averaged across experiments for a given condition (i.e., wild type, Pclo28) and plotted. For experiments with jasplakinolide, Fpoststim represents fluorescence after jasplakinolide treatment, as high-K+ stimulation did not further enhance EGFP-actin clustering in the presence of jasplakinolide.
Quantification of dendritic branching and puncta colocalization
Primary dendrites were counted manually based on MAP2 immunostaining. Spine density was calculated by counting number of Alexa 568 phalloidin-labeled spines per length MAP2-positive process (measured using ImageJ). Colocalization of EGFP-Synapsin1a with Homer, Piccolo, or FM4-64 was determined using the colocalization macro in ImageJ. This method typically underestimates the degree of colocalization by ∼10–15%.
Image processing
Representative images shown in the figures depict regions of interest (ROIs) selected from the original images. For instance, ROIs selected from images acquired on the scanning laser confocal microscope are typically 140 × 75 pixels (from the full-sized 640 × 480 pixel image). In addition, brightness/contrast are often enhanced to enable easier visualization of EGFP-actin/YFP-CaMKII/EGFP-Profilin2 and FM puncta. Therefore, the original image resolution and full dynamic range of fluorescence intensity are not always apparent from the images shown.
Results
Enhanced Synapsin1a dispersion and SV exocytosis at presynaptic boutons lacking Piccolo
In our previous study, we identified two phenotypes in presynaptic boutons of dissociated hippocampal neurons expressing a shRNA to eliminate Piccolo (Pclo28) (Leal-Ortiz et al., 2008): (1) enhanced EGFP-Synapsin1a (EGFP-Syn) dispersion from boutons into axons during electrical stimulation (Leal-Ortiz et al., 2008) (Fig. 2A,B), and (2) enhanced FM4-64 destaining, indicating more complete exocytosis of SVs (Leal-Ortiz et al., 2008) (Fig. 2D,E). We also found that, in both wild-type and Pclo28-expressing neurons, the extent of FM destaining at individual boutons was correlated with the extent of EGFP-Syn dispersion, suggesting that these processes were mechanistically linked (Leal-Ortiz et al., 2008). Here, we have explored the underlying cause of these phenotypes to elucidate the role of Piccolo at the synapse.
Figure 2.

Pclo28 phenotypes in dissociated hippocampal neurons. A, EGFP-Synapsin1a (EGFP-Syn) dispersion induced by 10 Hz, 90 s stimulation (at t = 0, 30, 60, and 90 s) in lentivirus-infected neurons expressing EGFP-Syn in the absence or presence of Pclo28 shRNA. Note the more pronounced dispersion of EGFP-Syn puncta in the Pclo28 background. Scale bar, 10 μm. B, Time course of EGFP-Syn dispersion in wild-type (black) and Pclo28-expressing (gray) boutons during 10 Hz, 90 s stimulation. Each time point represents an average of three curves obtained on the same day, each from a single coverslip containing ∼200 EGFP-Syn puncta. Rate of fluorescence loss (expressed as Fo − Ft/Fo for each time point t) is fit by a single exponential. Error bars show SEM (n = 3). C, Average extent of EGFP-Syn dispersion, expressed as percentage increase in EGFP-Syn dispersion for Pclo28 versus wt (n = 24 coverslips; >4 batches of neurons). The extent of EGFP-Syn dispersion in Pclo28 boutons is 55% greater than in wt boutons, as denoted by black line (***p < 0.0001, t test). D, FM4-64 destaining at EGFP-Syn puncta in wild-type or Pclo28 background. The arrows denote colocalized EGFP-Syn and FM4-64 puncta. FM destaining is more complete in Pclo28-expressing boutons. Scale bar, 10 μm. E, Time course of FM4-64 destaining at wild-type (black) and Pclo28 (gray) boutons during 10 Hz, 90 s stimulation. Each time point represents an average of two coverslips, each containing ∼200 EGFP-Syn puncta, all imaged the same day. Rate of fluorescence loss is fit by a single exponential. SEM bars are shown. F, Average extent of FM4-64 destaining, again expressed as percentage increase for Pclo28 versus wild type (n = 15 coverslips; >4 batches of neurons). The average extent of FM destaining is 15% greater in Pclo28 boutons than in wt boutons (**p < 0.005, t test).
Initially, we established a quantitative measure of these phenotypes. In previous studies, the activity-dependent dispersion of EGFP-Syn and destaining of FM4-64 within individual boutons were fit by single exponential decay curves and expressed as τ values, reflecting the time course of dispersion/destaining (Chi et al., 2001, 2003). This method proved useful in comparing the dispersion kinetics of wild-type EGFP-Syn with those of several phosphomutants, which affected the τ values of both dispersion and FM destaining (Chi et al., 2001, 2003). However, when we compared the time course of EGFP-Syn dispersion or FM destaining in wild-type versus Pclo28-expressing boutons using the same curve-fitting protocols, we found no significant differences between the τ values (data not shown). Thus, although the absolute rates of synapsin dispersion and SV exocytosis were significantly faster in boutons lacking Piccolo, the τ values were unchanged, indicating that this measurement could not quantify these differences. We therefore compared the extent of EGFP-Syn dispersion or FM destaining in Pclo28 versus wild-type boutons, as both of these processes are significantly enhanced in the absence of Piccolo (Fig. 2B,E). These values, expressed as percentage increase in extent of EGFP-Syn dispersion or FM destaining for Pclo28 boutons compared with wild-type boutons (Fig. 1), allowed us to express each set of dispersion/destaining curves obtained on a given day (Fig. 2B,E) as a single value (one point in Fig. 2C,F), and to compare these values across multiple experiments. This latter ability was important for our analyses, as absolute levels of dispersion/destaining in our cultures varied from week to week, while the ratios of dispersion/destaining in Pclo28 versus wild-type boutons remained relatively constant.
With this method, we found that Pclo28-expressing boutons exhibited highly significant increases in EGFP-Syn dispersion and FM destaining compared with wild-type boutons (55 and 15%, respectively; p < 0.0001 and p < 0.02; Fig. 2C,F). To assess our ability to “rescue” the Pclo28 phenotype by genetic and pharmacological manipulations, we also calculated the inverse result, percentage decrease in dispersion/destaining for wild-type versus Pclo28 boutons. These values (33% decrease in dispersion and 12% decrease in destaining for wild-type vs Pclo28) were used to define “complete rescue” of the Pclo28 phenotype (i.e., wild-type levels of dispersion or destaining, for subsequent experiments) (Figs. 3, 4).
Altered synapsin phosphorylation does not cause the Pclo28 phenotypes
One possible cause of the Pclo28 phenotypes is altered Synapsin1a phosphorylation (Chi et al., 2001, 2003). Activity-dependent phosphorylation of Synapsin1a by multiple kinases (Fig. 3A) has been shown to regulate its association with SVs and the actin cytoskeleton (Schiebler et al., 1986; Benfenati et al., 1992; Ceccaldi et al., 1995; Hosaka et al., 1999; Chi et al., 2001, 2003; Jovanovic et al., 2001). For instance, serine-to-alanine (S/A) phosphomutants that prevent PKA and/or CaMKII phosphorylation of EGFP-Synapsin1a exhibit decreased τ values of activity-dependent dispersion and FM destaining, suggesting that they dissociate more slowly from SVs and/or the actin cytoskeleton within presynaptic boutons (Chi et al., 2001, 2003).
In our previous study, we showed that activity-dependent phosphorylation of Synapsin1a was altered in Pclo28-expressing neurons (Leal-Ortiz et al., 2008), indicating that Piccolo could regulate synapsin phosphorylation. We also found that the CaMKII inhibitor KN62 normalized the Pclo28 phenotypes (Leal-Ortiz et al., 2008), suggesting that they were due to altered CaMKII phosphorylation of Synapsin1a. Therefore, we first examined whether the Synapsin1a S23A phosphomutant, which prevents CaMKII phosphorylation and has been reported to slow both dispersion and FM destaining kinetics in wild-type neurons (Chi et al., 2001, 2003), could suppress the Pclo28 phenotypes. Similar to previous reports, we found that the S23A mutant slowed the dispersion kinetics of EGFP-Syn in wild-type neurons (data not shown). Surprisingly, S23A did not decrease the extent of EGFP-Syn dispersion in the Pclo28 background (Fig. 3B–E), although it did slow the enhanced kinetics of FM destaining (Fig. 3F,G). To determine whether KN62 “rescued” the Pclo28 phenotypes by altering CaMKII-mediated phosphorylation of Synapsin1a, we treated neurons expressing EGFP-Syn/Pclo28 or EGFP-SynS23A/Pclo28 with KN62 before measuring dispersion. Intriguingly, we found that KN62 not only partially rescued the extent of wild-type Synapsin1a dispersion in boutons lacking Piccolo but also that of the S23A mutant (Fig. 3B,C), indicating that CaMKII phosphorylation of Synapsin1a does not mediate its enhanced dispersion in the absence of Piccolo. These data instead suggest that either CaMKII phosphorylation of a molecule other than Synapsin1a is altered in neurons lacking Piccolo, or that a lower affinity target of KN62 inhibition (i.e., CaMKI or IV, P2X7 receptors) could be mediating this effect (Chessell et al., 1998; Davies et al., 2000).
