

Abstract
siRNA-aptamer chimera is emerging as a highly promising approach for cell-type specific delivery of siRNA due to the outstanding targeting capability of aptamer and the compatibility of chimera with native ribonuclease (Dicer) processing. For efficient RNA interference (RNAi), however, additional challenges must be addressed, in particular how to get siRNA out of endosome after cell entry and how to preserve aptamer targeting specificity when chimeras are combined with delivery carriers. Here, we report a rationally designed nanoparticle vector that simultaneously displays large surface area for high siRNA payload, exposed aptamer for specific targeting, proton sponge effect for endosome escape, and fluorescence for imaging and quantification. A key concept of this work is to graft chimeras onto nanoparticle surface via a two-step process: first immobilizing siRNA onto nanoparticle via non-covalent interactions to facilitate intracellular unpackaging and reduce nanoparticle surface charge (avoiding non-specific electrostatic interactions between aptamers and nanoparticles), and then coupling siRNA and aptamer with retained conformation and high accessibility. Compared with conventional one-step adsorption of siRNA-aptamer chimeras onto nanoparticles with random orientations and conformations, which does not elicit much improved RNAi effect than non-targeted nanoparticle-siRNA complexes (~6-8% improvement of the total cell population), under the same RNA concentration our approach shows selective gene silencing and enables 34% more silenced cells of the total cell population over non-targeted nanoparticle-siRNA complexes. This remarkable difference in RNAi efficiency using nanoparticle-chimera complexes is directly related to cell uptake discrepancy resulted from aptamer conformation on nanoparticle surface (intact vs. random).
Keywords: Quantum dot, nanoparticle, siRNA, aptamer, chimera, gene silencing, targeting
siRNA is of considerable current interest in biology and medicine because it can elicit potent, target-specific knockdown of virtually any mRNA, creating new opportunities for functional genomics studies and medical treatment of tough diseases.1-3 Similar to other antisense approaches, applying siRNA technology in vivo, however, still faces major challenges, particularly the technical hurdles in selective delivery.4 To guide siRNA to diseased cells, targeting ligands such as small molecules, lipids, peptides, and proteins have been linked directly to siRNA or on the surface of siRNA carriers.5-10 Most recently, siRNA-aptamer chimera, employing only RNA molecules, has emerged as a promising approach for efficient delivery of siRNA to specific cell types, owing to its low immunogenicity, ease of chemical synthesis and modification, and the outstanding targeting specificity of aptamer. For example, chimeras composed of aptamer targeting prostate specific membrane antigen PSMA and siRNA targeting anti-apoptotic genes have been identified and optimized.11, 12 The aptamer block recognizes PSMA on prostate tumor cell surface and leads to chimera cell internalization, whereas the siRNA block enzymatically cleaved from the chimera causes cell death. Significantly reduced tumor mass was observed in mouse xenograft models of prostate tumor after administration of the chimera, though the exact mechanism of how chimera escape endosome after cell entry remains unclear.13 This limitation may help explain the fact that high concentrations of chimera were used in these pioneer studies to treat prostate tumors.11, 12
A solution to this problem and potential improvement of the chimera technology is to combine chimeras with nanocarriers with endosome rupturing capabilities. Recent advances in bionanotechnology and molecular engineering have produced a number of siRNA delivery strategies based on liposome, cationic polymers, and most recently inorganic nanoparticles such as gold, silica, magnetic, and semiconductor nanoparticles.14-18 In addition to the endosome rupture capability, inorganic nanoparticles often provide imaging functionalities as well due to their unique electronic, optical, and magnetic properties, which enable real-time imaging of siRNA delivery, distribution, and intracellular behavior.19 For example, we have previously reported the combination of quantum dots (QDs) with zwitterionic amphiphilic polymers for efficient in vitro siRNA delivery.20, 21 The intrinsic fluorescence of QDs and exquisitely designed energy transfer assays allow real-time observation of siRNA cell entry, endosome escape, and separation with the nanoparticle carrier.20 A common feature shared by these existing nanocarriers is that they are positively charged, which is essential for siRNA immobilization, condensation, and escape from endosome and lysosome. On the other hand, this high positive charge also creates a problem for targeted delivery of siRNA-aptamer chimeras. Since both the siRNA and aptamer blocks are negatively charged RNA molecules, they will non-specifically ‘collapse’ onto cationic materials and thus affect aptamer conformation and selective binding (Fig. 1a). Indeed, using planar microarray chips, Walter et al. have shown that aptamers interact with positively charged surface electrostatically, resulting in unfolding of aptamers and loss of binding activity.22
Figure 1.
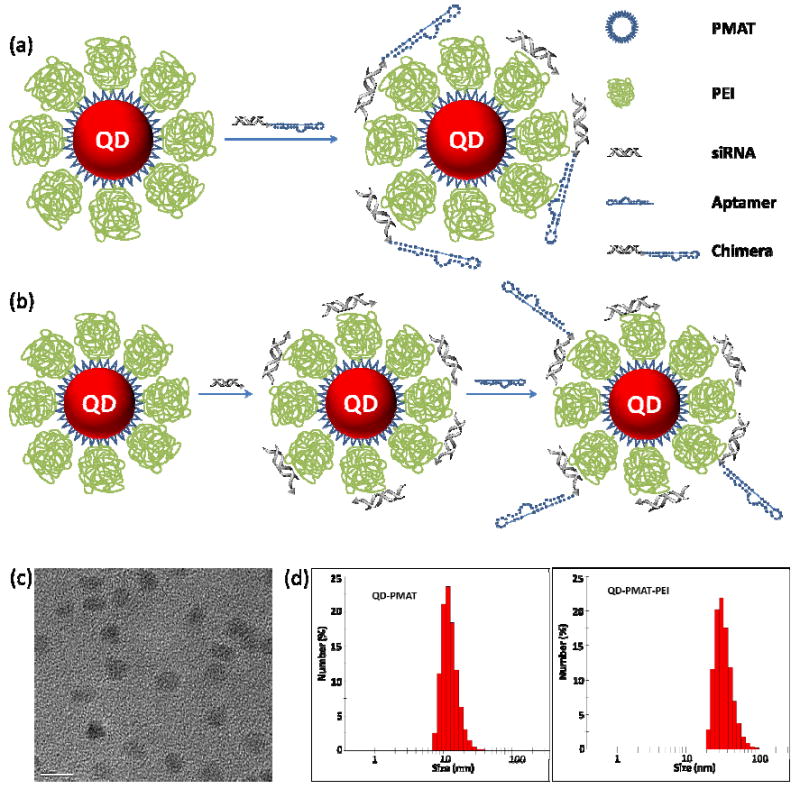
Schematic representation of cationic nanoparticles for targeted delivery of siRNA-aptamer chimera. (a) Immobilization of preformed siRNA-aptamer chimera onto positively charged QD-PMAT-PEI nanoparticles. The aptamer block collapsed on the carrier results in reduced binding activity. (b) Two-step immobilization of chimera on cationic nanoparticle surface. siRNA molecules with a thiol-reactive terminal group are first adsorbed on QD-PMAT-PEI surface to reduce the positive charge; subsequently aptamers with a single thiol group are brought in to form the siRNA-aptamer chimera on the nanoparticle surface. (c) TEM image of QD-PMAT-PEI nanoparticles (scale bar 10 nm). (d) Hydrodynamic diameter of QD-PMAT (12.1 ± 0.7 nm) and QD-PMAT-PEI (32.2 ± 4.5 nm) nanoparticles.
