

Abstract
Estrogen signaling is mediated by two estrogen receptors (ERs), ERα and ERβ, which have unique roles in the regulation of breast cancer cell proliferation. ERα induces proliferation in response to estrogen and ERβ inhibits proliferation in breast cancer cells, suggesting that ERβ selective ligands may be beneficial for promoting the anti-proliferative action of ERβ. Subtype selective ligands can be identified using transcriptional assays, but cell lines in which ERα or ERβ are independently expressed are required. Of the available reporter cell lines, none have been generated in breast cancer cells to identify subtype selective ligands. Here we describe the generation of two isogenic breast cancer cell lines, Hs578T-ERαLuc and Hs578T-ERβLuc, with stable integration of an estrogen responsive luciferase reporter gene. Hs578T-ERαLuc and Hs578T-ERβLuc cell lines are highly sensitive to estrogenic chemicals and ER subtype selective ligands, providing a tool to characterize the transcriptional potency and subtype selectivity of estrogenic ligands in the context of breast cancer cells. In addition to measuring reporter activity, ERβ target gene expression and growth inhibitory effects of ERβ selective ligands can be determined as biological endpoints. The finding that activation of ERβ by estrogen or ERβ selective natural phytoestrogens inhibits the growth of Hs578T-ERβ cells implies therapeutic potential for ERβ selective ligands in breast cancer cells that express ERβ.
Keywords: Estrogen receptors, subtype selectivity, phytoestrogens, breast cancer
1. Introduction
Estrogens regulate mammary gland growth and differentiation, ovary and uterus maturation, and bone homeostasis [1]. The physiological effects of estrogens are primarily mediated by two estrogen receptors (ERs), ERα and ERβ. Because of the broad range of ER target tissues and the ligand dependent activity of the receptors, synthetic and natural estrogens hold therapeutic promise in selectively targeting ERs. Therapies aimed at preventing ERα transcriptional activation are currently used for breast cancer treatment and osteoporosis prevention [2]. Though ERβ is not currently a therapeutic target, accumulating evidence suggests an anti-proliferative role for ERβ in breast cancer [3]. In the mammary gland, ERα and ERβ play opposing roles in regulating growth and differentiation in response to estrogens; ERα promotes proliferation while ERβ inhibits ERα-mediated proliferation [4–6]. Because the anti-proliferative action of ERβ may be enhanced by ligand-dependent activation, the paradigm of ER targeted therapies is expanding towards the development of ER subtype selective ligands [7].
Though ERα and ERβ share many structural and transcriptional features, ligands can display subtype selectivity. In classical ligand dependent transcriptional activation, the receptors dimerize upon ligand binding and undergo conformational changes to allow cofactor recruitment. The receptors directly bind DNA most often at estrogen response elements (EREs), consisting of a consensus GGTCAnnnTGACC sequence. ERα and ERβ have 97% identity within the DNA binding domains, and the receptors bind similar DNA sequences with high affinity. Genome wide binding studies in MCF7 breast cancer cells expressing ERα or ERβ independently have shown that ERα and ERβ bind similar sites in response to 17β-estradiol (E2); ~60% of ER binding sites contain full EREs and ~25% contain half EREs [8].
The ligand binding pockets of ERα and ERβ are relatively large, and the receptors bind a wide array of chemicals. The ligand binding domains of ERα and ERβ have 59% identity, and the receptors bind E2 with similar affinities. Despite similarities in their ligand binding domains, several ligands have modest selectivity for ERα or ERβ [9], and some synthetic ligands maintain high selectivity. For example, propyl pyrazole triol (PPT) is an ERα selective agonist that displays a 400-fold higher binding affinity for ERα compared to ERβ [10]. Estrogenic chemicals produced in plants, known as phytoestrogens, often display subtype selectivity for ERβ. For example, liquiritigenin is a flavanone derived from Glycyrrhizae uralensis that has been shown to have 20-fold higher binding affinity for ERβ and even greater selectivity in transcriptional assays [11]. Compounds such as liquiritigenin often show low binding affinities relative to E2, and ERβ selective ligands with higher affinity and greater selectivity are needed to fully elucidate the anti-proliferative role of ERβ in breast cancer.
Mammalian cell lines have been developed to enable screening for subtype selective ligands. HeLa cervical carcinoma cells have been used to create HELN-ERα and HELN-ERβ, two cell lines in which ERα or ERβ, respectively, are constitutively expressed with stable integration of a luciferase reporter downstream of an ERE [12]. Human embryonic kidney cells, HEK293, have also been created using a similar strategy in which ERα or ERβ are constitutively expressed and human placental alkaline phosphatase downstream of the vitellogenin ERE is stably integrated [13]. The only available breast cancer reporter cell line is T47D-KBLuc in which three tandem EREs upstream of a luciferase reporter have been stably integrated [14]. However, identification of subtype selective ligands is prohibited because T47D cells express both ERα and ERβ.
Here, we describe the generation of two isogenic reporter cell lines, Hs578T-ERαLuc and Hs578T-ERβLuc, that provide a tool to characterize the transcriptional potencies and subtype selectivity of estrogenic compounds in the context of breast cancer cells. These cell lines are highly sensitive to estrogenic ligands and subtype selective ligands and can be used to validate ER transcriptional activation by analysis of endpoints such as endogenous target gene regulation. Further, ERβ selective ligands are shown to induce ERβ-mediated reporter gene expression, endogenous gene regulation, and growth inhibition, suggesting that Hs578T-ERβLuc cells may be used to isolate ERβ selective ligands with desired biological effects.
2. Materials and Methods
2.1 Cell lines and reagents
Cosmosiin (apigenin 7-glucoside), dimethyl sulfoxide (DMSO), E2, and diethylstilbestrol (DES) were obtained from Sigma (St. Louis, MO); DPN, PPT, and ICI 182,780 were obtained from Tocris (Ellinsville, MO); liquiritigenin was obtained from Chromadex (Irvine, CA). Doxycycline (Dox) was obtained from Clontech. Hygromycin B, blasticidin S, zeocin, NaCl, sodium dodecyl sulfate (SDS), and dithiothreitol (DTT) were obtained from Research Products International (Mount Prospect, IL). Triton X-100 was obtained from Fisher (Fair Lawn, NJ); protease inhibitors were obtained from Roche Scientific (Basel, Switzerland); benzonase was obtained from Novagen (San Diego, CA). All other chemicals were obtained from Sigma (St. Louis, MO).
Cell culture media were obtained from Invitrogen (Carlsbad, CA). MCF7 and HEK293 cells were cultured in DMEM + 10% fetal bovine serum (FBS; Gemini Bio Products, West Sacramento, CA) at 37 °C and 5% CO2. Hs578T-ERα and Hs578T-ERβ were previously created by Secreto and coworkers [15]. These cells were cultured at 37 °C and 5% CO2 in DMEM/F12 supplemented with L-glutamine, 10% Tet-system approved FBS (Clontech Mountain View, CA), 500 mg/L Zeocin and 5 mg/L Blasticidin S.
2.2 Generation of Hs578T-ERαLuc and Hs578T-ERβLuc reporter cell lines
Stable reporter cell lines were created using a modified pGL4.32 reporter (Promega, Madison, WI) which contains the luc2P reporter and hygromycin resistance. The pGL4.32 vector was digested with Nhe1 and HindIII (New England Biolabs, Ipswich, MA) and three consensus EREs spaced by three nucleotides were cloned upstream of luc2P using the following oligonucleotides: 5′ –CTA GCG GTC ACA GTG ACC TGC GAG GTC ACA GTG ACC TGC GAG GTC ACA GTG ACC TGC GA – 3′ and 5′ – AGC TTC GCA GGT CAC TGT GAC CTC GCA GGT CAC TGT GAC CTC GCA GGT CAC TGT GAC CG – 3′. Successful cloning was verified by complete sequencing and the vector was designated pGL4.3xERE. Estrogen responsiveness was validated by batch transfecting HEK293 cells with 2 ng of CMX-ERα or CMX-ERβ, 45 ng pGL4.3xERE vector, and 40 ng CMX-β-galactosidase per well of a 48 well plate. Cells were incubated 24 hr to allow protein expression before the addition of the indicated ligands. After 24 hr of ligand treatment, cells were lysed, firefly luciferase substrate (Promega) was added, and luminescence was measured on a Victor X5 microplate reader (Perkin Elmer, Waltham, Massachusetts) using luminescence detection and a 700 nm filter. To normalize data for transfection efficiency, β-galactosidase expression was analyzed using the Tropix β-galactosidase detection kit (Applied Biosystems, Foster City, CA). Luciferase counts were normalized to β-gal counts in each well.
After characterizing the pGL4.3xERE stable reporter vector, Hs578T-ERα and Hs578T-ERβ cells were transfected with 10 μg of the vector and selected in 125 μg/mL hygromycin B for 4 weeks. Individual colonies were selected using 3 mm cloning discs, expanded, and screened for estrogen induced luciferase expression. One clone from each cell line was selected for further characterization, referred to here as Hs578T-ERαLuc and Hs578T-ERβLuc.