To investigate whether other Synapsin1a phosphorylation sites could contribute to the Pclo28 phenotypes, we assessed the ability of phosphomutants that prevent PKA (S1A) or nearly all (S12346A) Synapsin1a phosphorylation to rescue the enhanced dispersion/destaining. As with S23A, neither of these mutants rescued both phenotypes (Fig. 3D–G). S12346A partially rescued Synapsin1a dispersion but had no effect on FM destaining (Fig. 3E,G), while S1A, like S23A, had no effect on Synapsin1a dispersion but completely rescued the extent of FM destaining (Fig. 3E,G). Finally, we examined whether enhanced PKA or MAPK activity could be responsible for the Pclo28 phenotypes. In contrast to KN62, PKA and MAPK inhibitors (KT5720 and PD98, respectively) had no effect on EGFP-Syn dispersion (Fig. 3H,I), suggesting that, of the kinases examined, only CaMKII is linked to the Pclo28 phenotypes.
Activity-dependent F-actin assembly is disrupted in the absence of Piccolo
We next considered the possibility that Pclo28 phenotypes were due to altered presynaptic actin dynamics. Supporting this concept, actin depolymerization was shown to enhance EGFP-Syn dispersion and FM4-64 destaining in cultured hippocampal neurons (Sankaranarayanan et al., 2003), similar to Piccolo knockdown. We thus examined whether treating Pclo28-expressing neurons with the F-actin-stabilizing drug jasplakinolide could rescue these phenotypes. Intriguingly, we found that jasplakinolide largely rescued enhanced Synapsin1a dispersion and completely rescued the enhanced FM destaining (Fig. 4A–E). In contrast, jasplakinolide had no significant effect on dispersion/destaining in wild-type neurons (Fig. 4F,G). These results indicate that Pclo28 phenotypes are caused by impaired F-actin assembly within presynaptic boutons.
To directly assess F-actin polymerization in neurons lacking Piccolo, we monitored the dynamic behavior of EGFP-tagged β-actin (EGFP-actin). Previous studies showed that EGFP-actin clustered at presynaptic boutons in response to synaptic depolarization, and that this phenomenon represented F-actin polymerization (Colicos et al., 2001; Sankaranarayanan et al., 2003). For our experiments, we induced EGFP-actin clustering by stimulating neurons for 30–45 s with high-K+ (90 mm KCl) Tyrode's solution containing FM4-64, which allowed us to simultaneously label functional presynaptic boutons. We found that high-K+ treatment caused a robust recruitment of EGFP-actin to presynaptic sites in wild-type neurons, as indicated by the ∼60% colocalization of EGFP-actin puncta with FM4-64 as well as with Piccolo and synaptophysin immunostaining (Fig. 5A–C). High-resolution STED microscopy revealed that these clusters are composed of F-actin filaments, a majority of which appear to encircle presynaptic active zones and SV pools (Fig. 5D).
Figure 5.
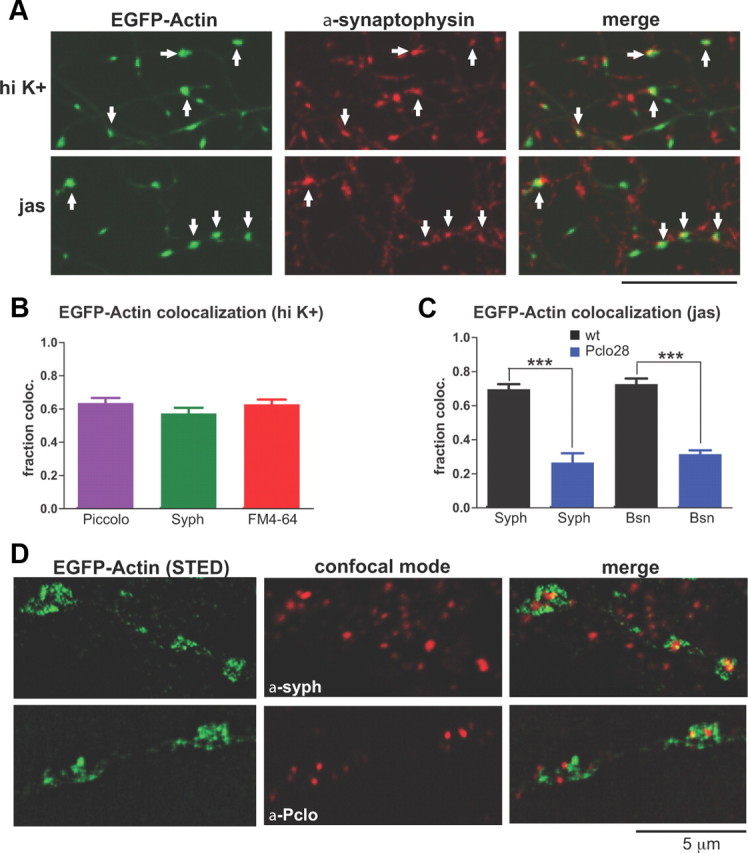
Colocalization of EGFP-actin puncta with presynaptic markers. A, Colocalization of axonal EGFP-actin puncta, induced by either high-K+ stimulation or jasplakinolide, with synaptophysin immunostaining (indicated by arrows). Scale bar, 10 μm. B, Quantification of EGFP-actin puncta colocalization with Piccolo or synaptophysin immunostaining, or with FM4-64. Approximately 60% of EGFP-actin puncta (induced by high-K+ stimulation for these experiments) are presynaptic based on colocalization with all three markers (n = 6 fields of view for Piccolo, 5 for synaptophysin, 9 for FM4-64). C, Quantification of EGFP-actin puncta colocalization with synaptophysin or Bassoon immunostaining in wild-type (black) or Pclo28-expressing (blue) neurons. Here, clustering was induced with jasplakinolide. Note that 70% of wild-type EGFP-actin puncta colocalize with synaptophysin and 73% with Bassoon, versus 27 and 32% of puncta, respectively, in Pclo28 neurons (n = 4 fields of view for each condition; ***p < 0.0005). Error bars show SEM. D, High-resolution images of high-K+-induced EGFP-actin puncta, taken using STED microscopy. Colocalization of EGFP-actin with synaptophysin and Piccolo immunostaining (acquired in confocal mode) are depicted in merged images. Note that EGFP-actin does not directly colocalize with Piccolo and synaptophysin puncta, but appears to surround them, indicating that a majority of presynaptic F-actin filaments form a meshwork encircling the active zone and SV pool. Scale bar, 5 μm.
To test whether Piccolo knockdown could inhibit activity-dependent EGFP-actin clustering, we infected neurons with either EGFP-actin or EGFP-actin/Pclo28 constructs, and compared the changes in fluorescence intensity and number/unit axon length of EGFP-actin clusters before and after 90 mm KCl depolarization. Strikingly, neurons lacking Piccolo had less punctate, more diffuse distributions of axonal EGFP-actin under basal conditions (Fig. 6B,C,E), and dramatically decreased clustering of presynaptic EGFP-actin following high-K+ depolarization (Fig. 6B,D,E). A similar attenuation of EGFP-actin clustering was seen in wild-type neurons pretreated with the actin depolymerizing drug latrunculin A (Fig. 6A,D,E) (Sankaranarayanan et al., 2003).
Figure 6.
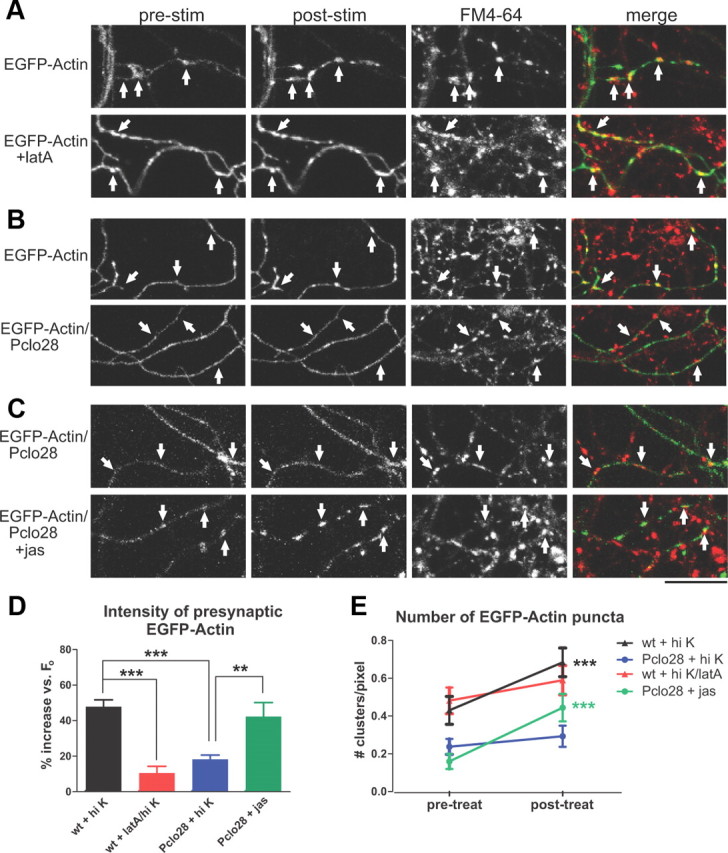
Depolarization-induced F-actin assembly is impaired in boutons lacking Piccolo. A, EGFP-actin in control and latrunculin A-treated wild-type axons before (pre) and after (post) stimulation with 90 mm KCl Tyrode's buffer plus FM4-64. The arrows indicate presynaptic sites based on FM4-64 labeling. EGFP-actin becomes more punctuate after stimulation; latrunculin A largely blocks this effect. B, EGFP-actin in wild-type or Pclo28-expressing axons before and after high-K+ stimulation. The arrows indicate presynaptic sites. Note the lack of activity-induced clustering in Pclo28 axons. C, EGFP-actin in control and jasplakinolide-treated Pclo28-expressing neurons. High-K+ stimulation does not induce EGFP-actin clustering in the absence of Piccolo, but jasplakinolide does. Scale bars: A–C, 15 μm. D, Percentage increase in EGFP-actin fluorescence intensity at presynaptic boutons following various treatments (wild type, n = 17 experiments; wild type plus latrunculin A, n = 8; Pclo28, n = 12; Pclo28 plus jasplakinolide, n = 7). Presynaptic EGFP-actin fluorescence increases 47.8% in wild-type neurons, but only 18.2% in the Piccolo knockdown and 10.5% in the presence of latrunculin (***p < 0.0001, t test). Jasplakinolide significantly increases EGFP-actin fluorescence intensity in boutons lacking Piccolo (42.3%; **p < 0.01, t test). E, Number of EGFP-actin puncta/unit axon length before (pretreat) and after (posttreat) high-K+ stimulation or jasplakinolide treatment (same n values as D). Axons lacking Piccolo have fewer EGFP-actin puncta before stimulation than wild-type axons (0.16 vs 0.43 puncta/pixel, respectively). High K+ induces new EGFP-actin puncta in wild-type neurons (from 0.43 to 0.68 clusters/pixel; ***p < 0.0001, paired t test), but not those treated with latrunculin A or lacking Piccolo. Jasplakinolide also induces significant EGFP-actin clustering in neurons expressing Pclo28 (from 0.16 to 0.44 clusters/pixel; ***p < 0.0001, paired t test).