Thus, we hypothesize that the control of aptamer orientation and 3D conformation on the surface of nanoparticles is critical in preserving its targeting functionality (at least for some aptamer sequences). Ideally, the siRNA block should become the anchor point for interaction with cationic nanoparticles for reduced enzymatic degradation and non-specific interaction with cells and tissues, whereas the aptamer block should stay on the outside with minimized interaction with nanoparticle surface. Towards this goal, here we present a two-step approach to link siRNA-aptamer chimeras onto nanoparticles with endosome-rupturing capability. In contrast to single-step adsorption of pre-formed chimeras onto nanoparticles (Fig. 1a), siRNA molecules (targeting eGFP) with a thiol-reactive terminal group are first adsorbed onto polyethylene imine (PEI)-coated nanoparticles electrostatically (Fig. 1b). This non-covalent interaction not only facilitates siRNA detaching from the nanocarrier inside cells but also helps neutralize some of the positive charges on nanoparticle surface. In the second step, aptamers (targeting PSMA) with a single thiol group away from the binding site are brought in to form siRNA-aptamer chimeras. Due to the reduced positive charge on nanoparticles, their interaction with aptamers is weakened, thus help retain aptamer flexibility, accessibility, and original binding activity. Note that QD is selected as a model system because of its excellent photoluminiscent properties for real-time imaging. We expect the design principles learned from these studies can be generalized because other types of nanoparticles can be readily made with similar surface chemistry.
RESULTS AND DISCUSSION
Water-soluble QDs are prepared by molecular self-assembly of hydrophobic QDs with amphiphilic copolymers (poly(maleic anhydride-alt-1-tetradecene), or PMAT).23 The resulting QDs are highly stable in aqueous solution (due to the high-density carboxylic acid groups of PMAT) with well-preserved optical properties. To capture siRNA and facilitate its escape from endosome, PEI is conjugated to water-soluble QDs due to PEI’s pH buffering capability, also known as the proton sponge effect.24 In contrast to prior reports on formation of QD-PEI cluster of approximately 200 nm by mixing PEI with carbodiimide compounds first,25 our approach by adding carbodiimide-activated QDs to PEI yields single non-aggregated QDs (TEM images in Fig. 1c). Dynamic light scattering measurements of purified QD-PEI particles shows an average hydrodynamic diameter of 32.2 ± 4.5 nm (Fig. 1d), which is ~10 nm greater than QD-PMAT (12.1 ± 0.7 nm) on both sides, indicating successful conjugation of PEI to QDs. To further confirm this result, we characterized the surface charge before and after PEI conjugation. The zeta potential of QDs reverses from negative 43.9 ± 5.3 mV to positive 27.0 ± 4.5 mV, confirming a positively charged PEI layer on QD surface.
To investigate the number of siRNA molecules that can be loaded on QDs, siRNA at a fixed amount is mixed with QDs at various molar ratios and probed with gel electrophoresis. As shown in Fig. 2a, when the molar ratio of siRNA/QD is below 50, no free siRNA band is detected after SYBR® gold staining. Further increase of the molar ratio to 100 results in a smeared stain of siRNA on the gel. These results indicate that at least 50 copies of siRNA can be stably condensed with QD-PEI complexes, a loading capacity that is ~5 times higher than that of the QD-zwitterionic amphiphilic polymer complexes we reported previously.20 This improvement is likely due to PEI’s higher density of positive charges and thicker coating on QD surface compared to the monolayer of amphiphilic polymers on QD surface.20 In general, high siRNA payload is desirable. For rational design of efficient siRNA delivery vehicles, however, additional parameters beyond the siRNA loading capacity must also be taken into account, such as the overall size of the final assembly that affects nanoparticle behaviors both in vitro and in vivo. For example, it is well documented that in plasma circulation, nanoparticle extravasation into tumors, clearance by kidney and the reticuloendothelial systems (RES), and transfer to the lymphatic systems are all directly correlated with particle sizes.26 Most recently, Jiang et al reported the size effect on receptor-mediated endocytosis and suggested that 40-60 nm is the optimal size range for enhanced uptake.27 As shown in Fig. 2b, quantitative DLS measurements reveal that QD-siRNA complexes with siRNA/QD ratio below 25 are close to this optimal size range for receptor mediated endocytosis. In contrast, above siRNA/QD ratio of 30, the particle size increases rapidly to hundreds of nanometers, indicating formation of large aggregates. When siRNA is in large excess, the particle size drops back down, a typical technique used in charge-mediated layer-by-layer self-assembly to avoid aggregation.16 Following size characterization, we evaluated QD-siRNA complexes in the optimal size range against the next design criterion, nanoparticle surface charge. As aforementioned, we hypothesize that the initial high positive charge of QD-PEI should be significantly reduced before addition of aptamers. Indeed, the positive charge of QD-PEI gradually decreases as the siRNA/QD ratio increases. The zeta potential drops to 19.6 ± 4.5 mV at ratio 10, and to 7.7 ± 6.3 mV at ratio 20, which is significantly lower than the original 27.0 mV of QD-PEI (Fig. 2b insert). We thus proceeded with the siRNA/QD ratio of 20 for the following experiments since this reduced positive charge should minimize interaction between QD-PEI and aptamer and help retain aptamer’s activity.
Figure 2.
