

Abstract
Matrix metalloproteinase 13 (MMP-13) has been implicated as the protease responsible for collagen degradation in cartilage during osteoarthritis (OA). Compounds that inhibit the metalloproteinase at the Zn binding site typically lack specificity among MMP family members. Analogs of the low-micromolar lead MMP-13 inhibitor 4, discovered through high-throughput screening, were synthesized to investigate structure activity relationships in this inhibitor series. Systematic modifications of 4 led to the discovery of MMP-13 inhibitors 20 and 24 which are more selective than 4 against other MMPs. Compound 20 is also approximately 5-fold more potent as an MMP-13 inhibitor than the original HTS-derived lead compound 4.
Articular cartilage consists primarily of extracellular matrix (ECM) and a small number of chondrocytes. In turn, cartilage ECM is composed of proteoglycans and collagen, especially fibrillar type II collagen. Type II collagen is extremely resistant to most proteinases because of its triple-helical structure. Several members of the matrix metalloproteinase (MMP) family catalyze the hydrolysis of collagen. MMP-13 is thought to be the most effective enzyme at cleaving type II collagen.1 Once collagen is cleaved, it denatures at body temperature to become gelatin.2 The cartilage degradation and denaturing process is thought to be a primary cause of osteoarthritis (OA). Thus, the development of a selective small-molecule inhibitor of MMP-13 has great therapeutic potential for halting the progression of OA.
A number of compounds have been described that inhibit MMP-13 and other metalloproteinases by chelating the active site Zn.3 However, these compounds typically display a of lack specificity among MMP family members and have exhibited toxicities in clinical trials, particularly musculoskeletal syndrome (MSS).3
Several selective non-chelating inhibitors of MMP-13 have been reported (for example, compounds 1, 2 and 3, Figure 1).4 Molecular modeling and X-ray co-crystal structures indicated that these compounds modulate the activity of MMP-13 by binding to the S1’ specificity pocket and preventing substrate from binding. The success of these compounds suggest that, by avoiding direct Zn interactions, one might be able to achieve inhibitor specificity among metalloproteinase subtypes and related enzyme families and thereby reduce toxicity.
Figure 1.
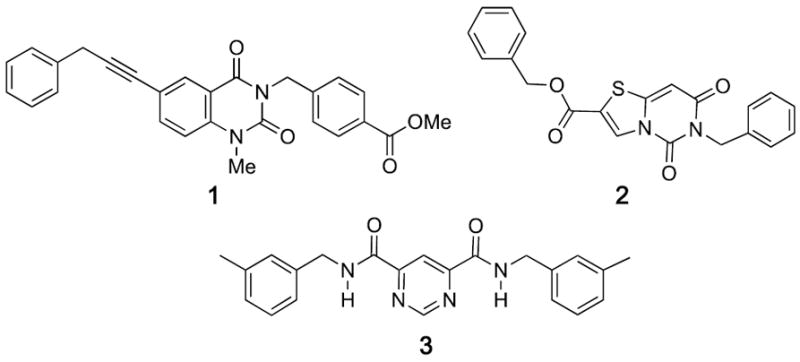
Non-chelating inhibitors of MMP-13.
Using high throughput screening (HTS) techniques, we identified several selective low micromolar inhibitors of MMP-13.5 One inhibitor (compound 4; Figure 2; also known as compound Q in the previous publication, and designated as a MMP-13 probe by the NIH MLPCN program; PubChem CID 2047223) exhibited properties suggesting that its mode of action was not via Zn chelation. Compound 4 possessed a smaller molecular scaffold than previously described MMP-13 inhibitors, yet inhibited only two of the previously tested MMPs, MMP-8 and MMP-13.5
Figure 2.