In wild-type neurons, jasplakinolide was shown to induce similar levels of presynaptic EGFP-actin clustering as stimulation and in fact occluded further EGFP-actin clustering via depolarization (Sankaranarayanan et al., 2003). Similarly, we found that jasplakinolide induced EGFP-actin clustering in Pclo28-expressing neurons (Fig. 6C–E), indicating that these neurons were still capable of F-actin polymerization. However, <40% of these clusters colocalized with presynaptic markers compared with ∼70% in wild-type neurons (Fig. 5C), further indicating that presynaptic F-actin assembly was specifically impaired in the absence of Piccolo.
CaMKII dynamics are altered in axons lacking Piccolo
Like actin, the important signaling molecule CaMKII, implicated in presynaptic and postsynaptic plasticity mechanisms, has been shown to undergo activity-dependent clustering at presynaptic boutons (Tao-Cheng et al., 2006). These findings suggest that presynaptic CaMKII dynamics could depend upon F-actin assembly and might be altered in the absence of Piccolo. To test this hypothesis, we first compared the activity-dependent clustering of YFP-CaMKIIα in control boutons versus those treated with latrunculin A. We found that, while high K+ induced strong clustering of YFP-CaMKIIα at presynaptic boutons, pretreatment with latrunculin A blocked this effect (Fig. 7A,C). We next examined the ability of YFP-CaMKIIα to undergo activity-dependent clustering in the absence of Piccolo. Intriguingly, YFP-CaMKIIα clustering was significantly reduced in Pclo28-expressing axons (Fig. 7A–C), indicating that Piccolo indeed facilitates the activity-dependent recruitment of CaMKII into presynaptic boutons via its regulation of F-actin assembly. This defect in presynaptic CaMKII clustering could explain the lower levels of activity-dependent, CaMKII-mediated Synapsin1a phosphorylation previously observed in neurons lacking Piccolo (Leal-Ortiz et al., 2008).
Figure 7.

CaMKIIα recruitment to presynaptic boutons is impaired in Piccolo knockdown neurons. A, YFP-CaMKIIα in control, latrunculin A-treated, and Pclo28-expressing axons, before (pre) and after (post) stimulation with 90 mm KCl Tyrode's buffer plus FM4-64. The arrows indicate presynaptic sites based on FM4-64 labeling. In wild-type axons, YFP-CaMKIIα becomes more punctuate after stimulation; latrunculin A and Pclo28 shRNA block this effect. Scale bar, 15 μm. B, Percentage increase in YFP-CaMKIIα fluorescence intensity at presynaptic boutons following high-K+ stimulation. Fluorescence increases 45% in wild-type neurons, but only 17% in the Piccolo knockdown and 7% in the presence of latrunculin (n = 8 for wt, 6 for Pclo28, 3 for latA; **p < 0.005, t test). C, Number of YFP-CaMKIIα puncta/unit axon length before (pretreat) and after (posttreat) high-K+ stimulation or latrunculin treatment (same n values as B). Pclo28-expressing axons have fewer YFP-CaMKIIα puncta before stimulation than wild-type axons (0.12 vs 0.20 puncta/pixel, respectively). High K+ induces new puncta in wild-type neurons (from 0.20 to 0.36 puncta/pixel; **p < 0.005, paired t test), but not those treated with latrunculin A or lacking Piccolo. Error bars show SEM.
Knockdown of Profilin2 phenocopies knockdown of Piccolo
To confirm that a specific defect in F-actin polymerization was responsible for the Pclo28 phenotypes, we examined phenotypes induced by knockdown of another molecule known to regulate presynaptic F-actin polymerization, Profilin2 (Pfn2). Profilins are ATP/ADP exchange factors that promote F-actin assembly in all eukaryotic cells (Cooley et al., 1992; Balasubramanian et al., 1994; Witke et al., 2001; Witke, 2004). Of the four profilin genes, only Pfn2 is brain specific and localized to presynaptic boutons (Di Nardo et al., 2000; Pilo Boyl et al., 2007), where its binding partners include Piccolo, synapsin, Dynamin 1, formin family members, and the Arp2/3 and WAVE (Wiskott–Aldrich syndrome protein family verprolin-homologous protein) complexes (Witke et al., 1998; Wang et al., 1999; Gareus et al., 2006; Pilo Boyl et al., 2007). Interestingly, Pfn2 knock-out mice exhibit enhanced Pvr due to decreased F-actin assembly, demonstrating a role for Pfn2 in presynaptic actin assembly and SV exocytosis (Pilo Boyl et al., 2007). We therefore assessed whether Pfn2 knockdown in cultured hippocampal neurons produced phenotypes similar to Piccolo knockdown. An shRNA against Pfn2 (Pfn380) was designed, coexpressed in lentivirus with either soluble EGFP (for Western blots) or EGFP-Syn (for live imaging), and tested in hippocampal neurons by Western blotting. These experiments revealed that Pfn380 efficiently eliminated Pfn2 from neurons without significantly affecting dendritic arborization or spine formation, the localization of EGFP-Syn at presynaptic boutons, or FM4-64 uptake (Fig. 8A–C,F,G). These findings are in general agreement with a previous study of Pfn2 knock-out mice (Gareus et al., 2006), but at odds with a more recent one demonstrating that knockdown of Pfn2a in hippocampal organotypic slices reduced dendritic arbor complexity and spine number (Michaelsen et al., 2010). We cannot fully explain the discrepancies between our dendritic data and that of Michaelsen et al. (2010), but suspect that they could arise from differences in experimental preparation (dissociated hippocampal cultures vs organotypic slices), shRNA design, or shRNA transfection method (lentivirus vs gene gun), any of which could impact the ability of other profilin isoforms or actin regulatory molecules to compensate for the loss of Pfn2 in dendrites. In any case, remaining experiments will focus on the consequences of Pfn2 knockdown in axons.
Figure 8.

Characterization of Pfn380 single knockdown and Pclo28/Pfn380 double knockdown. A, Left, Western blot of lysates from hippocampal neurons infected with soluble EGFP alone (FUGW) or EGFP plus Pfn380, probed with Profilin2 and tubulin antibodies. Pfn380 shRNA eliminates the majority of Profilin2 from these neurons. Right, Western blot of lysates from hippocampal neurons infected with soluble EGFP plus scrambled Pclo28 and Bsn16 shRNAs (FUGW/SC) or EGFP plus Pclo28 and Pfn380 shRNAs (Pclo28/Pfn380), probed with Piccolo, Profilin2, and tubulin antibodies. Pclo28 and Pfn380 eliminate the majority of Piccolo and Profilin2, respectively, from neurons. B, Quantification of the number of primary dendrites (labeled with MAP2 immunostaining) for neurons infected with EGFP-Syn alone (wt, black), plus Pfn380 (red), or plus Pclo/Pfn (purple). No significant difference was observed between wt and Pfn380 single or Pclo/Pfn double knockdowns, indicating that dendritic morphology is not affected by these manipulations. SEM bars are shown (n = 10 cells for wt, 7 for Pfn380, 6 for Pclo/Pfn). C, Quantification of dendritic spine density, measured as number of spines per pixel along MAP2-positive processes, for neurons infected with EGFP-Syn alone (wt, black), plus Pfn380 (red), or plus Pclo/Pfn (purple). No significant differences were observed between wild-type and Pfn380 single or Pclo/Pfn double knockdown, indicating that spine formation is not affected by these manipulations. SEM bars are shown (n = 4 fields of view for wt, 5 for Pfn380, 6 for Pclo/Pfn). D, Images depicting colocalization between EGFP-Syn and FM4-64 for wt and Pclo/Pfn-expressing neurons (indicated by arrows). Scale bar, 10 μm. E, Images depicting colocalization between EGFP-Syn and Homer for wt and Pclo/Pfn-expressing neurons. A subset of colocalized puncta is indicated by arrows. Merged image also contains MAP2 immunostaining to label dendrites (blue). Scale bar, 10 μm. F, Quantification of EGFP-Syn colocalization with FM4-64 for neurons expressing EGFP-Syn alone (wt, black), plus Pfn380 (red), or plus Pclo/Pfn (purple). Pfn380 and Pclo/Pfn boutons have a similar degree of colocalization with FM as wt boutons, indicating that SV exo/endocytosis is not significantly inhibited by Profilin2 or Pclo/Pfn knockdown. SEM bars are shown (n = 10 fields of view for each condition). G, Quantification of EGFP-Syn colocalization with Homer1 for neurons expressing EGFP-Syn alone (wt, black), plus Pfn380 (red), or plus Pclo/Pfn (purple). No significant differences were observed between the three conditions, indicating that the presynaptic localization of EGFP-Syn is not affected by Profilin2 or Pclo/Pfn knockdown. SEM bars are shown (n = 7 fields of view/condition for wt and Pfn380, 6 for Pclo/Pfn).