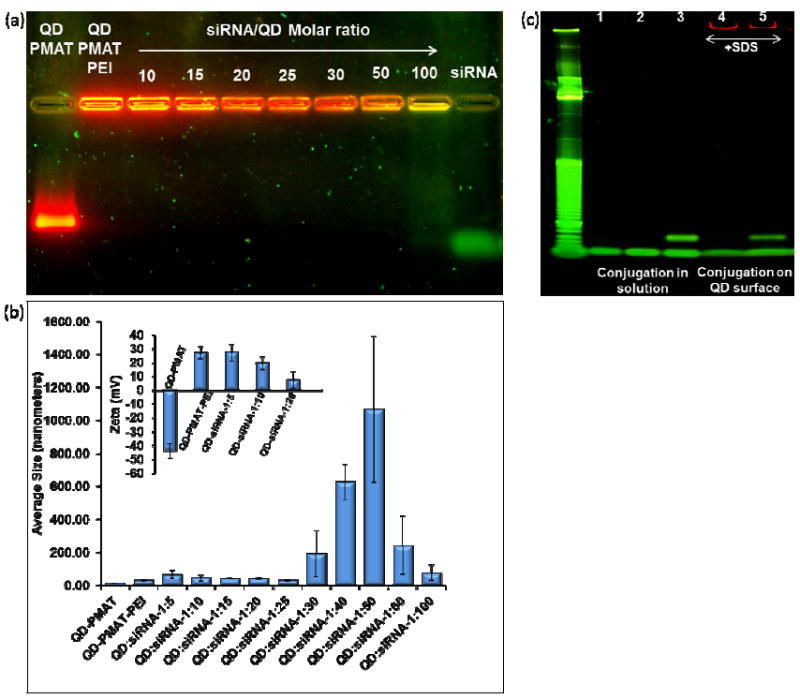
Characterization of the two-step QD-chimera formation process. (a) Determination of siRNA loading capacity on QD-PMAT-PEI surface. 20 pmole of siRNA is mixed with QDs at various molar ratios (10, 15, 20, 25, 30, 50, and 100). Free siRNA are undetectable up to 1:50 siRNA:QD molar ratio, however at 1:100 ratio the green flourescence of SYBR gold-stained siRNA can be seen indicating that at least 50 siRNA molecules can bind onto each QD. (b & insert) Hydrodynamic size and zeta potential measured by DLS of QD-PMAT-PEI nanoparticles before and after siRNA loading. The particle size of the complexes increases with increasing siRNA/QD molar ratio 5, 10, 15, 20, 25, 30, 40, 50, 60 and 100 as follows 66.3 ± 23.8 nm, 46.0 ± 18.9 nm, 42.4 ± 4.29 nm, 41.6 ± 5.0 nm, 31.4 ± 3.9 nm, 193.1 ± 141.3 nm, 625.7 ± 106.4 nm, 1067.8 ± 443.0 nm, 243 ± 177.5 nm, 76.5 ± 47.8 nm respectively. The zeta potential values for QD-PMAT and QD-PMAT-PEI are -43.9 ± 5.3 mV and 27.0 ± 4.5 mV. After siRNA binding at siRNA/QD molar ratio 5, 10, and 20, the values become 27.4 ± 6.1 mV, 19.6 ± 4.5 mV, and 7.7 ± 6.3 mV, respectively. The average values and error bars are calculated based on three runs. (c) Polyacrylamide gel (10%) characterization of thiol-aptamer conjugation with siRNA-SPDP after complexation on QD surface. Lane 1-5 are siRNA, aptamer, siRNA-aptamer chimera formed in solution, QD-siRNA with SDS, and chimera formed on QD surface with SDS, respectively. The DNA ladder on the left is a 10bp ladder (Track it Orange, Invitrogen). The yield of the siRNA –aptamer conjugation reaction does not change with or without QDs.
For targeted cell specific delivery, a 41-base RNA aptamer targeting PSMA is coupled to siRNA on nanoparticle surface, via the well-established thiol-disulfide exchange reaction.28 Previous research has identified PSMA as one of the most attractive cell surface markers for both prostate epithelial cells and neovascular endothelial cells.29 Accumulation and retention of PSMA targeting probes at the site of tumor growth is the basis of radioimmunoscintigraphic scanning (e.g., ProstaScint scan) and targeted therapy for human prostate cancer metastasis. By combining QD-antibody bioconjugate targeting PSMA and hyperspectral imaging, we have previously achieved molecular imaging of prostate tumors in mouse models.30 In order to overcome the shortcomings of antibody-based targeting such as immunogenicity, Coffey et al have identified two highly specific aptamer sequences for PSMA targeting,31 both of which have been proven to be effective in a variety of applications including formation of siRNA-aptamer chimeras for targeted prostate tumor treatment.11, 12 To conjugate PSMA aptamer onto siRNA molecules, RNA aptamer molecules with a terminal thiol group is added to (N - Succinimidyl 3-(2-pyridyldithio)-propionate (SPDP) modified siRNA on the surface of QDs. The starting molar ratio of QD/siRNA/aptamer in the reaction is kept at 1/20/10. The resulting siRNA-aptamer chimera is analyzed with polyacrylamide gel electrophoresis (PAGE) after detaching from QD surface due to treatment with sodium dodecyl sulfate (SDS). The 21bp double stranded siRNA and the 41-base RNA aptamer share similar mobility (Fig. 2c, lane 1&2). Compared with the control experiment where free siRNA and aptamer are conjugated (Fig. 2c, lane 3), the reaction on the surface of QD-PEI shows virtually the same yield (~71%, 7 out of 10 copies of aptamer reacted with siRNA) based on the approximate percentage of the chimera to free siRNA and aptamer (Fig. 2c, lane 5). Note that in lane 3 and 5, the band with lower mobility is siRNA-aptamer chimera, and the band with higher mobility is composed of a mixture of unreacted siRNA and aptamer. Therefore, the final assembly of QD-chimera on average should have a single QD, and ~20 copies of siRNA molecules with ~7 of them formed chimeras with aptamer, as schematically plotted in Fig. 1b. Further characterization of the fully assembled QD-chimera reveals a DLS size of 66.3 ± 2.2 nm and approximately neutral surface charge (-1.8 ± 0.6 mV).
To address the central hypothesis of this work, the functionality and targeting capability of the aptamer decorated on QD surface, we evaluated the cell uptake efficiency of QDs directed by the PSMA aptamer. For direct comparison of our two-step bioconjugation approach with the conventional methods where aptamers are linked to highly positively charged nanoparticles with random conformation and orientation, two key control groups are included in this study: (1) siRNA-SPDP complexed with QDs followed by addition of aptamers without thiol modification (Random 1); and (2) preformed siRNA-aptamer chimera mixed with QDs in a single step (Random 2). After 6 h incubation with PSMA-positive C4-2B cells, fluorescence microscopy reveals that compared to QD-siRNA without aptamer, random 1, and random 2 (Fig. 3b-d), the QD-chimera complexes prepared in the two-step process (Fig. 3e) have significantly improved cell uptake as a result of aptamer binding to PSMA antigen on the plasma membrane. This improved cell uptake indeed demonstrates that the internalization of the nanoparticle-chimera is affected by the molecular orientation and conformation of the PSMA aptamer.