2.3 Quantitative western blots and ligand binding assays
For quantitative western blots, cells were split in phenol red free DMEM/F12 + 5% SFS and treated with 50 ng/mL Dox or vehicle (water) 24 hr later. After 48 hr treatment, cells were collected by trypsinization, washed with Dulbecco’s phosphate buffer saline (Invitrogen), and lysed by suspension in lysis buffer (50 mM Tris pH 8.0, 400 mM NaCl, 10% glycerol, 0.5% triton X-100, protease inhibitors, and benzonase). After centrifugation, total protein was quantified using BioRad Protein Assay (BioRad), and 40 ug of protein was resolved using SDS-PAGE and 8% polyacrylamide gels. Proteins were transferred to a nitrocellulose membrane for 1.5 hr at 0.35 A. Membranes were blocked with 5% nonfat milk and incubated overnight with 1:1000 anti-FLAG-M2 antibody (Sigma) or 1:5000 anti-β-Actin (Sigma) at 4°C. Membranes were then incubated with IRDye 800CW goat-anti-mouse IgG secondary antibody (Licor Biosciences, Lincoln, NE) for 1 hr at room temperature and visualized on a Licor Odyssey near-infrared gel reader (Licor Biosciences).
For ligand binding assays, Hs578T-ERαLuc and Hs578T-ERβLuc cells were cultured in phenol red free DMEM/F12 + 10% 6x charcoal stripped FBS (SFS) for 3 days prior to the assay to remove residual estrogens from the cells. At 90% confluence, cells were collected, resuspended in phenol red free DMEM/F12 + 5% SFS, and plated at a density of 105 cells/well on a 24 well plate in the presence or absence of 50 ng/mL Dox. After 24 hr, cells were labeled in triplicate with 20 nM [3H]-E2 (89.2 Ci/mmol specific activity, Perkin Elmer) in the presence or absence of 450 μM DES cold competitor for 2 hr at 37 °C and 5% CO2. Labeled cells were washed 3 times with cold PBS + 0.1% BSA and lysed with 500 μL SDS lysis buffer (0.5% SDS, 0.05 M Tris-HCl pH 8.0, and 1 mM DTT). Total cell lysate (400 μL) was mixed with 5 mL liquid scintillation cocktail and [3H] bound radioactivity was liquid scintillation counted for 5 min. Two additional wells of each condition were used to count the cell number and determine the total protein using RC DC protein assay (BioRad, Hercules, CA).
2.4 Luciferase assays
Hs578T-ERαLuc and Hs578T-ERβLuc cells were cultured in phenol red free DMEM/F12 + 10% SFS for 3 days prior to the assay to remove residual estrogens from the cells. Cells were seeded in triplicate at a density of 104 cell/well on white 96 well tissue culture plates (Fisher) in phenol red free DMEM/F12 + 5% SFS treated with 50 ng/mL Dox. After 24 hr of Dox treatment, media were replaced with treated media containing vehicle (0.15% DMSO) or a range of serially diluted ligands. All treatments were conducted in the presence and absence of 100 nM ICI 182,780. After treatment for 24 hr, cells were washed with PBS and lysed with 35 μL lysis buffer (100 mM K2HPO4, 0.2% triton X-100, pH 7.8). Lysate (30 μL) was mixed 1:1 with luciferase substrate (Promega) and luminescence was measured on a Victor X5 microplate reader (Perkin Elmer, Waltham, Massachusetts) using luminescence detection and a 700 nm filter. Total protein (5 μL) was quantified using BioRad Protein Assay (BioRad). EC50 values were calculated using GraphPad Prism Software (Version 5.04, GraphPad Software Inc., San Diego, CA) and a three parameter log versus response nonlinear regression. Two tailed t-tests performed with GraphPad Prism Software were used to determine statistically significant differences from control treatments.
2.5 Gene expression analysis
For analysis of reporter induction by cosmosiin, Hs578T-ERαLuc and Hs578T-ERβLuc cells were split in phenol red free DMEM/F12 + 5% SFS and treated with 50 ng/mL Dox for 48 hr followed by treatment with DMSO (0.1%), 1 nM E2, or 1 μM cosmosiin for 4 or 24 hr. Total RNA was extracted using RNEasy Plus Kit according to manufacturer protocol (Qiagen, Valencia, CA). RNA (2 μg) was reverse transcribed using Superscript II RT according to manufacturer protocol (Invitrogen), and firefly luciferase (FLuc) expression was determined by reverse-transcription polymerase chain reaction using primers shown in Table 1.
Table 1.
Primer and Probe Sequences
| RPL13A | Primer 1 | 5′ - TGT TTG ACG GCA TCC CAC - 3′ |
| Primer 2 | 5′ - CTG TCA CTG CCT GGT ACT TC - 3′ | |
| Probe | 5′ - CTT CAG ACG CAC GAC CTT GAG GG - 3′ | |
| C3 | Primer 1 | 5′ - AAC TAC ATC ACA GAG CTG CG - 3′ |
| Primer 2 | 5′ - AAG TCC TCA ACG TTC CAC AG - 3′ | |
| Probe | 5′ - CGT TTC CCG AAG TGA GTT CCC AGA - 3′ | |
| JAG1 | Primer 1 | 5′ - GGA CTA TGA GGG CAA GAA CTG - 3′ |
| Primer 2 | 5′ - AAA TAT ACC GCA CCC CTT CAG - 3′ | |
| Probe | 5′ - TCA CAC CTG AAA GAC CAC TGC CG - 3′ | |
| ITGA6 | Primer 1 | 5′ - ACC CGA GAA GGA AAT CAA GAC - 3′ |
| Primer 2 | 5′ - CGC CAT CTT TTG TGG GAT TC - 3′ | |
| Probe | 5′ - TGG GTT GGA AGG GCT GTT TGT CA - 3′ | |
| FLuc | Primer 1 | 5′ – GGC TGA ATA CAA ACC ATC GG – 3′ |
| Primer 2 | 5′ – CTT TCT TGC TCA CGA ATA CGA – 3′ |
For quantitative real-time PCR analysis of endogenous target gene expression, Hs578T-ERα and Hs578T-ERβ cells were cultured in phenol red free DMEM/F12 + 10% SFS for 3 days prior to the assay to remove residual estrogens from the cells. Cells were split in phenol red free DMEM/F12 + 5% SFS and treated with 50 ng/mL Dox for 48 hr prior to ligand treatment. Cells were treated with Dox and ligands or vehicle (0.1% DMSO) for 24 hr, and total RNA was extracted using RNEasy Plus Kit according to manufacturer protocol (Qiagen). RNA (2 μg) was reverse transcribed as above, and quantitative PCR was performed using TaqMan Prime Time custom designed assays (IDT, Coralville, IA), FastStart Universal Probe Master Mix (Roche Scientific), and a CFX96 instrument (BioRad). Primer and probe sequences are shown in Table 1. Data were analyzed using the ΔΔCq method calculated by the CFX Manager Software (BioRad). Two tailed t-tests performed with GraphPad Prism Software were used to determine statistically significant differences from control treatments using data from three biological replicates.
2.6 Cell counting assays
Hs578T-ERα and Hs578T-ERβ cells were cultured in phenol red free DMEM/F12 + 10% SFS for 3 days prior to the assay to remove residual estrogens from the cells. Cells were seeded at a density of 15,000 cell/well in phenol red free DMEM/F12 + 5% SFS in triplicate in 6 well tissue culture dishes in the presence or absence of 50 ng/mL Dox. After 24 hr, the cells were treated with DMSO (0.1%) or compound in the presence or absence of 50 ng/mL Dox. Media were refreshed every 48 hr, and cells were counted after trypan blue exclusion using an automated cell counter (BioRad) according to manufacturer protocol.
3. Results
3.1 Generation of Hs578T-ERαLuc and Hs578T-ERβLuc reporter cell lines
In order to generate stable reporter breast cancer cell lines, we first cloned a construct encoding a selection marker and a luciferase reporter linked to EREs. The pGL4.32 vector (Promega) contains the luc2P gene and was modified to contain 3 tandem consensus EREs upstream of the minimal promoter (pGL4.3xERE, Fig. 1A). Upon complete sequencing, the estrogen responsiveness of the vector was validated in ER-negative HEK293 cells transfected with full length ERα (Fig. 1B) or ERβ (Fig. 1C). The pGL4.3xERE reporter showed extremely low background with a 65-fold induction in cells transfected with ERα. The ER antagonist ICI 182,780 abolished estrogen induced expression, reducing the luciferase signal to that of vehicle treated cells. Cells transfected with ERβ showed a 15-fold induction of luciferase upon E2 treatment; ICI 182,780 inhibited luciferase expression in both vehicle and estrogen treated cells. The minimal background luciferase expression and the selection marker conferred by the pGL4.3xERE vector made the vector suitable for creating stable reporter cells lines for the identification and characterization of ER selective agonists.