Using the high-K+/FM4-64 loading assay described above, we subsequently confirmed that presynaptic F-actin polymerization was impaired in neurons lacking Pfn2. As anticipated, these neurons exhibited more diffuse EGFP-actin expression in unstimulated axons, as well as dramatically reduced stimulation-induced clustering of EGFP-actin (Fig. 9A–C). Finally, we examined whether Pfn2 knockdown altered EGFP-Syn dispersion and FM destaining. We found that Pfn380-expressing boutons indeed had significantly enhanced activity-dependent EGFP-Syn dispersion and FM destaining kinetics (Fig. 9D–H), similar to those expressing Pclo28. These results strongly suggest that both phenotypes are due to defects in presynaptic F-actin assembly, rather than to other unrelated effects of Piccolo knockdown.
Figure 9.
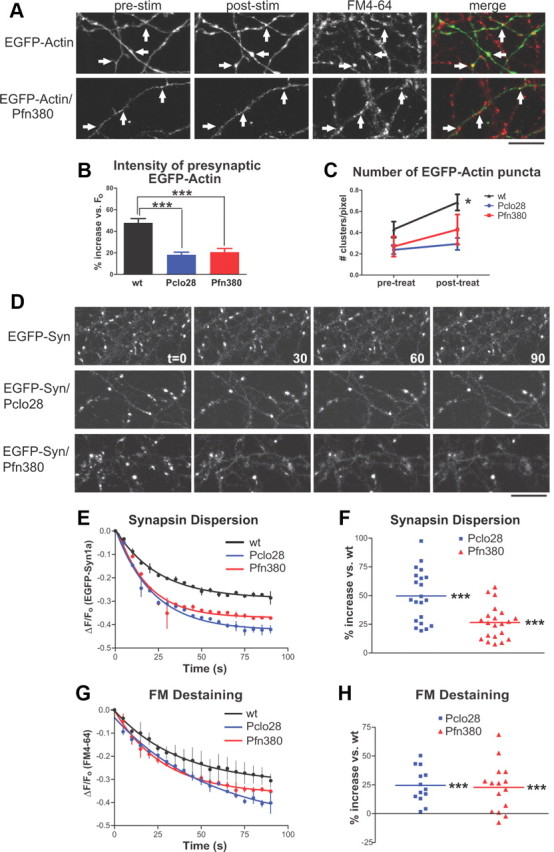
Profilin2 knockdown phenocopies Piccolo knockdown. A, EGFP-actin in axons of wild-type or Pfn380 neurons before (prestim) and after (poststim) 90 mm KCl plus FM4-64. The arrows indicate presynaptic sites based on FM4-64 labeling. Note the lack of activity-induced clustering in axons expressing Pfn380. B, Percentage increase in presynaptic EGFP-actin fluorescence intensity following high-K+ stimulation for wild-type, Pclo28, and Pfn380 boutons. Piccolo and Profilin2 knockdowns exhibit significantly attenuated presynaptic EGFP-actin fluorescence increases compared with wild-type neurons (47.8% for wt, n = 17, data from Fig. 6D; 18.2% for Pclo28, n = 12, data from Fig. 6D; 20.6% for Pfn380, n = 8; ***p < 0.0001, t test). C, Number of EGFP-actin puncta per unit axon length before (prestim) and after (poststim) high-K+ stimulation for wild-type, Pclo28, and Pfn380 boutons (same n values as B). Stimulation induces new EGFP-actin puncta in wild-type neurons (data from Fig. 6E), but not those lacking Piccolo (data from Fig. 6E) or Profilin2 *p < 0.05. D, EGFP-Syn dispersion in control, Pclo28, or Pfn380 neurons. Scale bars, 10 μm. E, Time course of EGFP-Syn dispersion in wild-type neurons (n = 2) and those lacking Piccolo (n = 2) or Profilin2 (n = 2). SEM bars are shown. F, Average extent of EGFP-Syn dispersion at boutons lacking Piccolo (n = 21) or Profilin2 (n = 21), expressed as percentage increase in dispersion versus wild type. Here, Pclo28-expressing boutons have a 52.3% increase in EGFP-Syn dispersion and those expressing Pfn380 have a 28.4% increase (***p < 0.0001, t test). G, Time course of FM destaining in wild-type neurons and those lacking Piccolo or Profilin2 (same n values as E). SEM bars are shown. H, Average extent of FM destaining at boutons expressing Pclo28 (n = 13) or Pfn380 (n = 15). Both exhibit similarly enhanced levels of FM destaining compared with wild type (24.5% for Pclo28, 22.7% for Pfn380; ***p < 0.0001, t test).
Piccolo and Profilin2 could function in the same molecular pathway
To explore whether Piccolo and Pfn2 could function together to regulate presynaptic F-actin assembly, we created EGFP/Pclo28/Pfn380 and EGFP-Syn/Pclo28/Pfn380 lentiviral constructs for double knockdown of Piccolo and Pfn2. We found that EGFP/Pclo28/Pfn380 effectively eliminated both proteins from neurons (Fig. 8A) and did not alter neuronal morphology, EGFP-Syn localization at presynaptic boutons, or FM4-64 uptake (Fig. 8B–G). Intriguingly, Pclo28/Pfn380 boutons exhibited similar degrees of EGFP-Syn dispersion and FM destaining to those lacking Piccolo or Pfn2 alone (Fig. 10A–C). These results are not due to saturated levels of actin depolymerization, as latrunculin treatment further enhanced EGFP-Syn dispersion in Pclo28 and Pfn380 single knockdowns, indicating that neither represents the maximum possible level of presynaptic actin depolymerization (data not shown). Our data indicate that Piccolo and Pfn2 could function in the same molecular pathway, as their combined knockdown did not cause a more severe phenotype.
Figure 10.
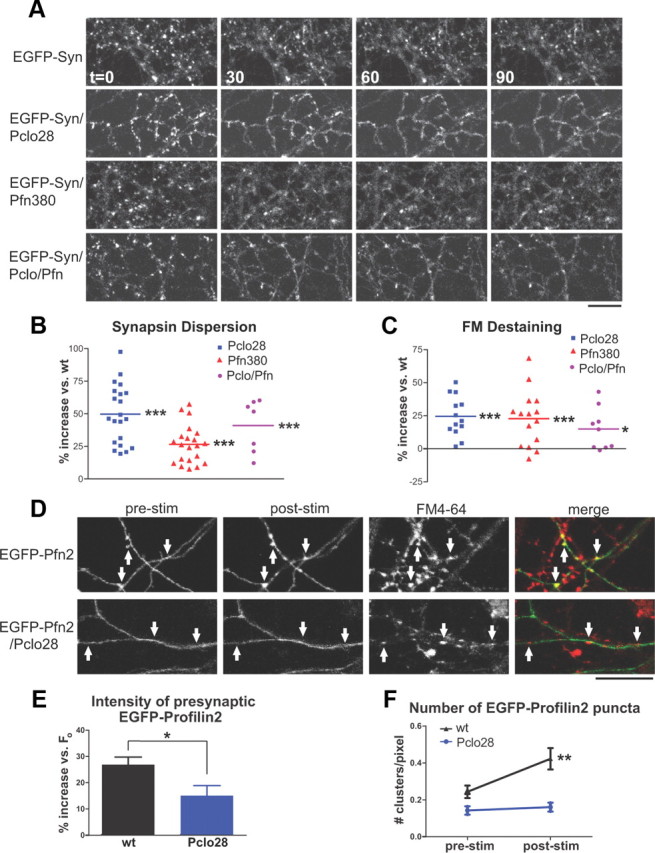
Piccolo and Profilin2 could lie in the same molecular pathway for F-actin assembly. A, EGFP-Syn dispersion in control neurons and those expressing Pclo28, Pfn380, or Pclo28/Pfn380 (Pclo/Pfn). Scale bars, 10 μm. B, Average extent of EGFP-Syn dispersion at boutons lacking Piccolo (n = 21), Profilin2 (n = 21), or both proteins (n = 7), expressed as percentage increase in dispersion versus wild type. Pclo28-expressing boutons have a 52.3% increase in EGFP-Syn dispersion, those expressing Pfn380 have a 28.4% increase, and those expressing Pclo/Pfn have a 40.9% increase (***p < 0.0001, t test). C, Average extent of FM destaining at boutons expressing Pclo28 (n = 13), Pfn380 (n = 15), or Pclo/Pfn (n = 9). All exhibit similarly enhanced levels of FM destaining compared with wild type (24.5% for Pclo28, 22.7% for Pfn380, 15% for Pclo/Pfn; ***p < 0.0001, *p < 0.05, t test). D, EGFP-Profilin2 in axons of wild-type and Pclo28 neurons before (prestim) and after (poststim) high-K+ stimulation. In wild-type neurons, EGFP-Profilin2 becomes more punctate after stimulation; this effect is absent in neurons lacking Piccolo. Scale bar, 15 μm. E, Percentage increase in presynaptic EGFP-Profilin2 fluorescence intensity following high-K+ stimulation for wild-type (n = 12) or Pclo28-expressing neurons (n = 13). Presynaptic EGFP-Profilin2 fluorescence shows little increase in neurons lacking Piccolo compared with wild-type neurons (15.0 vs 26.9%; *p < 0.05, t test). F, Number of EGFP-Profilin2 puncta per unit axon length before (prestim) and after (poststim) high-K+ stimulation for wild-type or Pclo28-expressing neurons (same n values as E). Note that axons lacking Piccolo exhibit fewer EGFP-Profilin2 puncta before stimulation than wild-type axons (0.14 vs 0.24 puncta/pixel). Stimulation induces new EGFP-Profilin2 puncta in wild-type neurons (from 0.24 to 0.42 puncta/pixel; **p < 0.005, paired t test), but not those lacking Piccolo. Error bars show SEM.