Figure 3.
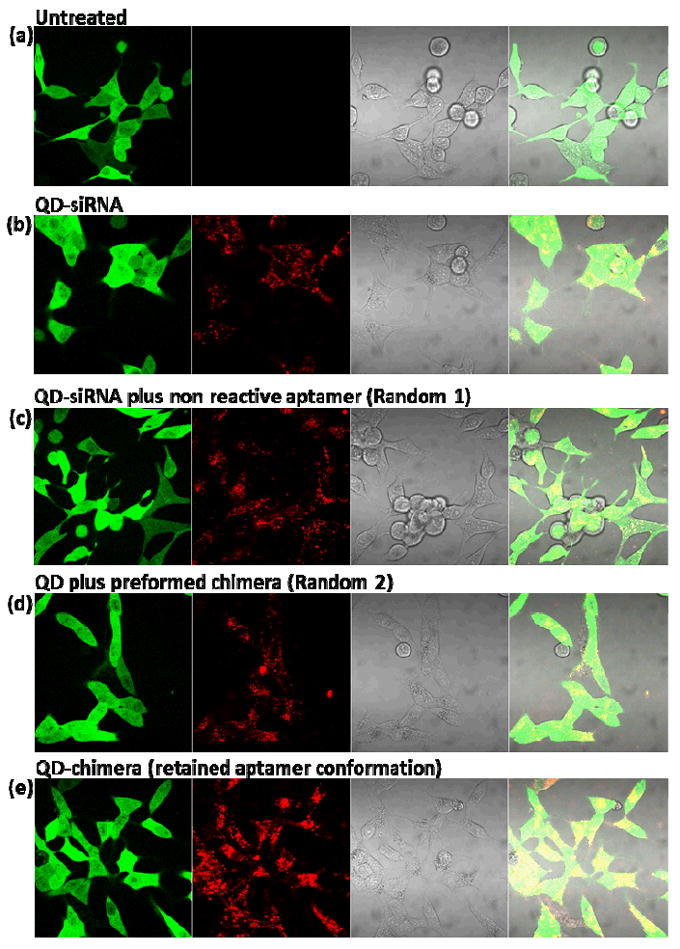
Intracellular uptake of QD-siRNA-aptamer in PSMA-positive eGFP-expressing C4-2B cells after 6h incubation. (a) Untreated C4-2B cells, (b-d) cell uptake of QD-siRNA without aptamer, QD-siRNA with non-reactive aptamer (Random 1), and QD with preformed chimera (Random 2), and (e) QD-chimera formed via the two-step process. The uptake of QD-chimera with intact aptamer conformation is remarkably higher than that in b-d. The red dot outside cells in panel d is likely a QD aggregate.
Next, using eGFP-expressing C4-2B cells as a model, we evaluated the gene silencing effect of this new class of nanoparticle-chimera complex with confocal microscopy and quantitative flow cytometry. Fig. 4a-e shows confocal images of the C4-2B cells treated with QD-siRNA without aptamer, random 1, random 2, and the QD-chimera. Qualitatively, it appears that the high uptake efficiency of QD-chimera complexes is directly correlated with efficient gene silencing (Fig. 4e), whereas QD-siRNA without aptamer (Fig. 4b) or with aptamer in unfavorable conformation (Fig. 4c&d) do not lead to significant suppression of GFP expression. The gene silencing efficiency and nanoparticle cell uptake are further quantitatively assessed by fluorescence activated cell sorting (FACS), where cells are evaluated with respect to the untreated eGFP-expressing cells (Fig. 4g). At the current gate value set with the GFP fluorescence intensity, the original untreated cells showed an eGFP-positive population of 82.7%. QD-siRNA without aptamer vs. QD-siRNA-aptamer complexes with random aptamer conformation show insignificant difference (6-8%) in gene silencing (GFP-positive cells 63.3% vs. 55.9%, and 54.7%, respectively, Fig. 4h-j). Remarkably, the GFP-positive cell group is reduced to 29.7% by the QD-chimera complexes prepared with the two-step procedure, which represents a ~34% drop compared to the non-targeted case (Fig. 4k). Taken together, these results clearly show the importance of aptamer conformation for efficiently delivery of siRNA-aptamer chimera. Positive control using cationic liposome (Lipofectamine™) to deliver preformed siRNA-aptamer chimera also shows excellent gene silencing efficiency (16% eGFP-positive cells) (Fig. 4l), confirming that the chemically conjugated siRNA-aptamer chimera is functional, similar to the chimera structures obtained with in vitro transcription.11, 12 It is also noteworthy that using our rationally designed nanoparticle vectors or Lipofectamine for chimera delivery, efficient gene expression suppression can be achieved at much lower chimera concentration than using chimera alone.11, 12
Figure 4.
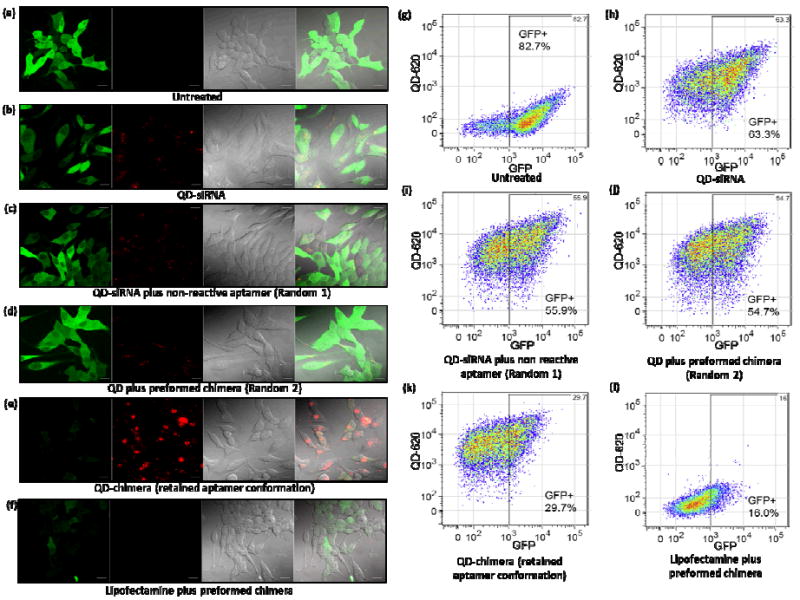
Evaluation of eGFP gene silencing in C4-2B cells with confocal microscopy and flow cytometry. (a) Untreated C4-2B cells only, (b) cells treated with QD-siRNA without aptamer, (c) cells treated with QD-siRNA and aptamer without terminal thiol group (random 1), (d) cells treated with QD and preformed chimera (random 2), (e) cells treated with QD-chimera with retained aptamer conformation, and (f) cells treated with Lipofectamine-chimera. Fluorescence imaging shows significant GFP reduction (green) and QD fluorescence (red) in (e). (g-k) Quantitative flow cytometry of eGFP silencing corresponding to the above fluorescence imaging studies. At the current gate value, the eGFP-positive cells are 82.7%, 63.3%, 55.9%, 54.7%, and 29.7%, respectively, revealing enhanced silencing is only observed when aptamer is in its native conformation. The flow cytometry study also confirms the microscopy result on enhanced uptake of QD-chimera with intact conformation. Compared to cells treated with QD-siRNA without targeting aptamer (h), cells in random 1 (i), random 2 (j), and QD-chimera (k) experiments on average uptake approximately 1.4, 1.4, and 2 times more particles revealed by the QD fluorescence channel. (l) Positive control using Lipofectamine confirms the activity of the chimera.