Figure 1.
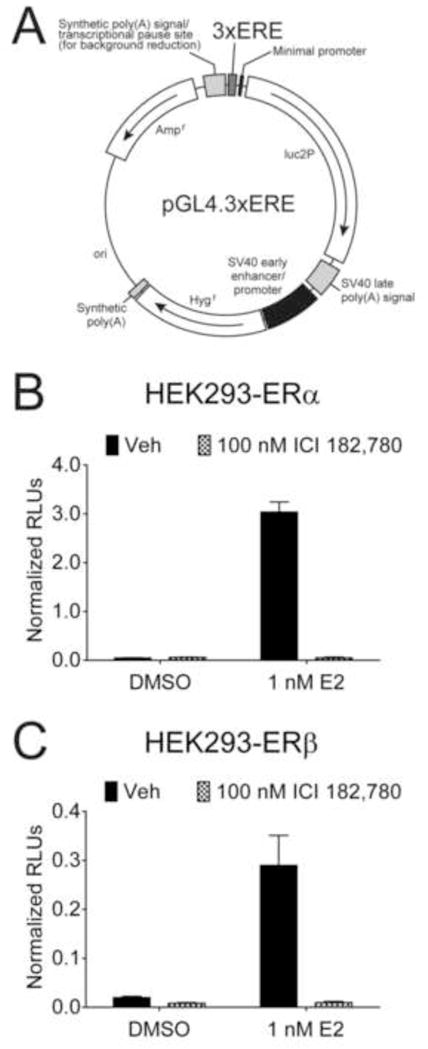
The pGL4.3xERE reporter construct is estrogen responsive. (A) Three tandem EREs were inserted upstream of the luc2P gene in the pGL4.32 luciferase reporter construct. HEK293 cells were batch transfected with the pGL4.3xERE reporter construct, a β-galactosidase construct, and full length ERα (B) or ERβ (C). After allowing 24 hr for protein expression, cells were treated in triplicate with vehicle (DMSO) or 1 nM E2 and vehicle or 100 nM ICI 182,780 (0.15 % final DMSO concentration) for an additional 24 hr. Raw luciferase units (RLUs) were normalized to β-galactosidase to normalize for transfection efficiency. Error bars represent standard deviations.
In order to create stable ER reporter breast cancer cell lines, an ER negative breast cancer cell line engineered to express either ERα or ERβ was necessary. Previously, Secreto and coworkers created such lines using Hs578T cells [15], a triple negative breast cancer cell line with a basal-like gene expression profile [16]. Hs578T cells lack expression of ERα and ERβ providing a clean background in which to express ERα or ERβ. Using the tetracycline inducible system, two cell lines were created in which ERα or ERβ are inducibly expressed (Hs578T-ERα and Hs578T-ERβ cells, respectively) [15]. Hs578T-ERα and Hs578T-ERβ cells were transfected with the pGL4.3xERE vector, and individual clones were isolated after hygromycin selection. Over 20 clones were screened for estrogen induced luciferase expression (data not shown). One clone from each cell line was selected for further characterization, referred to here as Hs578T-ERαLuc and Hs578T-ERβLuc. Additional ERα and ERβ reporter clones were used to verify reporter data obtained from Hs578T-ERαLuc and Hs578T-ERβLuc cells.
Hs578T-ERαLuc and Hs578T-ERβLuc cells were first characterized by assessing luciferase induction by ER ligands in the presence or absence of the full antagonist ICI 182,780 (Figure 2). Cells were treated with vehicle, 1 nM E2, 10 nM DPN (a reported ERβ selective agonist), or 10 nM PPT (a reported ERα selective agonist). PPT selectively activated luciferase expression in Hs578T-ERαLuc, but DPN activated the reporter in both Hs578T-ERαLuc and Hs578T-ERβLuc cells, though to a lesser extent in Hs578T-ERαLuc cells. Co-treatment with ICI 182,780 blocked luciferase induction in both cell lines (Fig. 2), and luciferase was not induced in the absence of Dox treatment (data not shown).
Figure 2.
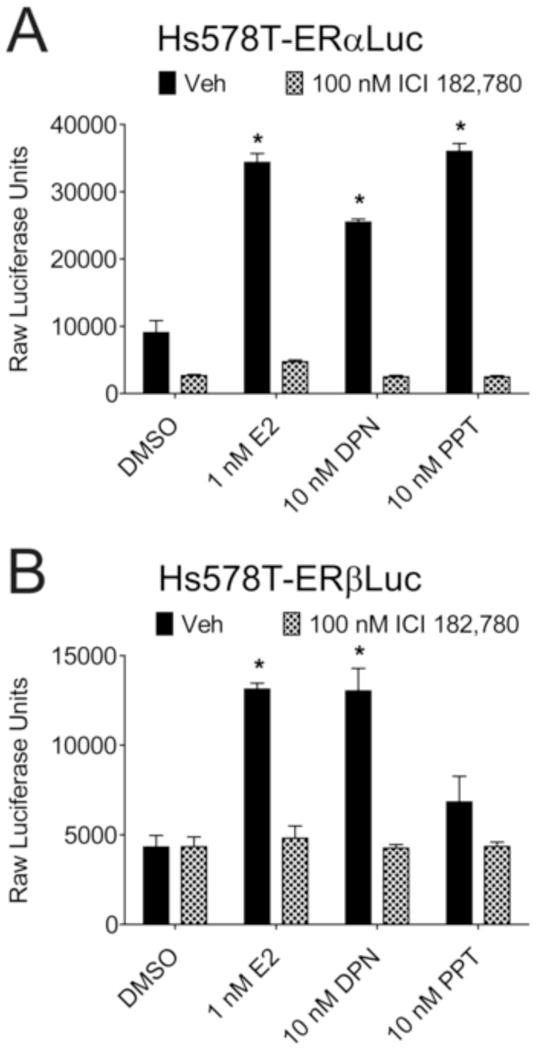
ER subtype selective ligands selectively induce luciferase in Hs578T-ERαLuc and Hs578T-ERβLuc cells. Hs578T-ERαLuc (A) and Hs578T-ERβLuc (B) cells were seeded in triplicate on 96 well plates in the presence of 50 ng/mL Dox to induce ER expression. After 24 hr, cells were treated with vehicle (DMSO), 1 nM E2, 10 nM DPN, or 10 nM PPT in the presence or absence of 100 nM ICI 182,780 (0.15% final DMSO concentration). Cells were lysed 24 hr after ligand treatment and raw luciferase units were counted. Error bars represent standard deviations. * p values < 0.05
Basal and E2-induced luciferase signals were much higher in Hs578T-ERαLuc cells when compared to Hs578T-ERβLuc cells, a trend observed in all luciferase assays. On average, Hs578T-ERβLuc cells expressed 630 luciferase units per mg protein and Hs578T-ERαLuc expressed 2900 luciferase units per mg protein at saturating E2 concentrations (0.1 nM or greater). A range of luciferase signals was observed among the clones screened (data not shown), suggesting the accessibility of the reporter in the chromatin may be responsible for differences in luciferase expression. In order to verify Hs578T-ERαLuc and Hs578T-ERβLuc cells had similar ER expression levels at the Dox concentration used throughout the study (50 ng/mL), quantitative western blots were used to compare ER expression in the parent cell lines and reporter cell lines (Fig. 3A). Western blots with FLAG antibody demonstrated similar ER expression in Hs578T-ERαLuc and Hs578T-ERβLuc cells and also confirmed expression levels similar to the parent cell lines. In addition, whole cell ligand binding assays were used to quantify the active receptor in each cell line (Fig. 3B). ERα positive MCF7 breast cancer cells expressed ~150,000 receptors/cell which was very similar to reported values [17]. Both Hs578T-ERαLuc and Hs578T-ERβLuc cells expressed ~120,000 receptors/cell after 50 ng/mL Dox treatment. The comparable number of ERs per cell suggests that differences in ER expression do not account for the higher luciferase signal observed Hs578T-ERαLuc cells. Higher luciferase expression in Hs578T-ERαLuc cells may be due to the accessibility of the reporter in the chromatin or the enhanced transcriptional activity of ERα, in agreement with previous findings that the transcriptional activity of ERα is greater than that of ERβ on ERE-containing reporters [18]. Finally, the reporter cell lines did not have an altered morphological phenotype compared to the parent cell lines (Fig. 3C), and no other phenotypic changes due to the integration of the luciferase reporter were observed in Hs578T-ERαLuc and Hs578T-ERβLuc cells.
Figure 3.