Profilin2 dynamics are altered in the absence of Piccolo
The ability of Pfn2 ability to promote F-actin assembly by catalyzing ATP-ADP exchange suggests that it would function downstream of Piccolo in a pathway for F-actin assembly. In this case, we would expect Pfn2 dynamics to be altered in the absence of Piccolo. To test this concept, we infected neurons with EGFP-Pfn2 or EGFP-Pfn2/Pclo28 constructs, and assessed presynaptic EGFP-Pfn2 clustering in response to the high-K+/FM4-64 loading protocol used to induce EGFP-actin clustering. Indeed, wild-type neurons exhibited pronounced presynaptic clustering of EGFP-Pfn2 following high-K+ stimulation (Fig. 10D–F). In contrast, Pclo28-expressing neurons exhibited both a more diffuse pattern of axonal EGFP-Pfn2 expression under basal conditions (Fig. 10D,F) and significantly reduced presynaptic clustering of EGFP-Pfn2 following depolarization (Fig. 10D–F). These data suggest that Pfn2 could lie downstream of Piccolo in a pathway for activity-dependent presynaptic F-actin assembly.
PcloΔEx14−/− mice exhibit normal F-actin assembly
A recent study described mice with a targeted deletion of exon 14 in the Pclo gene (PcloΔEx14), encoding 125 nt of the C2A domain (Mukherjee et al., 2010) (Fig. 11A). The authors reported a >95% reduction in Piccolo protein levels based on Western blots using a C-terminal antibody, yet no defects in neurotransmission or synaptic plasticity (Mukherjee et al., 2010). These data are clearly at odds with our experiments using Pclo28 shRNA. To reconcile the differences, we compared the protein expression patterns and primary synaptic phenotype of Pclo28-expressing neurons with those prepared from PcloΔEx14 mice (strain B6;129S6-Pclotm1Sud/J, created in the Südhof Laboratory and maintained at The Jackson Laboratory).
Figure 11.
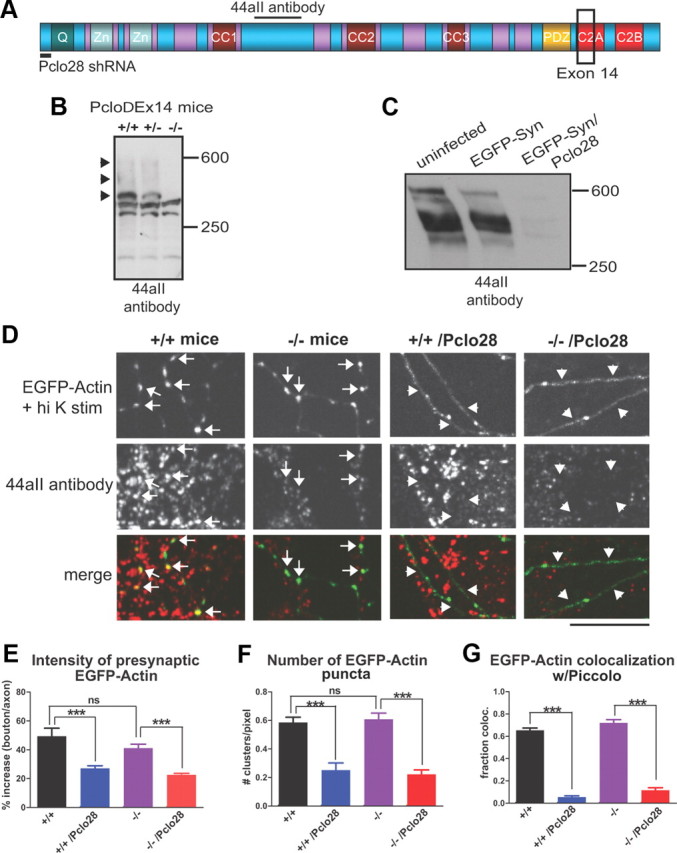
PcloΔEx14 mice exhibit normal presynaptic F-actin assembly. A, Schematic diagram of Piccolo depicting its multiple domains [Q, two Zinc finger (Zn), three coiled-coil (CC), PDZ, C2A, and C2B] and regions targeted by Pclo28 shRNA, exon 14 deletion, and the 44aII antibody. B, Western blot of brain homogenates from wild-type (+/+), heterozygous (+/−), and PcloΔEx14 (−/−) mice, probed with the 44aII antibody. The black arrowheads indicate major immunoreactive bands (∼560, 500, and 400 kDa) that are absent in −/− mice. C, Western blot of lysates from rat hippocampal neurons infected with EGFP-Synapsin1a in the absence or presence of Pclo28, probed with 44aII antibodies. Note the disappearance of all immunoreactive bands in lysates expressing Pclo28. D, EGFP-actin in axons of 10 DIV +/+ and −/− hippocampal neurons, in the absence or presence of Pclo28 shRNA, treated with high K+ (1 min) and immunostained with 44aII antibodies. For both genotypes, extensive axonal EGFP-actin clustering is induced by high K+, and clusters colocalize with presynaptic Piccolo immunoreactivity (arrows). Pclo28 eliminates EGFP-actin clustering and Piccolo immunoreactivity for both genotypes (arrowheads). Image gain is set approximately five times higher for EGFP-actin (but not Piccolo) in Pclo28-infected axons to enable its visualization, as it is largely diffuse. Scale bar, 10 μm. E, Intensity of high-K+-induced EGFP-actin puncta at presynaptic boutons, expressed as percentage increase in bouton/axon fluorescence (+/+, n = 5 images from 2 animals; −/−, n = 14 images, 2 animals; +/+/Pclo28, n = 8, 2 animals; −/−/Pclo28, n = 14, 2 animals). For both genotypes, presynaptic EGFP-actin fluorescence is >40% increased versus axon fluorescence, but only ∼20% increased in the presence of Pclo28 (***p < 0.0001, t test). F, Number of EGFP-actin puncta/unit axon length (n values same as E). For both genotypes, high-K+ stimulation induces EGFP-actin puncta densities of ∼0.6/pixel axon length, and Pclo28 reduces this value threefold (***p < 0.0001, t test). G, Fraction colocalization of EGFP-actin puncta with Piccolo immunostaining (n values same as E). For both genotypes, EGFP-actin puncta exhibit a similar degree of colocalization with Piccolo (∼0.65). Although Piccolo immunoreactivity is substantially weaker in −/− neurons, it is still readily detectable with 44aII antibodies. Pclo28 significantly reduces Piccolo immunoreactivity, and hence colocalization with axonal EGFP-actin clusters, in both genotypes (to <0.11; ***p < 0.0001, t test). Error bars show SEM.
Initially, we examined the expression pattern of Piccolo isoforms in total brain homogenates from wild-type, heterozygous, and PcloΔEx14 littermates by Western blot using our well characterized 44aII antibody (Cases-Langhoff et al., 1996; Fenster et al., 2000; Fenster and Garner, 2002) (Fig. 11A). This antibody was raised against a ∼1686 aa region of Piccolo that contains binding sites for two actin regulators, GIT1 and profilin (Fig. 11A) (Wang et al., 1999; Kim et al., 2003). In wild-type lysates, we observed a typical pattern of Piccolo-immunoreactive bands at ∼560, 500, 400, 350, 300, and 200 kDa (Fig. 11B), thought to arise by alternative splicing of the 350 kb, 26 exon Pclo gene (Fenster and Garner, 2002). In PcloΔEx14 lysates, we observed a selective loss of the 560, 500, and 400 kDa bands (Fig. 11B), supporting the concept that alternative splicing and not protein degradation of the 560 kDa protein gives rise to this pattern. In contrast, neurons expressing Pclo28, targeting a sequence in exon 1 (Fig. 11A), lose all immunoreactive bands (Fig. 11C). These data demonstrate that PcloΔEx14 mice are not Piccolo nulls, and suggest that the absence of synaptic phenotypes is not due to an inconsequential role for Piccolo in neurotransmitter release, but rather to the continued expression of multiple isoforms containing the central region of Piccolo.