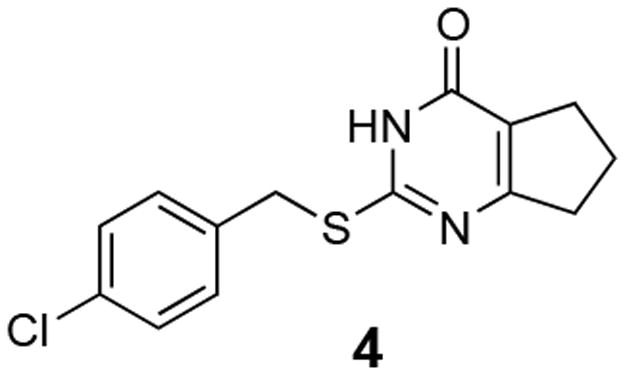
MMP-13 inhibitor 4 derived from HTS; PubChem CID 2047223
We pursued a two-pronged approach in our efforts to improve the selectivity and potency of MMP-13 inhibitor 4. First, structural analogs of 4 were identified by cheminformatic searches using Chemnavigator.com. Analogs containing the thiouracil scaffold with appropriate structural similarity to 4 were purchased from commercial suppliers. Second, analogs of 4 were synthesized starting with the condensation of thiourea (5) with a substituted β-ketoester 6 (Scheme 1). Alkylation of the thiocarbonyl unit of 7 with various alkylating agents to give the MMP-13 inhibitor scaffold 8.
Scheme 1.
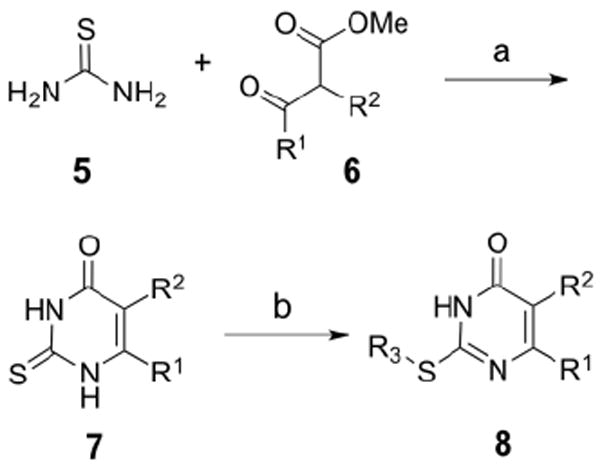
General scheme for synthesis of MMP-13 inhibitor analogs of 4: (a) DBU, MeCN, reflux, or Na, EtOH, 60 °C, 1-12 h. (b) acetone or DMF, Na2CO3, R3-X.
Analogs of compound 4 were initially evaluated for percent inhibition of MMP-13 using a FRET triple-helical peptide substrate assay as described previously.5 The triple-helical substrate (fTHP-15) has distinct conformational features that interact with protease secondary binding sites (exosites).5,6
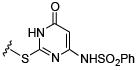
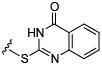
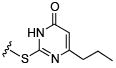
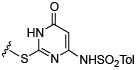
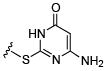
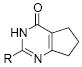
Initial efforts focused on modifications of the cyclopenene ring of 4 (Table 1). These structural changes resulted in a modest reduction of activity (compounds 10-15) to outright loss of MMP-13 inhibition (compounds 9 and 16-19). Replacement of the cyclopentenyl group by a propyl group in 9 resulted in loss of all MMP-13 inhibitory activity; however, analog 10 with a methyl group at this position retained modest activity. An analog with methyl groups at both available positions on the uracil ring (compound 35, Table 2) also displayed excellent MMP-13 inhibition activity. Amine and sulfonamide substituted analogs 11 and 12 retained modest MMP-13 inhibition, as did the fused thiophene derivative 15. However, incorporation of a fused phenyl ring (compound 16), a cyclohexyl (see 34 in Table 2) or a fused pyrazole ring (compound 17) resulted in inactive compounds. Attempts to extend substituents in compound 14 into regions of space that the side chains of 1-3 might occupy (assuming that 4 were to bind in the same site as 1-3) led to decreased activity compared to 4, while compounds 18 and 19 were inactive.
Table 1. Inhibition of MMP-13 activity by analogs of compound 4 with replacement of the original cyclopentenyl-fused uracil.