A remaining important issue is the cytotoxicity of the QD-PEI carrier, since the cationic polymer PEI is known for its high transfection efficiency but at the same time high toxicity. In our study, branched PEI (25 kD) is chemically conjugated to QD-PMAT. To ensure a fair toxicity comparison of free PEI and PEI on nanoparticle surface (since toxicity is concentration dependent), we first quantified the amount of PEI on nanoparticle surface using the O-pthalaldehyde (OPA), a sensitive assay for analysis of primary amines (OPA fluorescence wavelength is far away from that of QDs).32, 33 Based on this assay, we found that on average approximately 8 PEI molecules are coated on the surface of individual QDs. Cell viability is evaluated by cell titer-blue assay after 24 h incubation period. In contrast to free PEI that shows fast increasing toxicity above concentration of 10 μg/mL (0.4 μM) in C4-2B cells (Fig. 5a), QD-PMAT and QD-PMAT-PEI are generally non-toxic to cells in the concentration range probed (QD concentration 0-150 nM, for QD-PMAT-PEI the PEI concentration 0-1.2 μM) (Fig. 5b). This improved biocompatibility of surface bound PEI has been previously observed on silica and poly(methyl methacrylate) nanoparticle as well,34, 35 although the exact mechanism is largely unknown at this time and deserves systematic investigation in the future. Since the imaging and silencing experiments described above are performed at a QD concentration of 4 nM in the presence of siRNA (siRNA/QD molar ratio 20:1), we further probed the cytotoxicity of QD-PEI in the presence of siRNA at two QD concentration, 4 nM and a slightly elevated concentration of 10 nM (Fig. 5c). Regardless of the aptamer orientation, all the complexes at both concentrations do not result in significant toxicity with greater than 95% cell viability. The positive control Lipofectamine-RNA complexes used at the manufacturer-recommended quantity show high cell viability as well. These results indicate that the QD-chimera conjugates are non-toxic, which is perhaps not too surprising since GFP is a non-essential gene for cell survival.
Figure 5.
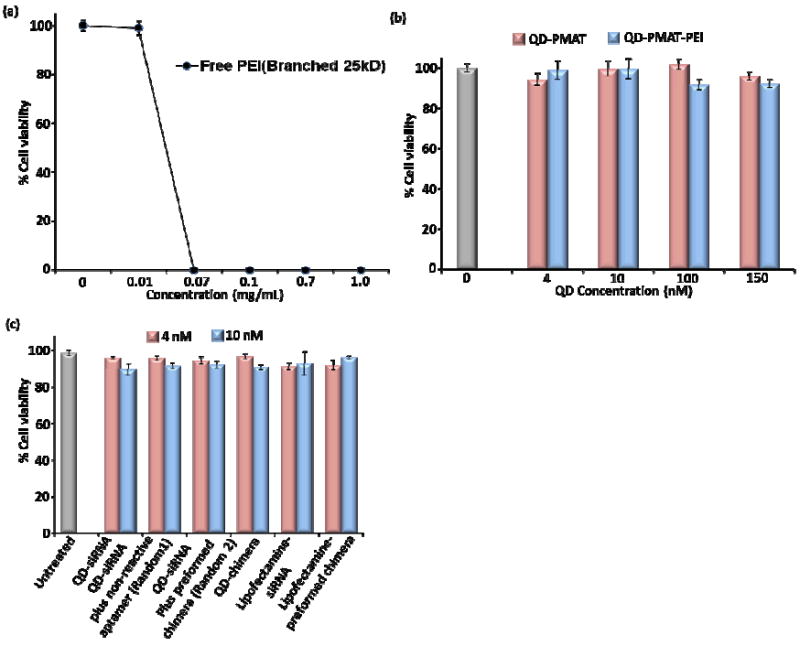
Dose-dependent cytoxicity of free PEI and QD-PMAT-PEI nanoparticles in C4-2B cells after 24 h treatment. (a) free PEI shows a sharp decline in cell viability above 0.4 μM. (b) QD-PMAT and QD-PMAT-PEI nanoparticles are not toxic to cells upto 150 nM based on QDs (for QD-PMAT-PEI the PEI concentration is 1.2 μM). (c) Comparison of cytotoxicity of QD-PMAT-PEI (4 and 10 nM) in the presence of siRNA. Cell viabilities after treatment with QD-siRNA, random 1, random 2, QD-chimera with preserved conformation, Lipofectamine with siRNA and Lipofectamine with preformed chimera, compared with untreated cells. The assembled complexes at the above concentrations are non-toxic to cells.
CONCLUSIONS
In conclusion, we have developed a new technology for linking siRNA-aptamer chimera to carrier nanoparticles, which simultaneously solved the technical hurdles previously encountered in chimera delivery such as endosome escape and aptamer orientation control. Since both siRNA and aptamer are RNA molecules and often share similar molecular weight, it is hard to design cationic delivery vehicles that selectively bind to the siRNA block, leaving the targeting aptamer block exposed. Based on a two-step process where siRNA molecules are first adsorbed onto nanoparticle surface electrostatically for nanoparticle surface charge reduction and easy intracellular unpackaging, and then coupled with aptamer for retained conformation and high accessibility, our approach shows significantly improved gene silencing over the conventional approaches based on simple mixing of siRNA-aptamer chimeras with cationic nanoparticles. To demonstrate the concept, QDs are used as a model system due to their unique optical properties and advantages of optical imaging (e.g., excellent sensitivity, resolution, multiplexing capability, and low cost) over other imaging modalities for cellular and small animal studies. They are the ideal tool for discovery and validation in cells and small animals, but their potential uses in humans as drug delivery vehicles are unclear at this time because bioconjugated CdSe-based QDs cannot be efficiently cleared from the body either as intact particles or as ions (for detailed discussions see recent review articles by Zrazhevskiy).36, 37 Fortunately, recent advances in nanotechnology have produced a large number of organic and inorganic nanoparticles with better in vivo biocompatibility than semiconductor QDs, and can be engineered in the same fashion for efficient and cell type-selective delivery of siRNA. Therefore, we expect the technology reported here can be generalized to help link siRNA-aptamer chimera with other types of nanoparticles in favorable conformation. Furthermore, as current research in biology and high-throughput screening produces more and more siRNA and aptamer sequences and combinations, we envision our technology will open new opportunities in targeted delivery and help develop individualized therapy based on siRNA-aptamer chimera.