To test this hypothesis, we examined whether the most prominent Pclo28 phenotype, impaired F-actin assembly, was present in hippocampal neurons cultured from PcloΔEx14 mice. Mouse neurons were infected with EGFP-actin or EGFP-actin/Pclo28 constructs at the time of plating, grown for 10 d, and then depolarized with high K+ for 1 min before fixation and immunostaining with 44aII antibody. Intriguingly, high-K+-induced EGFP-actin clustering was robust in both wild-type and PcloΔEx14 axons (Fig. 11D). No significant differences in presynaptic cluster intensity or number of clusters per unit length of axon were detected between the two genotypes (Fig. 11E,F). Moreover, while Piccolo levels were ∼75% lower in PcloΔEx14 neurons, remaining immunoreactivity was presynaptic and >70% colocalized with EGFP-actin clusters, similar to wild-type neurons (Fig. 11D,G). In contrast, Pclo28 expression eliminated both high-K+-induced EGFP-actin clustering and Piccolo immunoreactivity from axons of both mouse genotypes (Fig. 11D,G). These results clearly demonstrate that Piccolo isoforms containing the central domain are synaptically localized in PcloΔEx14 neurons, and can support activity-dependent F-actin assembly. Since Piccolo regulates neurotransmitter release via F-actin assembly, these data further illustrate the need for mice with a more complete genetic knock-out of Piccolo with which to study the synaptic and behavioral consequences of its loss of function.
Discussion
In this study, we have demonstrated that Piccolo regulates SV release by facilitating the activity-dependent assembly of presynaptic F-actin. Knockdown of Piccolo enhances activity-dependent Synapsin1a dispersion and SV exocytosis, and decreases presynaptic F-actin assembly and CaMKII recruitment. These phenotypes are rescued by F-actin stabilization and phenocopied by knockdown of Profilin2, a known regulator of activity-dependent F-actin assembly (Witke, 2004; Pilo Boyl et al., 2007). Importantly, our data also reveal that exon 14 deletion of PCLO eliminates only a subset of Piccolo isoforms that are not essential for presynaptic F-actin assembly, indicating that PcloΔEx14 mice are not an ideal model for assessing synaptic functions of Piccolo in the developing and mature nervous system.
Piccolo regulation of SV exocytosis
Piccolo functions together with Bassoon, RIMs, Munc13, and other proteins to structurally define the AZ and regulate neurotransmitter release (Garner et al., 2000; Fejtova and Gundelfinger, 2006; Schoch and Gundelfinger, 2006). Unlike RIM1α and Munc13, Piccolo appears not to regulate the docking and priming of SVs but rather to modulate SV release through a mechanism involving Synapsin1a (Leal-Ortiz et al., 2008). We initially considered the possibility that altered CaMKII phosphorylation of Synapsin1a caused the Piccolo knockdown phenotypes (Leal-Ortiz et al., 2008). Consistent with this hypothesis, the CaMKII inhibitor KN62 rescued these phenotypes, and CaMKII-mediated phosphorylation of Synapsin1a was impaired in neurons lacking Piccolo. In addition, the Synapsin1a S23A phosphomutant (lacking CaMKII phosphorylation) slowed SV exocytosis in wild-type hippocampal neurons (Chi et al., 2001, 2003) and rescued the enhanced SV exocytosis seen at boutons without Piccolo (Fig. 2). However, this mutant did not simultaneously rescue the enhanced Synapsin1a dispersion phenotype. Moreover, KN62 treatment attenuated the enhanced dispersion of the S23A mutant, suggesting that CaMKII substrates other than Synapsin1a were responsible for this phenotype. Since our data demonstrate that Synapsin1a dispersion and FM destaining are tightly linked, we hypothesized that an upstream molecule was responsible for coordinately regulating both events. One likely candidate, shown to modulate both activity-dependent SV exocytosis and Synapsin1a dispersion (Sankaranarayanan et al., 2003), was F-actin.
Roles of presynaptic F-actin
Three functions are ascribed to presynaptic F-actin. These include a barrier function at the active zone plasma membrane that limits SV fusion (Morales et al., 2000), a translocation function that regulates RP maintenance and RRP refilling during periods of sustained synaptic activity, often in association with Synapsin1a (Greengard et al., 1994; Hilfiker et al., 1999; Jensen et al., 2007; Cingolani and Goda, 2008), and an endocytic function that facilitates synaptic vesicle retrieval (Kuromi and Kidokoro, 1998; Shupliakov et al., 2002; Bloom et al., 2003; Richards et al., 2004). Whether these functions correspond to spatially and/or morphologically distinct pools of F-actin within presynaptic boutons remains unclear. Also unclear is the precise regulatory nature of each function, which appears to differ depending on synapse type. At central glutamatergic synapses, F-actin negatively regulates several aspects of SV exocytosis. For example, acute F-actin depolymerization in dissociated hippocampal neurons has been shown to enhance Pvr (Morales et al., 2000), presumably by dismantling an F-actin “barrier” that limits SV fusion at the AZ. In addition, a recent study performed in hippocampal slices suggests that F-actin functions to separate the RP from the RRP, thus limiting SV mobilization into the RRP and preserving synaptic efficacy during sustained activity (Jensen et al., 2007). Interestingly, this study also showed that mobilization of the RP was dependent on Synapsins 1 and 2, supporting the concept that both F-actin and synapsins are important mediators of RRP refilling during sustained activity (Jensen et al., 2007). These studies indicate that F-actin negatively regulates both SV translocation and fusion at glutamatergic synapses.
Which of these functions are regulated by Piccolo? Our experiments so far indicate that Piccolo regulates SV translocation. Activity-dependent Synapsin1a dispersion is dramatically enhanced in the absence of Piccolo, suggesting that Synapsin1a-associated functions of F-actin are compromised. In addition, SV exocytosis rates during 10 Hz, 90 s stimulation, shown to mobilize the RP (Jensen et al., 2007), are significantly enhanced at boutons lacking Piccolo, while F-actin levels are reduced, suggesting the absence of a regulatory barrier (i.e., F-actin) during SV translocation/RRP refilling. Whether Piccolo also regulates the AZ-associated barrier function or putative endocytic function of F-actin is unknown, although its binding partners Abp1and Profilin2 both have demonstrated roles in endocytosis (Kessels et al., 2001; Mise-Omata et al., 2003; Gareus et al., 2006), suggesting that Piccolo may also function in this process. Future studies will address these issues.
We have also uncovered a novel role for Piccolo-mediated F-actin assembly in facilitating the activity-dependent recruitment of CaMKIIα to presynaptic boutons. Since CaMKIIα is an important signaling molecule with multiple presynaptic substrates, including Synapsin1a and BK channels (Wang, 2008), it is possible that other aspects of presynaptic function and plasticity are affected by Piccolo loss. For instance, we have observed that the activity-dependent phosphorylation of Synapsin1a by CaMKII is significantly reduced in neurons lacking Piccolo, although the functional relevance of this defect is unclear. Other studies have reported that CaMKII is important for specific types of presynaptic plasticity (Ninan and Arancio, 2004; Wang, 2008), suggesting that such functions could be altered in the absence of Piccolo.
Piccolo as a scaffold for coordinating the dynamic assembly of F-actin
Several Piccolo binding partners are F-actin regulators, including Abp1, profilin, and GIT1 (Wang et al., 1999; Fenster et al., 2003; Kim et al., 2003), suggesting that Piccolo coordinates the dynamic assembly of F-actin by scaffolding actin regulatory proteins within presynaptic boutons. One prediction of this hypothesis is that knockdown of these binding partners will phenocopy Piccolo knockdown. Indeed, knockdown of Profilin2 mimics Piccolo knockdown, suggesting that these molecules lie in the same molecular pathway for F-actin assembly. Supporting this concept, double knockdown of Piccolo and Profilin2 gives a similar phenotype to single knockdown of either protein. Finally, EGFP-Profilin2 clustering in response to synaptic depolarization was absent at boutons lacking Piccolo, suggesting that Profilin2 functions downstream of Piccolo in this pathway. These data support the concept that Piccolo coordinates the recruitment of multiple proteins involved in activity-dependent F-actin assembly.
PcloΔEx14 mice
Mukherjee et al. (2010) recently described mice with a targeted deletion of exon 14 of the Pclo gene (PcloΔEx14), encoding part of the C-terminal C2A domain (Fig. 11A). The authors reported a >95% loss of Piccolo protein, but no defects in neurotransmission or synaptic plasticity, suggesting that Piccolo is dispensable for these processes (Mukherjee et al., 2010). These data are at odds with our studies using shRNAs to knockdown Piccolo. However, using well characterized antibodies that recognize a central region of Piccolo present in all known isoforms (Fig. 11A), we find that exon 14 deletion removes only a subset of isoforms (>400 kDa) from PcloΔEx14 mice, consistent with this region of Piccolo being alternatively spliced (Fenster and Garner, 2002). Importantly, remaining isoforms (∼300–400 kDa) are synaptically localized (Fig. 11D) and mediate activity-dependent F-actin assembly. Supporting this conclusion, Pclo28 expression in PcloΔEx14 neurons simultaneously eliminates these isoforms and activity-dependent F-actin assembly. Thus, while PcloΔEx14−/− mice may prove useful for studying a form of major depressive disorder linked to mutations in the C2A domain of Piccolo (Bochdanovits et al., 2009; Sullivan et al., 2009; Furukawa-Hibi et al., 2010), they are not optimal for assessing synaptic functions of Piccolo.