All structures in this table have the chlorobenzyl substituent of 4. Assays were performed in triplicate using 40 μM of each compound and fTHP-15 as substrate as described.5 The standard deviations were <10%.
| Compound | MMP-13 % Inhibition | Compound | MMP-13 % Inhibition | Compound | MMP-13 % Inhibition | |||
|---|---|---|---|---|---|---|---|---|
| 4 |
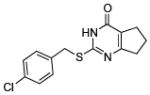
|
49.1 | 12 |
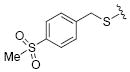
|
31.2 | 16 |
|
inactive |
| 9 |
|
inactive | 13 |
|
30.5 | 17 |
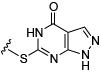
|
inactive |
| 10 |
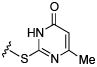
|
32.9 | 14 |
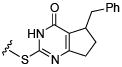
|
37.7 | 18 |
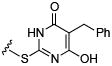
|
inactive |
| 11 |
|
37 | 15 |
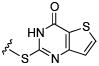
|
36.6 | 19 |
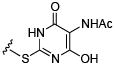
|
inactive |
Table 2. Inhibition of MMP-13 activity by analogs of compound 4 with different S-alkyl substituents.
Assays were performed in triplicate using 40 μM of each compound and fTHP-15 as substrate as described.5 The standard deviations were <10%.
 |
|
|||||||
|---|---|---|---|---|---|---|---|---|
| Compound, R = | MMP-13 % Inhibition | Compound, R = | MMP-13 % Inhibition | Additional Analogs | MMP-13 % Inhibition | |||
| 4 |
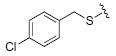
|
49.1 | 26 |
|
7.7 | 33 |
|
16.0 |
| 20 |
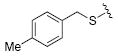
|
68.7 | 27 |
|
7.2 | |||
| 21 |
|
45.2 | 28 |
|
10.3 | 34 |
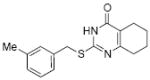
|
inactive |
| 22 |
|
46.7 | 29 |
|
17.1 | |||
| 23 |
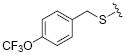
|
34.4 | 30 |
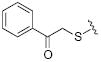
|
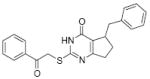
inactive | 35 |
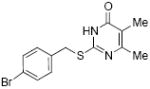
|
55.8 |
| 24 |
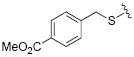
|
62.5 | 31 |
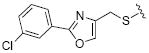
|
inactive | |||
| 25 |
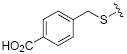
|
2.2 | 32 |
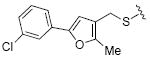
|
Inactive | |||
While the data in Table 1 suggested that modifications of the cyclopentenyl ring of 4 did not lead to increased MMP-13 inhibition, encouraging results emerged for two compounds (20 and 24) in the group of analogs presented in Table 2 with differentially substituted benzylic groups. The inhibition data for analogs 20 to 24 and 35 demonstrate that a variety of para substituents on the benzylic unit are compatible with MMP-13 inhibition, and that a meta substituent leads to decreased activity (compare 20 and 21). The carboxylic acid 25 suffered a severe loss of inhibition in comparison with the methyl ester 24, suggesting that a very polar or charged substituent is not permitted in this segment of the inhibitor binding pocket. Replacement of the benzylic aryl group with alkyl, alkenyl (an analog with R = H2C=CHCH2CH2CH2CH2S- analogous to 29 was inactive), and alkynyl chains (28, 29), or extending the alkyl chain between the sulfur and the aryl group (26, 27) proved detrimental to MMP-13 inhibitory activity. Attempts to introduce other polar or hydrogen-bonding substituents, as in compounds 30, 31, 32, and 33, also led to loss of activity in this inhibitor series.