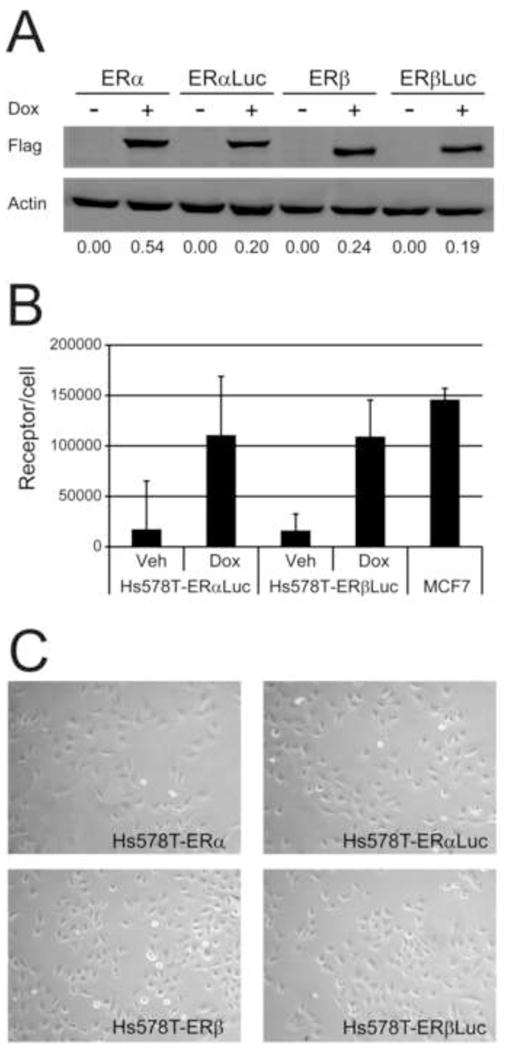
Hs578T-ERαLuc and Hs578T-ERβLuc cells express similar levels of ER. (A) Quantitative western blot with Hs578T-ERα (ERα), Hs578T-ERαLuc (ERαLuc), Hs578T-ERβ (ERβ), and Hs578T-ERβLuc (ERβLuc) treated with vehicle (-Dox) or 50 ng/mL Dox (+Dox). ER expression was detected using FLAG antibody and quantified by normalizing to b-actin using the Licor Odyssey near-infrared gel reader. The normalized integrated intensity for the FLAG signal is shown below the images. (B) Ligand binding assays confirmed the quantitative western blots. Hs578T-ERαLuc and Hs578T-ERβLuc cells were seeded in triplicate and treated with vehicle or 50 ng/mL Dox for 24 hr. Cells were labeled with 20 nM [3H]-E2 in the presence or absence of cold competitor for 2 hr, washed, and total cell lysate was assessed for bound radioactivity as described in Materials and Methods. MCF7 cells were included for comparison. Two additional wells of each cell line and condition were used to determine the cell number and the numbers of receptors per cell were calculated based on a 1:1 molar ratio of ligand to receptor. The average and standard deviation of three independent experiments are shown. (C) The morphology of Hs578T-ERαLuc and Hs578T-ERβLuc was similar to that of the parent Hs578T-ERα and Hs578T-ERβ cell lines. Representative phase-contrast microscopy images of each cell line (100X magnification).
3.2 Ligand selectivity of Hs578T-ERαLuc and Hs578T-ERβLuc reporter cell lines
We next assessed ligand subtype selectivity using these isogenic reporter cell lines. All luciferase data were normalized to the luciferase signal induced by a saturating concentration of E2 (0.1 nM) and expressed as the percent transactivation relative to 0.1 nM E2. Dose-response curves were obtained for E2, DPN, and PPT to characterize the sensitivity of the reporter cells to ER ligands (Fig. 4). Cells were treated with 10-fold dilutions of ligands and approximate EC50 concentrations for each ligand were calculated from 3 independent experiments (Table 2). The ratios of EC50 values obtained from Hs578T-ERαLuc cells and Hs578T-ERβLuc cells are also presented in Table 2 and provide a measure of the selectivity of the ligands. Higher α/β ratios indicate selectivity for ERβ.
Figure 4.
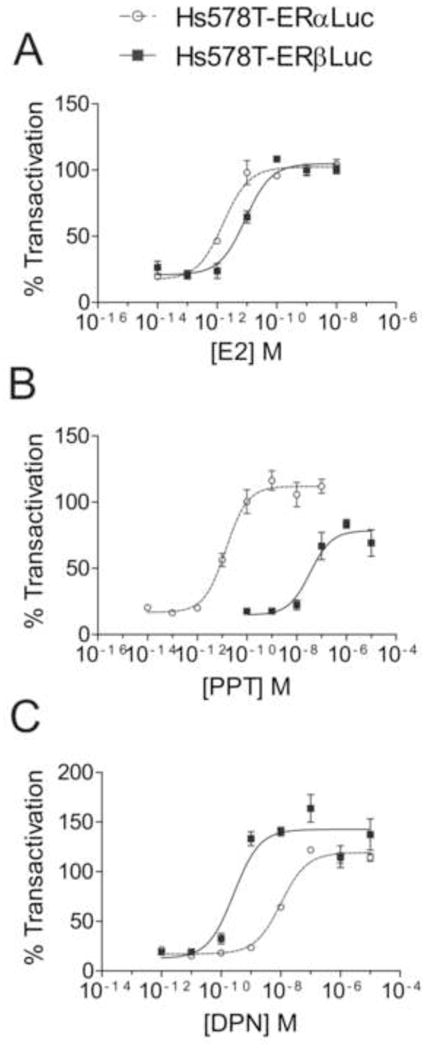
Hs578T-ERαLuc and Hs578T-ERβLuc show subtype selective activation. Dose response curves of E2 (A), PPT (B), and DPN (C). Hs578T-ERαLuc and Hs578T-ERβLuc were seeded in triplicate and treated with 50 ng/mL Dox for 24 hr. Cells were then treated with a range of ligand concentrations (0.15% final DMSO concentration) for 24 hr. Each plate contained DMSO, 0.1 nM E2, and 100 nM ICI 182,780 for controls. Luciferase signal was normalized to total protein in each well and expressed as a percent transactivation relative to signal obtained from saturating E2 treatment (0.1 nM). Each dose response experiment was conducted at least 3 times; data shown are from one representative experiment. EC50 values are shown in Table 2.
Table 2.
Average EC50 values for ER Ligands (M × 10−9)
| Hs578T-ERαLuc | Hs578T-ERβLuc | α/β | |
|---|---|---|---|
| E2 | 0.001 (0.0005) | 0.0065 (0.008) | 0.15 |
| DPN | 8.5 (3) | 0.26 (0.02) | 33 |
| PPT | 0.016 (0.001) | 26 (21) | 0.001 |
| Liquiritigenin | 100 (40) | 28 (2) | 3.6 |
Both cell lines were highly sensitive to estrogen (Fig. 4A). Hs578T-ERαLuc cells showed EC50 values near 1 pM; four additional Hs578T-ERαLuc clones showed similar sensitivities (data not shown). Hs578T-ERβLuc cells also showed EC50 values for estrogen in the pM range, though the average EC50 was 6.5-fold higher than that of Hs578T-ERαLuc cells. Similar differences in estrogen sensitivities have been observed in other ERE-luciferase reporter cell lines expressing ERα or ERβ [12–14], suggesting the difference in E2 sensitivity between Hs578T-ERαLuc and Hs578T-ERβLuc cells is due to differences in the transactivation of ERα and ERβ.
Next, dose responses to two highly selective ERα and ERβ agonists, PPT and DPN respectively, were analyzed using Hs578T-ERαLuc and Hs578T-ERβLuc cells. PPT showed nearly 1000-fold selectivity for ERα (Fig. 4B). Surprisingly, PPT could activate reporter expression in Hs578T-ERβLuc cells at concentrations greater than 100 nM, although it could not induce luciferase expression to the same extent as E2. It has been reported that PPT was unable to induce an estrogen responsive reporter in HEC-1 cells transfected with ERβ [10] or in HELN-ERα cells [12]. DPN was not as selective as PPT and could maximally activate luciferase expression Hs578T-ERαLuc cells at 100 nM (Fig. 4C). DPN fully activated ERβ at 10 nM. Though DPN has been shown to have a 50 to 70-fold higher binding affinity for ERβ [12, 19], comparison of EC50 values showed approximately 30-fold selectivity for ERβ in these reporter assays.
Next, the subtype selectivity of two natural phytoestrogens, liquiritigenin and cosmosiin, were analyzed using Hs578T-ERαLuc and Hs578T-ERβLuc cells (Fig. 5). Liquiritigenin is a phytoestrogen derived from Glycyrrhizae uralensis and the most active estrogenic component of MF101, an herbal supplement with therapeutic potential [11]. In the initial characterization of liquiritigenin, Mersereau and coworkers found liquiritigenin showed minimal activation of ERα at concentrations up to 2.5 μM in transcriptional assays in U2OS, HeLa, or WAR5 prostate cancer cells transfected with ERα [11]. Binding assays demonstrated that liquiritigenin had a 20-fold higher affinity for ERβ and selectivity was proposed to be due to selective recruitment of co-activators to ERβ, namely SRC-2 [11]. Comparison of EC50 values showed liquiritigenin had a 3.6-fold selectivity for ERβ, and maximal reporter induction was obtained by 100 nM liquiritigenin in Hs578T-ERβLuc cells and 1 μM in Hs578T-ERαLuc (Fig. 5A, Table 2).