In summary, we have demonstrated that Piccolo is an important regulator of presynaptic F-actin, functioning to coordinate its activity-dependent assembly from within the AZ and thereby modulating neurotransmitter release. Future studies will resolve the molecular pathway through which Piccolo regulates F-actin polymerization, and which functions of actin [i.e., initial synapse formation (Lucido et al., 2009), maturation (Zhang and Benson, 2001; Shen et al., 2006), or presynaptic plasticity (Jensen et al., 2007; Antonova et al., 2009)] are regulated by Piccolo.
Footnotes
This work was supported by NIH Grants NS39471 and NS353862 (C.C.G.). We thank Jacqueline Rodriguez for maintaining the Pclo mouse colony, Timothy Ryan for the EGFP-synapsin constructs, Ann Marie Craig for YFP-CaMKIIα, Walter Witke for EGFP-Profilin2, and Noam Ziv for providing OpenView image acquisition and analysis software.
The authors declare no competing financial interests.
References
- Antonova I, Lu FM, Zablow L, Udo H, Hawkins RD. Rapid and long-lasting increase in sites for synapse assembly during late-phase potentiation in rat hippocampal neurons. PLoS One. 2009;4:e7690. doi: 10.1371/journal.pone.0007690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balasubramanian MK, Hirani BR, Burke JD, Gould KL. The Schizosaccharomyces pombe cdc3+ gene encodes a profilin essential for cytokinesis. J Cell Biol. 1994;125:1289–1301. doi: 10.1083/jcb.125.6.1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benfenati F, Valtorta F, Rubenstein JL, Gorelick FS, Greengard P, Czernik AJ. Synaptic vesicle-associated Ca2+/calmodulin-dependent protein kinase II is a binding protein for synapsin I. Nature. 1992;359:417–420. doi: 10.1038/359417a0. [DOI] [PubMed] [Google Scholar]
- Bloom O, Evergren E, Tomilin N, Kjaerulff O, Löw P, Brodin L, Pieribone VA, Greengard P, Shupliakov O. Colocalization of synapsin and actin during synaptic vesicle recycling. J Cell Biol. 2003;161:737–747. doi: 10.1083/jcb.200212140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bochdanovits Z, Verhage M, Smit AB, de Geus EJ, Posthuma D, Boomsma DI, Penninx BW, Hoogendijk WJ, Heutink P. Joint reanalysis of 29 correlated SNPs supports the role of PCLO/Piccolo as a causal risk factor for major depressive disorder. Mol Psychiatry. 2009;14:650–652. doi: 10.1038/mp.2009.37. [DOI] [PubMed] [Google Scholar]
- Cases-Langhoff C, Voss B, Garner AM, Appeltauer U, Takei K, Kindler S, Veh RW, De Camilli P, Gundelfinger ED, Garner CC. Piccolo, a novel 420 kDa protein associated with the presynaptic cytomatrix. Eur J Cell Biol. 1996;69:214–223. [PubMed] [Google Scholar]
- Ceccaldi PE, Grohovaz F, Benfenati F, Chieregatti E, Greengard P, Valtorta F. Dephosphorylated synapsin I anchors synaptic vesicles to actin cytoskeleton: an analysis by videomicroscopy. J Cell Biol. 1995;128:905–912. doi: 10.1083/jcb.128.5.905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chessell IP, Michel AD, Humphrey PP. Effects of antagonists at the human recombinant P2X7 receptor. Br J Pharmacol. 1998;124:1314–1320. doi: 10.1038/sj.bjp.0701958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synapsin dispersion and reclustering during synaptic activity. Nat Neurosci. 2001;4:1187–1193. doi: 10.1038/nn756. [DOI] [PubMed] [Google Scholar]
- Chi P, Greengard P, Ryan TA. Synaptic vesicle mobilization is regulated by distinct synapsin I phosphorylation pathways at different frequencies. Neuron. 2003;38:69–78. doi: 10.1016/s0896-6273(03)00151-x. [DOI] [PubMed] [Google Scholar]
- Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Colicos MA, Collins BE, Sailor MJ, Goda Y. Remodeling of synaptic actin induced by photoconductive stimulation. Cell. 2001;107:605–616. doi: 10.1016/s0092-8674(01)00579-7. [DOI] [PubMed] [Google Scholar]
- Cooley L, Verheyen E, Ayers K. chickadee encodes a profilin required for intercellular cytoplasm transport during Drosophila oogenesis. Cell. 1992;69:173–184. doi: 10.1016/0092-8674(92)90128-y. [DOI] [PubMed] [Google Scholar]
- Davies SP, Reddy H, Caivano M, Cohen P. Specificity and mechanism of action of some commonly used protein kinase inhibitors. Biochem J. 2000;351:95–105. doi: 10.1042/0264-6021:3510095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dillon C, Goda Y. The actin cytoskeleton: integrating form and function at the synapse. Annu Rev Neurosci. 2005;28:25–55. doi: 10.1146/annurev.neuro.28.061604.135757. [DOI] [PubMed] [Google Scholar]
- Di Nardo A, Gareus R, Kwiatkowski D, Witke W. Alternative splicing of the mouse profilin II gene generates functionally different profilin isoforms. J Cell Sci. 2000;113:3795–3803. doi: 10.1242/jcs.113.21.3795. [DOI] [PubMed] [Google Scholar]
- Engqvist-Goldstein AE, Drubin DG. Actin assembly and endocytosis: from yeast to mammals. Annu Rev Cell Dev Biol. 2003;19:287–332. doi: 10.1146/annurev.cellbio.19.111401.093127. [DOI] [PubMed] [Google Scholar]
- Evergren E, Benfenati F, Shupliakov O. The synapsin cycle: a view from the synaptic endocytic zone. J Neurosci Res. 2007;85:2648–2656. doi: 10.1002/jnr.21176. [DOI] [PubMed] [Google Scholar]
- Fejtova A, Gundelfinger ED. Molecular organization and assembly of the presynaptic active zone of neurotransmitter release. Results Probl Cell Differ. 2006;43:49–68. doi: 10.1007/400_012. [DOI] [PubMed] [Google Scholar]
- Fenster SD, Garner CC. Gene structure and genetic localization of the PCLO gene encoding the presynaptic active zone protein Piccolo. Int J Dev Neurosci. 2002;20:161–171. doi: 10.1016/s0736-5748(02)00046-1. [DOI] [PubMed] [Google Scholar]
- Fenster SD, Chung WJ, Zhai R, Cases-Langhoff C, Voss B, Garner AM, Kaempf U, Kindler S, Gundelfinger ED, Garner CC. Piccolo, a presynaptic zinc finger protein structurally related to bassoon. Neuron. 2000;25:203–214. doi: 10.1016/s0896-6273(00)80883-1. [DOI] [PubMed] [Google Scholar]
- Fenster SD, Kessels MM, Qualmann B, Chung WJ, Nash J, Gundelfinger ED, Garner CC. Interactions between Piccolo and the actin/dynamin-binding protein Abp1 link vesicle endocytosis to presynaptic active zones. J Biol Chem. 2003;278:20268–20277. doi: 10.1074/jbc.M210792200. [DOI] [PubMed] [Google Scholar]
- Furukawa-Hibi Y, Nitta A, Fukumitsu H, Somiya H, Furukawa S, Nabeshima T, Yamada K. Overexpression of piccolo C2A domain induces depression-like behavior in mice. Neuroreport. 2010;21:1177–1181. doi: 10.1097/WNR.0b013e3283411685. [DOI] [PubMed] [Google Scholar]
- Gareus R, Di Nardo A, Rybin V, Witke W. Mouse profilin 2 regulates endocytosis and competes with SH3 ligand binding to dynamin 1. J Biol Chem. 2006;281:2803–2811. doi: 10.1074/jbc.M503528200. [DOI] [PubMed] [Google Scholar]
- Garner CC, Kindler S, Gundelfinger ED. Molecular determinants of presynaptic active zones. Curr Opin Neurobiol. 2000;10:321–327. doi: 10.1016/s0959-4388(00)00093-3. [DOI] [PubMed] [Google Scholar]
- Greengard P, Benfenati F, Valtorta F. Synapsin I, an actin-binding protein regulating synaptic vesicle traffic in the nerve terminal. Adv Second Messenger Phosphoprotein Res. 1994;29:31–45. doi: 10.1016/s1040-7952(06)80005-4. [DOI] [PubMed] [Google Scholar]
- Hilfiker S, Pieribone VA, Czernik AJ, Kao HT, Augustine GJ, Greengard P. Synapsins as regulators of neurotransmitter release. Philos Trans R Soc Lond B Biol Sci. 1999;354:269–279. doi: 10.1098/rstb.1999.0378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirokawa N, Sobue K, Kanda K, Harada A, Yorifuji H. The cytoskeletal architecture of the presynaptic terminal and molecular structure of synapsin 1. J Cell Biol. 1989;108:111–126. doi: 10.1083/jcb.108.1.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosaka M, Hammer RE, Südhof TC. A phospho-switch controls the dynamic association of synapsins with synaptic vesicles. Neuron. 1999;24:377–387. doi: 10.1016/s0896-6273(00)80851-x. [DOI] [PubMed] [Google Scholar]