METHODS
Unless specified, chemicals were purchased from Sigma-Aldrich (St. Louis, MO) and used without further purification. SPDP crosslinker and DTT were purchased from Pierce (Rockford, IL). Hydrophobic CdSe/ZnS core-shell QDs (620nm emission) were a gift from Oceannanotech LLC (Springdale, Arkansas). Human prostate cancer cell line C4-2B (a lineage derived LNCaP subline) stably transfected with GFP was received from our collaborator, Prof. Bob Vessella, and was maintained in G418 antibiotics (0.02 mg/mL) supplemented RPMI-1640 medium with 10% FBS (Gibco, Carlsbad, CA). siRNA targeting eGFP with a 5′ amine on the sense strand was purchased from Dharmacon (Lafayette, CO). The sequences of the sense and antisense strands are 5′-N6-CAAGCUGACCCUGAAGUUCUU-3′ and 5′-GAACUUCAGGGUCAGCUUGCC-3′, respectively. Lipofectamine™ reagent was obtained from Invitrogen Co. (Carlsbad, CA). A UV-2450 spectrophotometer (Shimadzu, Columbia, MD) and a Fluoromax4 fluorometer (Horiba Jobin Yvon, Edison, NJ) were used to characterize the absorption and emission spectra of fluorescent materials. A tabletop ultracentrifuge (Beckman TL120) was used for nanoparticle purification and isolation. The dry and hydrodynamic radii of QDs were measured on a FEI TECNAI G2 F20 S-TWIN electron microscope and a nanoparticle size analyzer (NanoZS, Worcestershire, United Kingdom). Confocal fluorescence micrographs were obtained on a Zeiss confocal microscope (LSM 510, Germany) equipped with DPSS, argon, and He/Ne lasers with lines at 405, 458, 488, 543, and 633 nm. Electrophoretic gel images were acquired with a macro-imaging system (Lightools Research, Encinitas, CA).
Preparation and characterization of QD-PMAT-PEI
To solubilize hydrophobic CdSe/ZnS core-shell QDs coated with tri-n-octylphosphine oxide (TOPO), QDs were mixed with amphiphillic polymer, PMAT, in chloroform at a molar ratio of 1:500. The solvent was slowly evaporated, resulting in the formation of a thin film of QD-polymer mixture. The dried film was dissolved in 50 mM borate buffer (pH 8.5) and filtered through 0.2 μm syringe filter. The obtained solution was subjected to three rounds of ultracentrifugation at 55,000 rpm for 1 h to remove excess PMAT polymers. The purified water-soluble QDs were characterized for absorption, fluorescence emission, hydrodynamic size and zeta potential. The carboxyl groups on the surface of water-soluble QD-PMAT (1 nmole, in 2 mL PBS) were activated with 1-ethyl-3-(3-dimethylaminopropyl) carbodiimide (EDAC, 5 μmole) for 15 minutes and the QDs were added to a PEI solution (0.5 μmole in 3 ml PBS). The reaction was stirred for 4h at ambient temperature, and excess PEI was removed by three rounds of ultracentrifugation (35,000 rpm for 30 minutes), filtered with 0.2 μm syringe filter and resuspended in cell culture grade PBS (Dulbecco).
Binding of siRNA to QD-PMAT-PEI
To determine the binding ratio of siRNA to QDs, 5′ amine modified siRNA against eGFP (20 pmole) was mixed with 2, 1.33, 1, 0.8, 0.66, 0.4, 0.2 pmole QD-PMAT-PEI and incubated for 20 minutes to obtain QD:siRNA molar ratios of 1:10, 1:15, 1:20, 1:25, 1:30, 1:50, and 1:100. The complexes were loaded on agarose gel for electrophoretic analysis and stained with SYBR gold dye to detect siRNA.
Modification of siRNA with SPDP and aptamer with thiol group
2 nmole double stranded siRNA and 1 nmole aptamer with a 5′ amine were each dissolved in PBS buffer and activated with SPDP crosslinker (20 nmole) initially dissolved in DMSO for 2 h. excess SPDP crosslinker was removed by three rounds of centrifugation using a membrane ultrafiltration filter tube (Vivaspin, MWCO:3000). For thiol modification of aptamer, the SPDP modified aptamer was treated with DTT dissolved in PBS (500 mM, 50 μl) containing 10 mM EDTA for 1 h and excess DTT was again purified away with ultrafiltration filters.
QD-chimera complex assembly and characterization
siRNA-aptamer chimera was prepared by mixing 80 pmole SPDP-modified siRNA with 40 pmole thiol-modified aptamer and incubated overnight. To assemble the QD-siRNA-aptamer complexes, the molar ratio of QD:siRNA:aptamer 1:20:10 was used. First 4 pmole QD-PMAT-PEI was mixed with 80 pmole siRNA-SPDP and incubated for 15 minutes for siRNA binding to QD surface. After incubation either thiol-modified or unmodified aptamer (40 pmole) was added to the above QD-PMAT-PEI-siRNA complex and incubated overnight to form the QD-chimera (retained aptamer conformation) and QD-siRNA plus non-reactive aptamer (random 1). The QD-chimera complex was further purified on membrane ultrafiltration tube (Vivaspin, MWCO:5000). For the assembly of QD-siRNA plus preformed chimera (random 2), first the SPDP modified siRNA (80 pmole) was reacted with thiol modified aptamer (40 pmole) overnight and washed once on membrane ultrafiltration filter tube (Vivaspin, MWCO:5000) to remove displacement side product of the reaction (pyridine 2-thione), the obtained preformed siRNA-aptamer chimera was mixed directly with 4 pmole QD-PMAT-PEI nanoparticles. The conjugates were characterized by 15% PAGE and stained with SYBR gold to detect siRNA. To release the chimera from positively charged QDs, 1% SDS was used.