Figure 5.
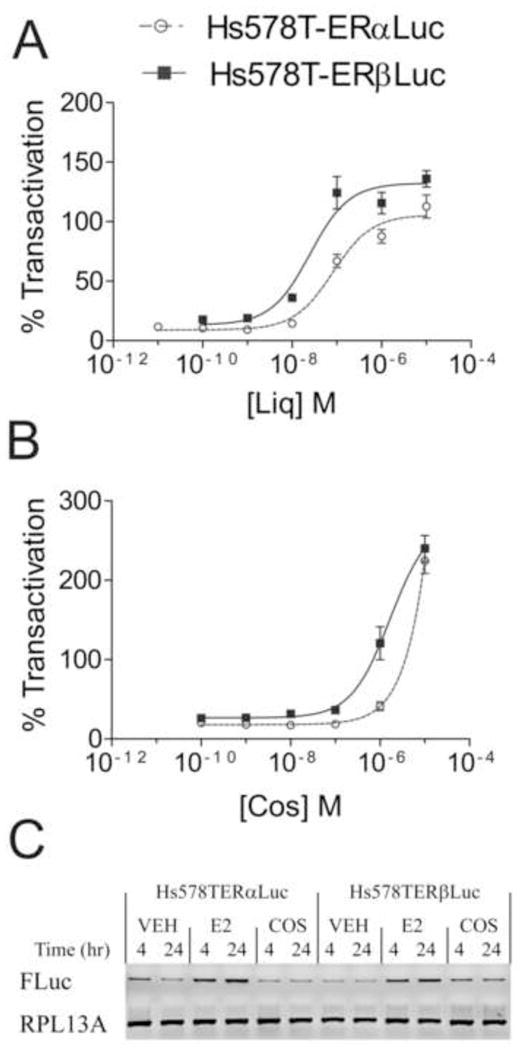
Liquiritigenin (Liq) and cosmosiin (Cos) induce reporter expression in Hs578T-ERαLuc and Hs578T-ERβLuc. Dose response curves of liquiritigenin (A) and cosmosiin (B). Hs578T-ERαLuc and Hs578T-ERβLuc were seeded in triplicate and treated with 50 ng/mL Dox for 24 hr. Cells were then treated with a range of ligand concentrations (0.15% final DMSO concentration) for 24 hr. Each plate contained DMSO, 0.1 nM E2 and 100 nM ICI 182,780 for controls. Luciferase signal was normalized to total protein in each well and expressed as a percent transactivation relative to signal obtained from saturating E2 treatment (0.1 nM). Each dose response experiment was conducted at least 3 times; data shown are from one representative experiment. EC50 values are shown in Table 2. EC50 values for cosmosiin could not be determined because of supramaximal reporter induction. The supramaximal induction by cosmosiin was not due to supramaximal transcription of the luciferase reporter (C). Hs578T-ERαLuc and Hs578T-ERβLuc cells were treated with 50 ng/mL Dox for 48 hr followed by treatment with DMSO (0.1%), 1 nM E2 or 1 μM cosmosiin for 4 or 24 hr. Firefly luciferase (FLuc) expression was determined by RT-PCR. RPL13A expression was used to ensure equal loading.
Cosmosiin, or apigenin 7-glucoside, is a flavone found in chamomile [20] that was identified as an ER agonist that selectively induces ERα/β and ERβ/β dimers as measured by bioluminescence resonance energy transfer (BRET) assays (unpublished data). It has a 3-fold higher binding affinity for ERβ as measured by competitive ligand binding assays (IC50 ERα 15.9 μM, IC50 ERβ 3.3 μM, unpublished data). Interestingly, cosmosiin induced luciferase expression to a much greater extent than E2, an effect described as supramaximal induction [21]. Even at concentrations up to 10 μM, cosmosiin did not saturate the luciferase output, and EC50 values could not be reasonably calculated (Fig. 5B). Another Hs578T-ERβLuc clone treated with cosmosiin also showed supramaximal induction (data not shown). Cosmosiin did not induce luciferase expression in Dox-treated cells co-treated with ICI 182,780 or cells not treated with Dox (data not shown), indicating the supramaximal induction was due to ERβ activation. To determine if the supramaximal induction truly represented enhanced transcriptional activation, the transcript levels of luciferase were assessed after 4 and 24 hr treatments of E2 and cosmosiin (Fig. 5C). Cosmosiin did not induce luciferase expression to a greater extent than E2 in either Hs578T-ERαLuc or Hs578T-ERβLuc cells, indicating alternative mechanisms are responsible for the supramaximal effect.
3.3 Selective regulation of ERα and ERβ target genes by ERβ selective ligands
We next sought to validate the subtype selectivity of DPN, PPT, liquiritigenin and cosmosiin by assessing regulation of endogenous ER target genes. Estrogen responsive target genes of ERα and ERβ were previously identified in Hs578T-ERα and Hs578T-ERβ cells [15], and two ERβ target genes and one ERα target gene were selected for analysis. Cells were treated with 50 ng/mL Dox for 48 hr to induce expression of the receptors and further treated with the corresponding ligands for 24 hr. Complement component 3 (C3, NM_000064) was up-regulated in Hs578T-ERβ cells upon E2 treatment (Fig. 6A). DPN and liquiritigenin were capable of inducing C3 expression to a comparable level as E2 at concentrations that fully activate ERβ with minimal ERα activation, as measured by reporter assays (Fig. 6A). Cosmosiin induced C3 expression at 1 μM, but not to the same extent as E2, demonstrating cosmosiin does not fully activate the receptor at this concentration. PPT slightly induced C3 expression compared to DMSO in Hs578T-ERβ cells, although PPT induced expression of C3 to a much lesser degree compared to E2. Repression of the ERβ target gene Jagged 1 (JAG1, NM_000214) occurred to a similar degree by E2, DPN, liquiritigenin and cosmosiin, although 100 nM liquiritigenin and 1 μM cosmosiin do not fully repress JAG1 expression compared to E2, DPN or 1 μM liquiritigenin (Fig. 6B). Although the ERα selective agonist PPT slightly induced C3 expression in Hs578T-ERβ cells, it had no effect on JAG1 repression, demonstrating incomplete ERβ activation by PPT. To further validate the subtype selectivity observed in reporter assays, expression of the ERα target gene alpha-6 integrin (ITGA6, NM_000210) was determined after treatment of Hs578T-ERα cells with E2, DPN, PPT, liquiritigenin and cosmosiin. As shown in Figure 6C, ITGA6 was up-regulated by E2 and PPT treatment, but DPN and liquiritigenin did not fully activate its expression at concentrations that showed selectivity in reporter assays (10 nM and 100 nM, respectively). At 1 μM, liquiritigenin and cosmosiin were capable of activating ERα, and ITGA6 expression was induced in Hs578T-ERα cells.
Figure 6.
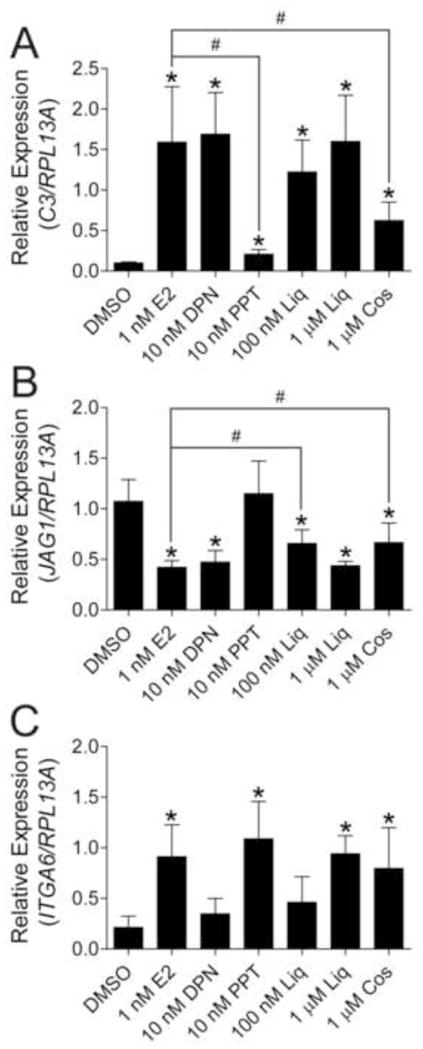
ERβ selective ligands selectively regulate ER target genes. Hs578T-ERα and Hs578T-ERβ cells were treated with 50 ng/mL Dox for 48 hr to induce ER expression followed by treatment with the corresponding ligands for 24 hr. Total RNA was assayed for expression of the ERβ target genes C3 and JAG1 in Hs578T-ERβ cells (A, B respectively) and the ERα target gene ITGA6 in Hs578T-ERα (C) cells by quantitative reverse-transcription polymerase chain reaction. Target gene expression was calculated using the ΔΔCq method by normalizing to the ribosomal protein RPL13A. Data represent the average and standard deviation of three biological replicates. * p values < 0.05 compared to DMSO control, # p values < 0.05 compared to E2 treatment
Therefore, the subtype selectivity of DPN and liquiritigenin observed in reporter cell lines was validated by subtype selective regulation of endogenous target genes. Cosmosiin, however, activated expression of an Hs578T-ERα endogenous gene target at concentrations that only slightly activated luciferase reporter expression in Hs578T-ERαLuc cells.