- Jensen V, Walaas SI, Hilfiker S, Ruiz A, Hvalby Ø. A delayed response enhancement during hippocampal presynaptic plasticity in mice. J Physiol. 2007;583:129–143. doi: 10.1113/jphysiol.2007.131300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic JN, Sihra TS, Nairn AC, Hemmings HC, Jr, Greengard P, Czernik AJ. Opposing changes in phosphorylation of specific sites in synapsin I during Ca2+-dependent glutamate release in isolated nerve terminals. J Neurosci. 2001;21:7944–7953. doi: 10.1523/JNEUROSCI.21-20-07944.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessels MM, Engqvist-Goldstein AE, Drubin DG, Qualmann B. Mammalian Abp1, a signal-responsive F-actin-binding protein, links the actin cytoskeleton to endocytosis via the GTPase dynamin. J Cell Biol. 2001;153:351–366. doi: 10.1083/jcb.153.2.351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Ko J, Shin H, Lee JR, Lim C, Han JH, Altrock WD, Garner CC, Gundelfinger ED, Premont RT, Kaang BK, Kim E. The GIT family of proteins forms multimers and associates with the presynaptic cytomatrix protein Piccolo. J Biol Chem. 2003;278:6291–6300. doi: 10.1074/jbc.M212287200. [DOI] [PubMed] [Google Scholar]
- Kuromi H, Kidokoro Y. Two distinct pools of synaptic vesicles in single presynaptic boutons in a temperature-sensitive Drosophila mutant, shibire. Neuron. 1998;20:917–925. doi: 10.1016/s0896-6273(00)80473-0. [DOI] [PubMed] [Google Scholar]
- Leal-Ortiz S, Waites CL, Terry-Lorenzo R, Zamorano P, Gundelfinger ED, Garner CC. Piccolo modulation of Synapsin1a dynamics regulates synaptic vesicle exocytosis. J Cell Biol. 2008;181:831–846. doi: 10.1083/jcb.200711167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li YC, Bai WZ, Zhou L, Sun LK, Hashikawa T. Nonhomogeneous distribution of filamentous actin in the presynaptic terminals on the spinal motoneurons. J Comp Neurol. 2010;518:3184–3192. doi: 10.1002/cne.22374. [DOI] [PubMed] [Google Scholar]
- Lucido AL, Suarez Sanchez F, Thostrup P, Kwiatkowski AV, Leal-Ortiz S, Gopalakrishnan G, Liazoghli D, Belkaid W, Lennox RB, Grutter P, Garner CC, Colman DR. Rapid assembly of functional presynaptic boutons triggered by adhesive contacts. J Neurosci. 2009;29:12449–12466. doi: 10.1523/JNEUROSCI.1381-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaelsen K, Murk K, Zagrebelsky M, Dreznjak A, Jockusch BM, Rothkegel M, Korte M. Fine-tuning of neuronal architecture requires two profilin isoforms. Proc Natl Acad Sci U S A. 2010;107:15780–15785. doi: 10.1073/pnas.1004406107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mise-Omata S, Montagne B, Deckert M, Wienands J, Acuto O. Mammalian actin binding protein 1 is essential for endocytosis but not lamellipodia formation: functional analysis by RNA interference. Biochem Biophys Res Commun. 2003;301:704–710. doi: 10.1016/s0006-291x(02)02972-8. [DOI] [PubMed] [Google Scholar]
- Morales M, Colicos MA, Goda Y. Actin-dependent regulation of neurotransmitter release at central synapses. Neuron. 2000;27:539–550. doi: 10.1016/s0896-6273(00)00064-7. [DOI] [PubMed] [Google Scholar]
- Mukherjee K, Yang X, Gerber SH, Kwon HB, Ho A, Castillo PE, Liu X, Südhof TC. Piccolo and bassoon maintain synaptic vesicle clustering without directly participating in vesicle exocytosis. Proc Natl Acad Sci U S A. 2010;107:6504–6509. doi: 10.1073/pnas.1002307107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninan I, Arancio O. Presynaptic CaMKII is necessary for synaptic plasticity in cultured hippocampal neurons. Neuron. 2004;42:129–141. doi: 10.1016/s0896-6273(04)00143-6. [DOI] [PubMed] [Google Scholar]
- Phillips GR, Huang JK, Wang Y, Tanaka H, Shapiro L, Zhang W, Shan WS, Arndt K, Frank M, Gordon RE, Gawinowicz MA, Zhao Y, Colman DR. The presynaptic particle web: ultrastructure, composition, dissolution, and reconstitution. Neuron. 2001;32:63–77. doi: 10.1016/s0896-6273(01)00450-0. [DOI] [PubMed] [Google Scholar]
- Pilo Boyl P, Di Nardo A, Mulle C, Sassoè-Pognetto M, Panzanelli P, Mele A, Kneussel M, Costantini V, Perlas E, Massimi M, Vara H, Giustetto M, Witke W. Profilin2 contributes to synaptic vesicle exocytosis, neuronal excitability, and novelty-seeking behavior. EMBO J. 2007;26:2991–3002. doi: 10.1038/sj.emboj.7601737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards DA, Rizzoli SO, Betz WJ. Effects of wortmannin and latrunculin A on slow endocytosis at the frog neuromuscular junction. J Physiol. 2004;557:77–91. doi: 10.1113/jphysiol.2004.062158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sankaranarayanan S, Atluri PP, Ryan TA. Actin has a molecular scaffolding, not propulsive, role in presynaptic function. Nat Neurosci. 2003;6:127–135. doi: 10.1038/nn1002. [DOI] [PubMed] [Google Scholar]
- Schiebler W, Jahn R, Doucet JP, Rothlein J, Greengard P. Characterization of synapsin I binding to small synaptic vesicles. J Biol Chem. 1986;261:8383–8390. [PubMed] [Google Scholar]
- Schoch S, Gundelfinger ED. Molecular organization of the presynaptic active zone. Cell Tissue Res. 2006;326:379–391. doi: 10.1007/s00441-006-0244-y. [DOI] [PubMed] [Google Scholar]
- Shen W, Wu B, Zhang Z, Dou Y, Rao ZR, Chen YR, Duan S. Activity-induced rapid synaptic maturation mediated by presynaptic cdc42 signaling. Neuron. 2006;50:401–414. doi: 10.1016/j.neuron.2006.03.017. [DOI] [PubMed] [Google Scholar]
- Shupliakov O, Bloom O, Gustafsson JS, Kjaerulff O, Low P, Tomilin N, Pieribone VA, Greengard P, Brodin L. Impaired recycling of synaptic vesicles after acute perturbation of the presynaptic actin cytoskeleton. Proc Natl Acad Sci U S A. 2002;99:14476–14481. doi: 10.1073/pnas.212381799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Südhof TC. The synaptic vesicle cycle. Annu Rev Neurosci. 2004;27:509–547. doi: 10.1146/annurev.neuro.26.041002.131412. [DOI] [PubMed] [Google Scholar]
- Sullivan PF, de Geus EJ, Willemsen G, James MR, Smit JH, Zandbelt T, Arolt V, Baune BT, Blackwood D, Cichon S, Coventry WL, Domschke K, Farmer A, Fava M, Gordon SD, He Q, Heath AC, Heutink P, Holsboer F, Hoogendijk WJ, et al. Genome-wide association for major depressive disorder: a possible role for the presynaptic protein piccolo. Mol Psychiatry. 2009;14:359–375. doi: 10.1038/mp.2008.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tao-Cheng JH, Dosemeci A, Winters CA, Reese TS. Changes in the distribution of calcium calmodulin-dependent protein kinase II at the presynaptic bouton after depolarization. Brain Cell Biol. 2006;35:117–124. doi: 10.1007/s11068-007-9012-5. [DOI] [PubMed] [Google Scholar]
- Waites CL, Specht CG, Härtel K, Leal-Ortiz S, Genoux D, Li D, Drisdel RC, Jeyifous O, Cheyne JE, Green WN, Montgomery JM, Garner CC. Synaptic SAP97 isoforms regulate AMPA receptor dynamics and access to presynaptic glutamate. J Neurosci. 2009;29:4332–4345. doi: 10.1523/JNEUROSCI.4431-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Kibschull M, Laue MM, Lichte B, Petrasch-Parwez E, Kilimann MW. Aczonin, a 550-kD putative scaffolding protein of presynaptic active zones, shares homology regions with Rim and Bassoon and binds profilin. J Cell Biol. 1999;147:151–162. doi: 10.1083/jcb.147.1.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZW. Regulation of synaptic transmission by presynaptic CaMKII and BK channels. Mol Neurobiol. 2008;38:153–166. doi: 10.1007/s12035-008-8039-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witke W. The role of profilin complexes in cell motility and other cellular processes. Trends Cell Biol. 2004;14:461–469. doi: 10.1016/j.tcb.2004.07.003. [DOI] [PubMed] [Google Scholar]
- Witke W, Podtelejnikov AV, Di Nardo A, Sutherland JD, Gurniak CB, Dotti C, Mann M. In mouse brain profilin I and profilin II associate with regulators of the endocytic pathway and actin assembly. EMBO J. 1998;17:967–976. doi: 10.1093/emboj/17.4.967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witke W, Sutherland JD, Sharpe A, Arai M, Kwiatkowski DJ. Profilin I is essential for cell survival and cell division in early mouse development. Proc Natl Acad Sci U S A. 2001;98:3832–3836. doi: 10.1073/pnas.051515498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai R, Olias G, Chung WJ, Lester RA, tom Dieck S, Langnaese K, Kreutz MR, Kindler S, Gundelfinger ED, Garner CC. Temporal appearance of the presynaptic cytomatrix protein bassoon during synaptogenesis. Mol Cell Neurosci. 2000;15:417–428. doi: 10.1006/mcne.2000.0839. [DOI] [PubMed] [Google Scholar]
- Zhang W, Benson DL. Stages of synapse development defined by dependence on F-actin. J Neurosci. 2001;21:5169–5181. doi: 10.1523/JNEUROSCI.21-14-05169.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]