Cellular uptake of QD-siRNA-aptamer conjugates
For cell uptake studies, eGFP-expressing C4-2B cells were seeded in glass-bottom dishes (Mat-tek) and were grown overnight before addition of QD-siRNA-aptamer complexes. The QD-siRNA-aptamer complexes were assembled as indicated above, and diluted in cell culture media at final QD concentration of 4 nM and incubated for 6 h. Confocal images were obtained using 63x oil immersion objective on a Carl Zeiss LSM 510 Meta microscope.
Microscopy and flow cytometry studies of eGFP down regulation in C4-2B cells
PSMA-positive eGFP-expressing C4-2B cells were seeded in 12-well plates at a density of 5 × 104 cells and maintained for 24 h before treated with QD (or Lipofectamine)-chimera complex. For the QD studies, QD-siRNA without aptamer, QD-chimera (intact), random 1, and random 2 complexes were added in 1 ml complete RPMI media to the cells. For transfection with Lipofectamine RNAimax, 3 μl/well transfection reagent (following manufacturers protocol) was diluted in 100 μl OPTIMEM, mixed with preformed chimera (100 μl) for 15 min, diluted in complete media (800 μl), and added to cells. The media was changed with fresh medium after 6 h and cells were further incubated for 42 h. After 48 h the cells were analyzed by a FACScan flow cytometer (~ 10,000 live cell events were collected for each sample). For confocal imaging, QDs were excited with a 405 nm laser. Fluorescence was detected passing a long pass emission filter (560 nm). eGFP was excited with a 488 nm laser detected passing a band pass filter (505-530 nm).
Cytotoxicity by the CellTiter-blue assay
Cell viability was assessed with standard CellTiter-blue assay, C4-2B cells (15 × 103) were seeded in 96 well plates and grown until 70% confluence. Cells were treated with the nanoparticle-RNA complexes discussed above for 24 h, washed with PBS, followed by addition of 20 μl the CellTiter-blue reagent. Cell viability was assessed by the absorbance of the converted dye at excitation 570 nm and emission 590 nm on a TECAN Infinite M200 microplate reader.
Acknowledgments
This work was supported in part by NIH (R01CA140295), NSF (0645080), and the UW Department of Bioengineering. X.H.G. thanks the NSF for a Faculty Early Career Development award (CAREER). We are also grateful to Prof. Bob Vessella for his eGFP-expressing C4-2B cells, and Dr. Andrew Wang at Oceannanotech for the highly emissive QDs.
References
- 1.Dykxhoorn DM, Novina CD, Sharp PA. Killing the Messenger: Short RNAs that Silence Gene Expression. Nat Rev Mol Cell Biol. 2003;4:457–467. doi: 10.1038/nrm1129. [DOI] [PubMed] [Google Scholar]
- 2.Hannon GJ. RNA interference. Nature. 2002;418:244–251. doi: 10.1038/418244a. [DOI] [PubMed] [Google Scholar]
- 3.Scherer LJ, Rossi JJ. Approaches for the Sequence-Specific Knockdown of mRNA. Nat Biotechnol. 2003;21:1457–1465. doi: 10.1038/nbt915. [DOI] [PubMed] [Google Scholar]
- 4.Whitehead KA, Langer R, Anderson DG. Knocking Down Barriers: Advances in SiRNA Delivery. Nat Rev Drug Discov. 2009;8:129–138. doi: 10.1038/nrd2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nishina K, Unno T, Uno Y, Kubodera T, Kanouchi T, Mizusawa H, Yokota T. Efficient in vivo Delivery of SiRNA to the Liver by Conjugation of Alpha-Tocopherol. Mol Ther. 2008;16:734–740. doi: 10.1038/mt.2008.14. [DOI] [PubMed] [Google Scholar]
- 6.Soutschek J, Akinc A, Bramlage B, Charisse K, Constien R, Donoghue M, Elbashir S, Geick A, Hadwiger P, Harborth J, et al. Therapeutic Silencing of an Endogenous Gene by Systemic Administration of Modified SiRNAs. Nature. 2004;432:173–178. doi: 10.1038/nature03121. [DOI] [PubMed] [Google Scholar]
- 7.Wolfrum C, Shi S, Jayaprakash KN, Jayaraman M, Wang G, Pandey RK, Rajeev KG, Nakayama T, Charrise K, Ndungo EM, et al. Mechanisms and Optimization of in vivo Delivery of Lipophilic SiRNAs. Nat Biotechnol. 2007;25:1149–1157. doi: 10.1038/nbt1339. [DOI] [PubMed] [Google Scholar]
- 8.Chiu YL, Ali A, Chu CY, Cao H, Rana TM. Visualizing a Correlation between SiRNA Localization, Cellular Uptake, and RNAi in Living Cells. Chem Biol. 2004;11:1165–1175. doi: 10.1016/j.chembiol.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 9.Moschos SA, Jones SW, Perry MM, Williams AE, Erjefalt JS, Turner JJ, Barnes PJ, Sproat BS, Gait MJ, Lindsay MA. Lung Delivery Studies Using SiRNA Conjugated to TAT(48-60) and Penetratin Reveal Peptide Induced Reduction in Gene Expression and Induction of Innate Immunity. Bioconjugate Chem. 2007;18:1450–1459. doi: 10.1021/bc070077d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jeong JH, Mok H, Oh YK, Park TG. SiRNA Conjugate Delivery Systems. Bioconjugate Chem. 2009;20:5–14. doi: 10.1021/bc800278e. [DOI] [PubMed] [Google Scholar]
- 11.McNamara JO, Andrechek ER, Wang Y, Viles KD, Rempel RE, Gilboa E, Sullenger BA, Giangrande PH. Cell Type-Specific Delivery of SiRNAs with Aptamer-siRNA Chimeras. Nat Biotechnol. 2006;24:1005–1015. doi: 10.1038/nbt1223. [DOI] [PubMed] [Google Scholar]
- 12.Dassie JP, Liu XY, Thomas GS, Whitaker RM, Thiel KW, Stockdale KR, Meyerholz DK, McCaffrey AP, McNamara JO, 2nd, Giangrande PH. Systemic Administration of Optimized Aptamer-SiRNA Chimeras Promotes Regression of PSMA-Expressing Tumors. Nat Biotechnol. 2009;27:839–849. doi: 10.1038/nbt.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levy-Nissenbaum E, Radovic-Moreno AF, Wang AZ, Langer R, Farokhzad OC. Nanotechnology and Aptamers: Applications in Drug Delivery. Trends Biotechnol. 2008;26:442–449. doi: 10.1016/j.tibtech.2008.04.006. [DOI] [PubMed] [Google Scholar]