3.4 Growth inhibition of Hs578T-ERβ cells by liquiritigenin and cosmosiin
We next characterized the growth effects of liquiritigenin and cosmosiin in Hs578T-ERα and Hs578T-ERβ cells. It was previously shown that E2 inhibits the growth of Hs578T-ERβ cells [15], supporting the notion that the anti-proliferative action of ERβ may be activated by estrogenic ligands. We tested whether 100 nM liquiritigenin, a concentration at which ERβ was selectively activated, and 1 μM cosmosiin could also inhibit the growth of Hs578T-ERβ cells. Hs578T-ERα and Hs578T-ERβ cells were treated with vehicle (DMSO), 1 nM E2, 100 nM liquiritigenin or 1 μM cosmosiin in the presence or absence of 50 ng/mL Dox (with or without ER, respectively) for a total of 5 days. When ERα and ERβ were not expressed (- Dox), the compounds had no effect on the growth of the cells (Fig. 7A, B). In contrast, E2, liquiritigenin, and cosmosiin inhibited the growth of Hs578T-ERβ cells when ERβ was expressed (+ Dox, Fig. 7D), and there was an approximately 50% reduction in the number of cells after 5 days of treatment with all three compounds (Fig. 7F). Hs578T-ERα cells showed slight inhibition with E2 and liquiritigenin treatment when ERα was expressed (Fig. 7C), but there was not a statistically significant effect after 5 days of treatment as measured by 2 independent experiments (Fig. 7E). However, ERα expression in ER negative cells often leads to growth inhibition [22, 23], and it is likely that activation of ERα inhibits the growth of Hs578T-ERα cells. This suggests that 100 nM liquiritigenin partially activates ERα despite minimal regulation of ITGA6 at this concentration.
Figure 7.
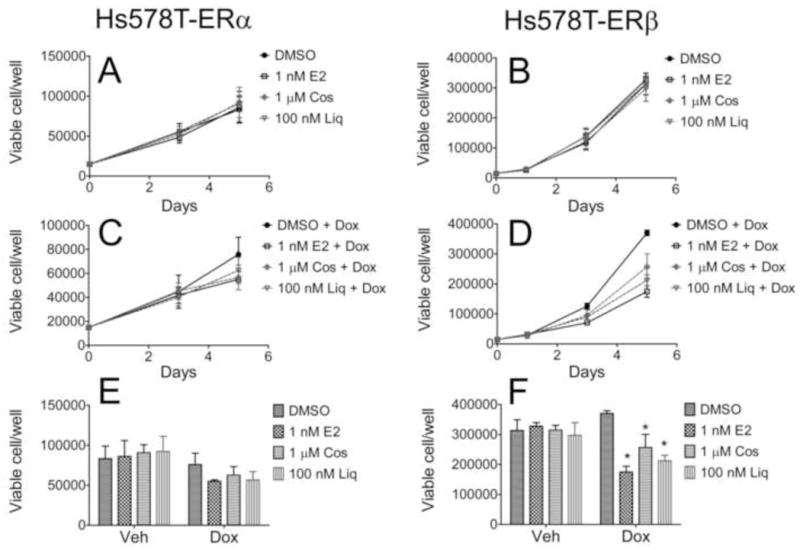
Cosmosiin (Cos) and liquiritigenin (Liq) inhibit the growth of Hs578T-ERβ cells. Hs578T-ERα (A, C, E) and Hs578T-ERβ cells (B, D, F) were seeded in 6 well plates and treated with vehicle (A, B) or 50 ng/mL Dox (C, D). After 24 hr, the cells were treated with vehicle (0.1% DMSO) or the indicated ligands, and treatments were refreshed every 48 hr. Cells were counted at the times indicated using trypan blue exclusion. Comparisons of the cell number on day 5 are represented in panels E (Hs578T-ERα) and F (Hs578T-ERβ). Data represent two independent experiments. * p values < 0.05
4. Discussion
ERα is an established therapeutic target for breast cancer treatment, but the development of subtype selective estrogenic ligands has gained interest with the identification of ERβ [1]. ERβ opposes the actions of ERα suggesting that it may be a potential therapeutic target. Exogenous ERβ expression in ERα positive breast cancer cells impaired E2 stimulated proliferation [24] and tumor growth in xenografts [25]. In support of the anti-proliferative role of ERβ, MCF7 cells were more proliferative when ERβ was knocked down [6]. Activation of ERβ by subtype selective ligands may enhance ERβ growth repression without stimulating proliferation through ERα; indeed ERβ selective ligands inhibited growth of HC11 mouse mammary cells [5]. Here, we have also shown that ERβ ligands can inhibit the growth of breast cancer cells when ERβ is expressed. In breast cancer, however, ERβ expression is thought to decline during progression [26–28] so ligands aimed at targeting ERβ must be highly selective and used only in patients that lack ERα or those with low ERα:ERβ ratios of expression. The rate of ERβ positivity in breast cancer has been reported to range from 13% to 83% [29–32]. In order to effectively target ERβ for cancer treatment, there is an imminent need to: a) identify ERβ selective ligands with minimal side effects and better in vivo efficacy and selectivity, and b) design clinical trials to recruit patients with low ERα:ERβ ratios in earlier stages of disease progression.
Although ERβ selective ligands have not yet been used for cancer treatment, the therapeutic value of ERβ has been assessed in other diseases. Two of the most promising ERβ selective therapies are the ERβ selective ligand ERB-041 and the herbal extract MF-101 [33]. Clinical trials have been completed to determine the efficacy of ERB-041 for treatment of Crohn’s disease, endometriosis, interstitial cystitis, and rheumatoid arthritis. Although results have not been published for most of the clinical trials, results of the rheumatoid arthritis trial showed ERB-041 was well tolerated but did not improve arthritis symptoms [34]. MF-101 also showed a relatively safe profile and reduced the frequency of hot flashes in a phase II clinical trial for treatment of post-menopausal symptoms [35]. Liquiritigenin is the most active estrogenic component of MF-101[11], suggesting ERβ selective ligands may prove useful for treating post-menopausal symptoms.
Strategies to identify ER subtype selective ligands include competitive ligand binding, dimerization, transcriptional reporter, and proliferation assays [21, 36]. Competitive ligand binding assays provide insight into binding affinities and are useful for high throughput small molecule screening [37], but they are limited because ligands can act as agonists or antagonists and binding affinity does not often reflect transcriptional potency. BRET assays to measure receptor dimerization have been used to identify subtype selective ligands [38], but also cannot differentiate between agonists or antagonists [39]. Agonists can be characterized using proliferation assays in MCF7 cells, which are highly sensitive and provide a biologically relevant endpoint in the context of estrogen-sensitive cells [40]. However, this assay is limited by a lack of specificity, as non-estrogenic mitogens can stimulate proliferation, and cannot be used to detect subtype selective agonists.
Transcriptional assays can differentiate between agonists and antagonists, overcoming limitations of binding and dimerization assays. Mammalian reporter cell lines useful for identifying subtype selective ligands have been created from HeLa cervical carcinoma cells [12] and HEK293 kidney cells [13]. HELN-ERα and HELN-ERβ were generated from HeLa cells in two steps: 1) stable integration of ERE-luciferase to generate HELN cells, 2) stable expression of ERα or ERβ to generate HELN-ERα and HELN-ERβ [12]. 293/hERα and 293/hERβ cells were generated by a similar strategy. Only one breast cancer reporter cell line, T47D-KBLuc, is available to characterize agonists in the context of breast cancer cells [14], but both ERα and ERβ are expressed, preventing identification of subtype selective ligands.