The four most potent MMP-13 inhibitors in this structural series (4, 20, 24, and 35) have para subsituents on the benzylic side chain. The only deviation from the orginal heterocyclic scaffold that did not significantly lessen inhibition was the exchange of the cyclopentene-fused uracil ring system of 4 with the dimethyl substitute uracil in 35. Counter screening of these compounds was performed against five other members of the MMP family, specifically MMP-1, MMP-2, MMP-3, MMP-8, and MMP-14/MT1-MMP, to evaluate their MMP selectivities. Since inhibition of MMP-13 collagenolytic activity is desired for OA applications, counter screening of other collagenolytic MMPs (MMP-1, MMP-2, MMP-8, and MMP-14) was pursued, along with a non-collagenolytic MMP (MMP-3) as a control. Due to its relatively high activity against MMP-8 (20% inhibition at 40 μM; data not shown), compound 35 was not further examined. The only MMP that was inhibited significantly by all three compounds was MMP-13 (Table 3). Compounds 20 and 24 were better inhibitors of MMP-13 than the original inhibitor 4. Compound 20 did not inhibit MMP-1 or MMP-8, and showed low levels of inhibition of MMP-2, MMP-9, and MMP-14 (Table 3). This is distinct from compound 4, which inhibited MMP-8. Compound 24 was less selective than compound 20, inhibiting MMP-8 and showing greater levels of inhibition of MMP-14 (Table 3).
Table 3. Inhibition of MMP-1, MMP-2, MMP-8, MMP-9, MMP-13, and MMP-14 activity by compounds 4, 20 and 24.
Assays were performed using 40 μM of each compound and fTHP-15 as substrate as described.5 Enzyme concentrations were 10 nM for MMP-2, MMP-9, and MMP-14, 2.3 nM for MMP-13, 4 nM for MMP-1, and 2 nM for MMP-8.
| Target | Inhibition (%) at [inhibitor] = 40 μM | ||
|---|---|---|---|
| 4 | 20 | 24 | |
| MMP-1 | 0 | 0 | 0 |
| MMP-2 | 25 ± 11 | 19 ± 2.1 | 26 ± 9.2 |
| MMP-8 | 17 ± 3.0 | 0 | 8.0 ± 5.0 |
| MMP-9 | 10 ± 4.0 | 7.0 ± 3.0 | 16 ± 4.0 |
| MMP-13 | 51 ± 0 | 67 ± 2.0 | 61 ± 1.0 |
| MMP-14 | 22 ± 5.0 | 20 ± 6.0 | 31 ± 9.0 |
Single inhibitor kinetic assays were performed with MMP-13 and fTHP-15 using methods described previously5,7 to determine the inhibition constant (Ki) and modality of compounds 20 and 24. Lines of best fit crossing on the X axis indicated a non-competitive mode of inhibition of MMP-13-mediated hydrolysis of the fTHP by compound 20 (Figure 3, left). The Ki value was calculated to be 824 ± 171 nM. Lines of best fit crossing on the X axis also indicated a non-competitive mode of inhibition of MMP-13-mediated hydrolysis of the fTHP by compound 24 (Figure 3, right). The Ki value was calculated to be 1,526 ± 260 nM. The Ki value for compound 4 was 3,800 nM (Table 4).
Figure 3.
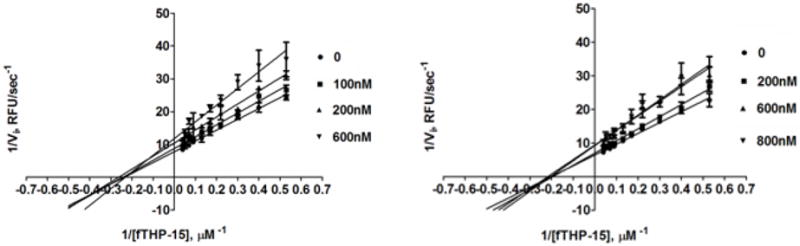
Lineweaver-Burk plot of MMP-13 inhibition of fTHP-15 hydrolysis by compound 20 (left) or 24 (right).