In this report, we described the development of a new set of breast cancer reporter cell lines to characterize subtype selective estrogenic ligands. Hs578T-ERαLuc and Hs578T-ERβLuc cells were highly sensitive to E2 with EC50 values of 1 pM and 6.5 pM, respectively (Fig. 4A). Similar E2 sensitivity was observed in T47D-KBLuc cells, which showed an approximate EC50 of 3 pM [14]. Hs578T-ERαLuc and Hs578T-ERβLuc cells were more sensitive to E2 than HELN-ER and 293/ER reporter cells, but all reporter cell lines showed greater E2 sensitivity in ERα expressing cells. HELN-ERα cells were approximately 3 times more sensitive to E2 than HELN-ERβ cells (EC50 of 0.017 nM and 0.068 nM, respectively) [12] and 293/hERα cells were approximately 4 times more sensitive to E2 than cells expressing ERβ (EC50 of 50 pM and 200 pM, respectively) [13]. Although Hs578T-ERαLuc and Hs578T-ERβLuc cells were not created using the same strategy as HELN-ER or 293/hER reporter cells and likely have unique genomic integration of the reporter, similar sensitivities observed in all reporter cell lines suggest that this does not inhibit comparison of subtype selectivity.
Reporter assays with two ER subtype selective ligands confirmed that Hs578T-ERαLuc and Hs578T-ERβLuc cells could be used to differentiate between ERα and ERβ selective ligands. The ERβ selective agonist DPN maintained 33-fold selectivity in Hs578T-ERLuc cells (EC50 of 0.26 nM for ERβ and 8.5 nM for ERα, Table 2). Dose response assays with the ERα selective agonist PPT revealed the sensitivity of Hs578T-ERβLuc cells (Fig. 4B). Although PPT was unable to activate reporter expression in HEC-1 cells transfected with ERβ [12], PPT did activate reporter expression in Hs578T-ERβLuc cells at high concentrations, although not to the full extent induced by E2. PPT reporter activation was blocked by ICI 182,780 co-treatment (Fig. 2A) and did not occur in the absence of Dox treatment (data not shown), verifying reporter activation was mediated by ERβ. Despite activation of ERβ at high concentrations, PPT could not fully activate reporter expression in Hs578T-ERβLuc cells and maintained 1000-fold selectivity for ERα.
Subtype selectivity of two natural phytoestrogens, cosmosiin and liquiritigenin, was also assessed in Hs578T-ERαLuc and Hs578T-ERβLuc cells. Liquiritigenin maintained selectivity for ERβ but to a lesser extent than expected, as it has been shown to minimally activate ERα in other cell lines [11]. The discrepancy in the selectivity of liquiritigenin may be due to the enhanced sensitivity of Hs578T-ERαLuc cells, differences in cofactor expression in Hs578T cells, or purity of the compound (our studies utilized commercially available liquiritigenin and Mersereau and coworkers [11] used extract from G. uralensis). The selectivity of cosmosiin could not be assessed using luciferase assays due to supramaximal induction (Fig. 5B). Supramaximal activation of estrogen responsive reporters have been described in many systems [21]. Here, we showed that supramaximal induction by cosmosiin was not due to enhanced transcriptional activation of the reporter (Fig. 5C). Despite limitations of the reporter system, the subtype selectivity of cosmosiin could be characterized by assessing target gene regulation in Hs578T-ERα and Hs578T-ERβ cells. While DPN and liquiritigenin maintained similar extents of selectivity as measured by reporter assays, cosmosiin activated both ERα and ERβ as measured by endogenous gene regulation (Fig. 6). Cosmosiin and liquiritigenin induced similar growth inhibitory effects as E2 in Hs578T-ERβ cells, indicating the phytoestrogens could elicit ERβ activation to a similar extent as E2 (Fig. 7).
Hs578T-ERαLuc and Hs578T-ERβLuc cells have several advantages for identifying ERβ selective agonists in comparison to available mammalian reporter cell lines. First, the Hs578T reporter cell lines have inducible expression of ERα and ERβ, allowing determination of off-target reporter activation by assessing reporter expression in the absence of Dox. Second, Hs578T-ERαLuc and Hs578T-ERβLuc cells are highly sensitive to estrogenic ligands. Third, endogenous gene regulation can be used to validate subtype selectivity. Finally, growth inhibition assays using Hs578T-ERβ cells in the presence and absence of Dox can be used to determine the biological endpoint of ERβ activation and validate specificity of ligands to ensure they do not have off-target cytotoxic effects. High throughput screening may be possible using Hs578T-ERαLuc and Hs578T-ERβLuc cells, and luciferase assay optimization using Hs578T-ERβLuc cells has shown a Z factor of 0.5 (data not shown), an acceptable range for high throughput screening [41]. Therefore, Hs578T-ERαLuc and Hs578T-ERβLuc cells are useful for the identification and characterization of ER subtype selective ligands that may hold therapeutic promise.
Acknowledgments
This work was supported by the National Institute of Environmental Health and Safety (Grant T32 ES007015), the National Institutes of Health (Grants R01CA125387, R03MH089442, CA125387), the Shaw Scientist Award from the Greater Milwaukee Foundation, the Department of Defense Breast Cancer Research Program (Grants BC100252, Era of Hope Scholar Award), and the UWCCC (Multi-IT Grant). We gratefully acknowledge Linda Schuler, Nancy Thompson, and Serife Ayaz-Guner for critical review of the manuscript.
Abbreviations
- BRET
bioluminescence resonance energy transfer
- Cos
cosmosiin
- Dox
doxycycline
- DPN
diarylpropionitrile
- E2
17β-estradiol
- ER
estrogen receptor
- PPT
propyl pyrazole triol
- ERE
estrogen response element
- ICI
ICI 182,780
- Liq
liquiritigenin
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Nilsson S, Gustafsson JA. Estrogen Receptors: Therapies Targeted to Receptor Subtypes. Clin Pharmacol Ther. 2011;89:44–55. doi: 10.1038/clpt.2010.226. [DOI] [PubMed] [Google Scholar]
- 2.Swaby R, Sharma C, Jordan V. SERMs for the treatment and prevention of breast cancer. Rev Endocr and Metab Disord. 2007;8:229–39. doi: 10.1007/s11154-007-9034-4. [DOI] [PubMed] [Google Scholar]
- 3.Hartman J, Ström A, Gustafsson JA. Estrogen receptor beta in breast cancer--Diagnostic and therapeutic implications. Steroids. 2009;74:635–41. doi: 10.1016/j.steroids.2009.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Chang EC, Frasor J, Komm B, Katzenellenbogen BS. Impact of estrogen receptor beta on gene networks regulated by estrogen receptor alpha in breast cancer cells. Endocrinology. 2006;147:4831–42. doi: 10.1210/en.2006-0563. [DOI] [PubMed] [Google Scholar]
- 5.Helguero LA, Faulds MH, Gustafsson JA, Haldosen LA. Estrogen receptors alpha (ERα) and beta (ERβ) differentially regulate proliferation and apoptosis of the normal murine mammary epithelial cell line HC11. Oncogene. 2005;24:6605–16. doi: 10.1038/sj.onc.1208807. [DOI] [PubMed] [Google Scholar]
- 6.Treeck O, Lattrich C, Springwald A, Ortmann O. Estrogen receptor beta exerts growth-inhibitory effects on human mammary epithelial cells. Breast Cancer Res Treat. 2010;120:557–65. doi: 10.1007/s10549-009-0413-2. [DOI] [PubMed] [Google Scholar]
- 7.Minutolo F, Macchia M, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor β ligands: Recent advances and biomedical applications. Med Res Rev. 2011;31:364–442. doi: 10.1002/med.20186. [DOI] [PubMed] [Google Scholar]
- 8.Charn TH, Liu ET-B, Chang EC, Lee YK, Katzenellenbogen JA, Katzenellenbogen BS. Genome-Wide Dynamics of Chromatin Binding of Estrogen Receptors {alpha} and {beta}: Mutual Restriction and Competitive Site Selection. Mol Endocrinol. 2010;24:47–59. doi: 10.1210/me.2009-0252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kuiper GG, Carlsson B, Grandien K, Enmark E, Haggblad J, Nilsson S, et al. Comparison of the ligand binding specificity and transcript tissue distribution of estrogen receptors alpha and beta. Endocrinology. 1997;138:863–70. doi: 10.1210/endo.138.3.4979. [DOI] [PubMed] [Google Scholar]
- 10.Stauffer SR, Coletta CJ, Tedesco R, Nishiguchi G, Carlson K, Sun J, et al. Pyrazole ligands: Structure-affinity/activity relationships and estrogen receptor-β-selective agonists. J Med Chem. 2000;43:4934–47. doi: 10.1021/jm000170m. [DOI] [PubMed] [Google Scholar]