Table 4. IC50 or Ki values (μM) for inhibition of MMP-1, MMP-8, and MMP-13 by compounds 4, 20, and 24.
| Target | Inhibitor | ||
|---|---|---|---|
| 4 | 20 | 24 | |
| MMP-1 (IC50) | >40 | >40 | >40 |
| MMP-8 (IC50) | >40 | >40 | >40 |
| MMP-13 (Ki) | 3.8 | 0.8 | 1.5 |
An RP-HPLC-based secondary screen was performed to determine whether the compounds of interest were artifactual inhibitors of MMP-13.5 Hydrolyzed substrate was examined before and after inhibitor addition to evaluate fluorescence interference. Compounds 20 and 24 were inhibitors in the RP-HPLC secondary screen (data not shown). Thus, the results from the FRET assay (Table 3) were not due to fluorescence artifacts.
The identification of a selective, non-competitive MMP-13 inhibitor (compound 4) led to the present medicinal chemistry characterization of the 2-(arylmethylthio)-cyclopentapyrimidin-4-one scaffold. Two compounds (2-[(4-methylphenyl)methyl sulfanyl]-1,5,6,7-tetrahydrocyclopenta[d]pyrimidin-4-one; 20 and methyl 4-[(4-oxo-1,5,6,7-tetrahydrocyclo penta[d]pyrimidin-2-yl)sulfanylmethyl] benzoate; 24) demonstrated improved potency (as measured by Ki) and selectivity compared to the previously reported compound 4. Most significantly, compound 20 did not inhibit MMP-8, whereas compound 4 did. The lack of inhibition of MMP-8 is desirable, as MMP-8 is a prominent anti-target.8 Additionally, MSS has been attributed to inhibition of MMP-1 and ADAM17/TACE,9 and compounds 4, 20, and 24 do not inhibit MMP-1 (Table 3) or TACE (data not shown). Initial clinical trials with MMP inhibitors were disappointing, with one of the problematic features being a lack of selectivity.3b,10 Compounds 20 and 24 are selective exosite-binding inhibitors of MMP-13, and thus represent leads for the development of the next generation in metalloproteinase therapeutics.
We initially assumed that compound 4 would bind within the same S1’ specificity pocket as compound 1, and designed multiple analogs to achieve interactions present in the crystal structure of compound 1 bound to MMP-13.4e However, because none of these changes led to substantially improved activity in the compound 4 analogs, we suspected that compound 4 and its analogs did not bind in the S1’ pocket as did compound 1. Preliminary hydrogen-deuterium exchange mass spectrometry experiments6 found protection of MMP-13 from exchange by compound 1 at the previously observed site of compound 1 binding,4e but no protection from exchange by compound 20 at this site (M. Chalmers, P. Griffin, P. Hodder, G. Fields, and W. R. Roush, unpublished results). Dual inhibition kinetic assays4f demonstrated non-mutually exclusive binding between compound 4 and either compound 1 or 3 (D. Minond, T. P. Spicer, W. R. Roush, G. B. Fields, and P. S. Hodder, manuscript in preparation). We are pursuing detailed biochemical characterization and competition experiments to further evaluate the unique binding of compound 4 and its analogs, and will report those results in due course.
Supplementary Material
Acknowledgments
This research was supported by the NIH Molecular Library Screening Center Network grant U54MH074404 (Dr. Hugh Rosen, Principal Investigator) and NIH R01CA098799 (G.B.F.). We thank Dr. Patrick Griffin and Dr. Michael Chalmers, of the Scripps Florida Department of Molecular Therapeutics, for the preliminary hydrogen-deuterium exchange mass spectrometry experiments designed to identify the binding site for the MMP-13 exosite inhibitors 4, 20, and 24.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tardif G, Reboul P, Pelletier J-P, Martel-Pelletier J. Mod Rheumatol. 2004;14:197. doi: 10.1007/s10165-004-0292-7. [DOI] [PubMed] [Google Scholar]
- 2.Takaishi H, Kimura T, Dalal S, Okada Y, D’Armiento J. Curr Pharm Biotech. 2008;9:47. doi: 10.2174/138920108783497659. [DOI] [PubMed] [Google Scholar]
- 3.(a) Aureli L, Gioia M, Cerbara I, Monaco S, Fasciglione GF, Marini S, Ascenzi P, Topai A, Coletta M. Curr Med Chem. 2008;15:2192. doi: 10.2174/092986708785747490. [DOI] [PubMed] [Google Scholar]; (b) Jacobsen JA, Major Jourden JL, Miller MT, Cohen SM. Biochim Biophys Acta. 2010;1803:72. doi: 10.1016/j.bbamcr.2009.08.006. [DOI] [PubMed] [Google Scholar]
- 4.(a) Chen JM, Nelson FC, Levin JI, Mobilio D, Moy FJ, Nilakantan R, Zask A, Powers R. J Am Chem Soc. 2000;122:9648. [Google Scholar]; (b) Engel CK, Pirard B, Schimanski S, Kirsch R, Habermann J, Klinger O, Schlotte V, Weithmann KU, Wendt KU. Chem Biol. 2005;12:181. doi: 10.1016/j.chembiol.2004.11.014. [DOI] [PubMed] [Google Scholar]; (c) Blagg JA, Noe MC, Wolf-Gouveia LA, Reiter LA, Laird ER, Chang SP, Danley DE, Downs JT, Elliott NC, Eskra JD, Griffiths RJ, Hardink JR, Hauget AI, Jones CS, Liras JL, Lopresti-Morrow LL, Mitchell PG, Pandit J, Robinson RP, Subramanyam C, Vaughn-Bowser ML, Yocum SA. Bioorg Med Chem Lett. 2005;15:1807. doi: 10.1016/j.bmcl.2005.02.038. [DOI] [PubMed] [Google Scholar]; (d) Reiter LA, Freeman-Cook KD, Jones CS, Martinelli GJ, Antipas AS, Berliner MA, Datta K, Downs JT, Eskra JD, Forman MD, Greer EM, Guzman R, Hardink JR, Janat F, Keene NF, Laird ER, Liras JL, Lopresti-Morrow LL, Mitchell PG, Pandit J, Robinson RP, Sperger D, Vaughn-Bowser ML, Waller DM, Yocum SA. Bioorg Med Chem Lett. 2006;16:5822. doi: 10.1016/j.bmcl.2006.08.066. [DOI] [PubMed] [Google Scholar]; (e) Johnson AR, Pavlovsky AG, Ortwine DF, Prior F, Man C-F, Bornemeier DA, Banotai CA, Mueller WT, McConnell P, Yan C, Baragi V, Lesch C, Roark WH, Wilson M, Datta K, Guzman R, Han H-K, Dyer RD. J Biol Chem. 2007;282:27781. doi: 10.1074/jbc.M703286200. [DOI] [PubMed] [Google Scholar]; (f) Gooljarsingh LT, Lakdawala A, Coppo F, Luo L, Fields GB, Tummino PJ, Gontarek RR. Protein Sci. 2008;17:66. doi: 10.1110/ps.073130208. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Li JJ, Nahra J, Johnson AR, Bunker A, O’Brien P, Yue W-S, Ortwine DF, Man C-F, Baragi V, Kilgore K, Dyer RD, Han H-K. J Med Chem. 2008;51:835. doi: 10.1021/jm701274v. [DOI] [PubMed] [Google Scholar]; (h) Heim-Riether A, Taylor SJ, Liang S, Gao DA, Xiong Z, August EM, Collins BK, Farmer BT, II, Haverty K, Hill-Drzewi M, Junker H-D, Margarit SM, Moss N, Neumann T, Proudfoot JR, Smith Keenan L, Sekul R, Zhang Q, Li J, Farrow NA. Bioorg Med Chem Lett. 2009;19:5321. doi: 10.1016/j.bmcl.2009.07.151. [DOI] [PubMed] [Google Scholar]; (i) Baragi VM, Becher G, Bendele AM, Biesinger R, Bluhm H, Boer J, Deng H, Dodd R, Essers M, Feuerstein T, Gallagher BM, Jr, Gege C, Hochgürtel M, Hofmann M, Jaworski A, Jin L, Kiely A, Korniski B, Kroth H, Nix D, Nolte B, Piecha D, Powers TS, Richter F, Schneider M, Steeneck C, Sucholeiki I, Taveras A, Timmermann A, Van Veldhuizen J, Weik J, Wu X, Xia B. Arthritis Rheum. 