- 11.Mersereau JE, Levy N, Staub RE, Baggett S, Zogric T, Chow S, et al. Liquiritigenin is a plant-derived highly selective estrogen receptor beta agonist. Mol Cell Endocrinol. 2008;283:49–57. doi: 10.1016/j.mce.2007.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Escande A, Pillon A, Servant N, Cravedi J-P, Larrea F, Muhn P, et al. Evaluation of ligand selectivity using reporter cell lines stably expressing estrogen receptor alpha or beta. Biochem Pharmacol. 2006;71:1459. doi: 10.1016/j.bcp.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 13.Barkhem T, Carlsson B, Nilsson Y, Enmark E, Gustafsson J-Å, Nilsson S. Differential Response of Estrogen Receptor α and Estrogen Receptor β to Partial Estrogen Agonists/Antagonists. Mol Pharmacol. 1998;54:105–12. doi: 10.1124/mol.54.1.105. [DOI] [PubMed] [Google Scholar]
- 14.Wilson VS, Bobseine K, Gray LE., Jr Development and characterization of a cell line that stably expresses an estrogen-responsive luciferase reporter for the detection of estrogen receptor agonist and antagonists. Toxicol Sci. 2004;81:69–77. doi: 10.1093/toxsci/kfh180. [DOI] [PubMed] [Google Scholar]
- 15.Secreto FJ, Monroe DG, Dutta S, Ingle JN, Spelsberg TC. Estrogen receptor alpha/beta isoforms, but not betacx, modulate unique patterns of gene expression and cell proliferation in Hs578T cells. J Cell Biochem. 2007;101:1125–47. doi: 10.1002/jcb.21205. [DOI] [PubMed] [Google Scholar]
- 16.Kao J, Salari K, Bocanegra M, Choi YL, Girard L, Gandhi J, et al. Molecular profiling of breast cancer cell lines defines relevant tumor models and provides a resource for cancer gene discovery. PLoS One. 2009;4:e6146. doi: 10.1371/journal.pone.0006146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brünner N, Boulay V, Fojo A, Freter CE, Lippman ME, Clarke R. Acquisition of hormone-independent growth in MCF-7 cells is accompanied by increased expression of estrogen-regulated genes but without detectable DNA amplifications. Cancer Res. 1993;53:283–90. [PubMed] [Google Scholar]
- 18.Cowley SM, Parker MG. A comparison of transcriptional activation by ERα and ERβ. J Steroid Biochem Mol Biol. 1999;69:165–75. doi: 10.1016/s0960-0760(99)00055-2. [DOI] [PubMed] [Google Scholar]
- 19.Meyers MJ, Sun J, Carlson KE, Marriner GA, Katzenellenbogen BS, Katzenellenbogen JA. Estrogen receptor-β potency-selective ligands: Structure-activity relationship studies of diarylpropionitriles and their acetylene and polar analogues. J Med Chem. 2001;44:4230–51. doi: 10.1021/jm010254a. [DOI] [PubMed] [Google Scholar]
- 20.Srivastava JK, Gupta S. Antiproliferative and apoptotic effects of chamomile extract in various human cancer cells. J Agric Food Chem. 2007;55:9470–8. doi: 10.1021/jf071953k. [DOI] [PubMed] [Google Scholar]
- 21.Montaño M, Bakker EJ, Murk AJ. Meta-analysis of Supramaximal Effects in In Vitro Estrogenicity Assays. Toxicol Sci. 2010;115:462–74. doi: 10.1093/toxsci/kfq056. [DOI] [PubMed] [Google Scholar]
- 22.Garcia M, Derocq D, Freiss G, Rochefort H. Activation of estrogen receptor transfected into a receptor-negative breast cancer cell line decreases the metastatic and invasive potential of the cells. Proc Natl Acad Sci U S A. 1992;89:11538–42. doi: 10.1073/pnas.89.23.11538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang W, Smith R, Burghardt R, Safe SH. 17 beta-Estradiol-mediated growth inhibition of MDA-MB-468 cells stably transfected with the estrogen receptor: cell cycle effects. Mol Cell Endocrinol. 1997;133:49–62. doi: 10.1016/s0303-7207(97)00142-1. [DOI] [PubMed] [Google Scholar]
- 24.Hodges-Gallagher L, Valentine C, Bader S, Kushner P. Estrogen receptor beta increases the efficacy of antiestrogens by effects on apoptosis and cell cycling in breast cancer cells. Breast Cancer Res Treat. 2008;109:241–50. doi: 10.1007/s10549-007-9640-6. [DOI] [PubMed] [Google Scholar]
- 25.Ström A, Hartman J, Foster JS, Kietz S, Wimalasena J, Gustafsson JA. Estrogen receptor β inhibits 17β-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc Natl Acad Sci U S A. 2004;101:1566–71. doi: 10.1073/pnas.0308319100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bardin A, Boulle N, Lazennec G, Vignon F, Pujol P. Loss of ER{beta} expression as a common step in estrogen-dependent tumor progression. Endocr Relat Cancer. 2004;11:537–51. doi: 10.1677/erc.1.00800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shaw JA, Udokang K, Mosquera JM, Chauhan H, Jones JL, Walker RA. Oestrogen receptors alpha and beta differ in normal human breast and breast carcinomas. J Pathol. 2002;198:450–7. doi: 10.1002/path.1230. [DOI] [PubMed] [Google Scholar]
- 28.Skliris GP, Munot K, Bell SM, Carder PJ, Lane S, Horgan K, et al. Reduced expression of oestrogen receptor beta in invasive breast cancer and its re-expression using DNA methyl transferase inhibitors in a cell line model. J Pathol. 2003;201:213–20. doi: 10.1002/path.1436. [DOI] [PubMed] [Google Scholar]
- 29.Marotti JD, Collins LC, Hu R, Tamimi RM. Estrogen receptor-[beta] expression in invasive breast cancer in relation to molecular phenotype: results from the Nurses’ Health Study. Mod Pathol. 2009;23:197. doi: 10.1038/modpathol.2009.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Honma N, Horii R, Iwase T, Saji S, Younes M, Takubo K, et al. Clinical importance of estrogen receptor-beta evaluation in breast cancer patients treated with adjuvant tamoxifen therapy. J Clin Oncol. 2008;26:3727–34. doi: 10.1200/JCO.2007.14.2968. [DOI] [PubMed] [Google Scholar]
- 31.O’Neill PA, Davies MP, Shaaban AM, Innes H, Torevell A, Sibson DR, et al. Wild-type oestrogen receptor beta (ERbeta1) mRNA and protein expression in Tamoxifen-treated post-menopausal breast cancers. Br J Cancer. 2004;91:1694–702. doi: 10.1038/sj.bjc.6602183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mann S, Laucirica R, Carlson N, Younes PS, Ali N, Younes A, et al. Estrogen receptor beta expression in invasive breast cancer. Hum Pathol. 2001;32:113–8. doi: 10.1053/hupa.2001.21506. [DOI] [PubMed] [Google Scholar]
- 33.Mohler ML, Narayanan R, Coss CC, Hu K, He Y, Wu Z, et al. Estrogen receptor β selective nonsteroidal estrogens: seeking clinical indications. Expert Opin Ther Pat. 2010;20:507–34. doi: 10.1517/13543771003657164. [DOI] [PubMed] [Google Scholar]
- 34.Roman-Blas JA, Castañeda S, Cutolo M, Herrero-Beaumont G. Efficacy and safety of a selective estrogen receptor β agonist, ERB-041, in patients with rheumatoid arthritis: a 12-week, randomized, placebo-controlled, phase II study. Arthritis Care Res (Hoboken) 2010;62:1588–93. doi: 10.1002/acr.20275. [DOI] [PubMed] [Google Scholar]
- 35.Grady D, Sawaya GF, Johnson KC, Koltun W, Hess R, Vittinghoff E, et al. MF101, a selective estrogen receptor beta modulator for the treatment of menopausal hot flushes: a phase II clinical trial. Menopause. 2009;16:458–65. doi: 10.1097/gme.0b013e31818e64dd. [DOI] [PubMed] [Google Scholar]
- 36.Shanle EK, Xu W. Endocrine Disrupting Chemicals Targeting Estrogen Receptor Signaling: Identification and Mechanisms of Action. Chem Res Toxicol. 2010;24:6–19. doi: 10.1021/tx100231n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Bolger R, Wiese TE, Ervin K, Nestich S, Checovich W. Rapid screening of environmental chemicals for estrogen receptor binding capacity. Environ Health Perspect. 1998;106:551–7. doi: 10.1289/ehp.98106551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Powell E, Huang SX, Xu Y, Rajski SR, Wang Y, Peters N, et al. Identification and characterization of a novel estrogenic ligand actinopolymorphol A. Biochem Pharmacol. 2010;80:1221–9. doi: 10.1016/j.bcp.2010.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Powell E, Xu W. Intermolecular interactions identify ligand-selective activity of estrogen receptor a/b dimers. Proc Natl Acad Sci U S A. 2008;105:19012–7. doi: 10.1073/pnas.0807274105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Silva E, Scholze M, Kortenkamp A. Activity of xenoestrogens at nanomolar concentrations in the E-Screen assay. Environmental Health Perspectives. 2007;115:91–7. doi: 10.1289/ehp.9363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang JH, Chung TD, Oldenburg KR. A Simple Statistical Parameter for Use in Evaluation and Validation of High Throughput Screening Assays. J Biomol Screen. 1999;4:67–73. doi: 10.1177/108705719900400206. [DOI] [PubMed] [Google Scholar]