2009;60:2008. doi: 10.1002/art.24629. [DOI] [PubMed] [Google Scholar]; (j) Piecha D, Weik J, Kheil H, Becher G, Timmermann A, Jaworski A, Burger M, Hofmann MW. Inflamm Res. 2010;59:379. doi: 10.1007/s00011-009-0112-9. [DOI] [PubMed] [Google Scholar]; (k) Schnute ME, O’Brien PM, Nahra J, Morris M, Roark WH, Hanau CE, Ruminski PG, Scholten JA, Fletcher TR, Hamper BC, Carroll JN, Patt WC, Shieh HS, Collins B, Pavlovsky AG, Palmquist KE, Aston KW, Hitchcock J, Rogers MD, McDonald J, Johnson AR, Munie GE, Wittwer AJ, Man C-F, Settle SL, Nemirovskiy O, Vickery LE, Agawal A, Dyer RD, Sunyer T. Bioorg Med Chem Lett. 2010;20:576. doi: 10.1016/j.bmcl.2009.11.081. [DOI] [PubMed] [Google Scholar]; (l) Gao DA, Xiong Z, Heim-Riether A, Amodeo L, August EM, Cao X, Ciccarelli L, Collins BK, Harrington K, Haverty K, Hill-Drzewi M, Li X, Liang S, Margarit SM, Moss N, Nagaraja N, Proudfoot J, Roman R, Schlyer S, Keenan LS, Taylor S, Wellenzohn B, Wiedenmayer D, Li J, Farrow NA. Bioorg Med Chem Lett. 2010;20:5039. doi: 10.1016/j.bmcl.2010.07.036. [DOI] [PubMed] [Google Scholar]; (m) Jüngel A, Ospelt C, Lesch M, Thiel M, Sunyer T, Schorr O, Michel BA, Gay RE, Kolling C, Flory C, Gay S, Neidhart M. Ann Rheum Dis. 2010;69:898. doi: 10.1136/ard.2008.106021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lauer-Fields JL, Minond D, Chase PS, Baillargeon PE, Saldanha SA, Stawikowska R, Hodder P, Fields GB. Bioorg Med Chem. 2009;17:990. doi: 10.1016/j.bmc.2008.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lauer-Fields JL, Chalmers MJ, Busby SA, Minond D, Griffin PR, Fields GB. J Biol Chem. 2009;284:24017. doi: 10.1074/jbc.M109.016873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ki values were determined by non-linear regression (hyperbolic equation) analysis using the mixed inhibition model which allows for simultaneous determination of the mechanism of inhibition.10 The mechanism of inhibition was determined using the “alpha” parameter derived from the mixed-model inhibition by GraphPad Prism. The mechanism of inhibition was additionally confirmed by Lineweaver-Burk plots (Figure 3).
- 8.Morrison CJ, Butler GS, Rodríguez D, Overall CM. Curr Opin Cell Biol. 2009;21:645. doi: 10.1016/j.ceb.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Becker DP, Barta TE, Bedell LJ, Boehm TL, Bond BR, Carroll J, Carron CP, DeCrescenzo GA, Easton AM, Freskos JN, Funckes-Shippy CL, Heron M, Hockerman S, Howard CP, Kiefer JR, Li MH, Mathis KJ, McDonald JJ, Mehta PP, Munic GE, Sunyer T, Swearingen CA, Villamil CI, Welsch D, Williams JM, Yu Y, Yao J. J Med Chem. 2010;53:6653. doi: 10.1021/jm100669j. [DOI] [PubMed] [Google Scholar]
- 10.Overall CM, Kleifeld O. Br J Cancer. 2006;94:941. doi: 10.1038/sj.bjc.6603043. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.