- 14.Medarova Z, Pham W, Farrar C, Petkova V, Moore A. In Vivo Imaging of SiRNA Delivery and Silencing in Tumors. Nat Med. 2007;13:372–377. doi: 10.1038/nm1486. [DOI] [PubMed] [Google Scholar]
- 15.Derfus AM, Chen AA, Min DH, Ruoslahti E, Bhatia SN. Targeted Quantum Dot Conjugates for SiRNA Delivery. Bioconjugate Chem. 2007;18:1391–1396. doi: 10.1021/bc060367e. [DOI] [PubMed] [Google Scholar]
- 16.Elbakry A, Zaky A, Liebl R, Rachel R, Goepferich A, Breunig M. Layer-by-Layer Assembled Gold Nanoparticles for SiRNA Delivery. Nano Lett. 2009;9:2059–2064. doi: 10.1021/nl9003865. [DOI] [PubMed] [Google Scholar]
- 17.Meng H, Liong M, Xia T, Li Z, Ji Z, Zink JI, Nel AE. Engineered Design of Mesoporous Silica Nanoparticles to Deliver Doxorubicin and P-glycoprotein SiRNA to Overcome Drug Resistance in a Cancer Cell Line. ACS Nano. 2010;4:4539–4550. doi: 10.1021/nn100690m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xia T, Kovochich M, Liong M, Meng H, Kabehie S, George S, Zink JI, Nel AE. Polyethyleneimine Coating Enhances the Cellular Uptake of Mesoporous Silica Nanoparticles and Allows Safe Delivery of SiRNA and DNA Constructs. ACS Nano. 2009;3:3273–3286. doi: 10.1021/nn900918w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Qi L, Gao X. Emerging Application of Quantum Dots for Drug Delivery and Therapy. Exp Opin on Drug Delivery. 2008;5:263–267. doi: 10.1517/17425247.5.3.263. [DOI] [PubMed] [Google Scholar]
- 20.Qi L, Gao X. Quantum Dot-Amphipol Nanocomplex for Intracellular Delivery and Real-Time Imaging of SiRNA. ACS Nano. 2008;2:1403–1410. doi: 10.1021/nn800280r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yezhelyev MV, Qi L, O’Regan RM, Nie S, Gao X. Proton-Sponge Coated Quantum Dots for SiRNA Delivery and Intracellular Imaging. J Am Chem Soc. 2008;130:9006–9012. doi: 10.1021/ja800086u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Walter JG, Kolkpinar OZ, Friehs K, Stahl F, Scheper T. Systematic Investigation of Optimal Aptamer Immobilization for Protein Microarray Applications. Anal Chem. 2008;80:7372–7378. doi: 10.1021/ac801081v. [DOI] [PubMed] [Google Scholar]
- 23.Pellegrino T, Manna L, Kudera S, Liedl T, Koktysh D, Rogach AL, Keller S, Raddler J, Natile G, Parak WJ. Hydrophobic Nanocrystals Coated with an Amphiphilic Polymer Shell a General Route to Water Soluble Nanocrystals. Nano Letters. 2004;4:703–707. [Google Scholar]
- 24.Boussif O, Lezoualc’h F, Zanta MA, Mergny MD, Scherman D, Demeneix B, Behr JP. A Versatile Vector for Gene and Oligonucleotide Transfer into Cells in Culture and in vivo: Polyethylenimine. Proc Natl Acad Sci USA. 1995;92:7297–7301. doi: 10.1073/pnas.92.16.7297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee H, Kim IK, Park TG. Intracellular Trafficking and Unpacking of SiRNA/Quantum Dot-PEI Complexes Modified with and without Cell Penetrating Peptide: Confocal and Flow Cytometric FRET Analysis. Bioconjugate Chem. 2010;21:289–295. doi: 10.1021/bc900342p. [DOI] [PubMed] [Google Scholar]
- 26.Moghimi SM, Hunter AC, Murray JC. Long-Circulating and Target-Specific Nanoparticles: Theory to Practice. Pharmacol Rev. 2001;53:283–318. [PubMed] [Google Scholar]
- 27.Jiang W, KimBetty YS, Rutka JT, Chan WCW. Nanoparticle-Mediated Cellular Response is Size-Dependent. Nat Nanotechnol. 2008;3:145–150. doi: 10.1038/nnano.2008.30. [DOI] [PubMed] [Google Scholar]
- 28.Kam NW, Liu Z, Dai H. Functionalization of Carbon Nanotubes via Cleavable Disulfide Bonds for Efficient Intracellular Delivery of SiRNA and Potent Gene Silencing. J Am Chem Soc. 2005;127:12492–12493. doi: 10.1021/ja053962k. [DOI] [PubMed] [Google Scholar]
- 29.Chang SS, Reuter VE, Heston WDW, Gaudin PB. Metastatic Renal Cell Carcinoma Neovasculature Expresses Prostate-Specific Membrane Antigen. Urology. 2001;57:801–805. doi: 10.1016/s0090-4295(00)01094-3. [DOI] [PubMed] [Google Scholar]
- 30.Gao X, Cui Y, Levenson RM, Chung LWK, Nie S. In vivo Cancer Targeting and Imaging with Semiconductor Quantum Dots. Nat Biotechnol. 2004;22:969–976. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- 31.Lupold SE, Hicke BJ, Lin Y, Coffey DS. Identification and Characterization of Nuclease-Stabilized RNA Molecules that Bind Human Prostate Cancer Cells via the Prostate-Specific Membrane Antigen. Cancer Res. 2002;62:4029–4033. [PubMed] [Google Scholar]
- 32.Benson JR, Hare PE. O-phthalaldehyde: Fluorogenic Detection of Primary Amines in the Picomole Range. Comparison with Fluorescamine and Ninhydrin. Proc Natl Acad Sci USA. 1975;72:619–622. doi: 10.1073/pnas.72.2.619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Roth M. Fluorescence Reaction for Aamino Acids. Anal Chem. 1971;43:880–882. doi: 10.1021/ac60302a020. [DOI] [PubMed] [Google Scholar]
- He WT, Xue YN, Peng N, Liu WM, Zhuo RX, Huang SW. One-Pot Preparation of Polyethylenimine-Silica Nanoparticles as Serum-Resistant Gene Delivery Vectors: Intracellular Trafficking and Transfection. J Mater Chem. 2011;21:10496–10503. doi: 10.1016/j.jconrel.2011.08.052. [DOI] [PubMed] [Google Scholar]
- 35.Zhu J, Tang A, Law LP, Feng M, Ho KM, Lee DKL, Harris FW, Li P. Amphiphilic Core Shell Nanoparticles with Poly(ethylenimine) Shells as Potential Gene Delivery Carriers. Bioconjugate Chem. 2004;16:139–146. doi: 10.1021/bc049895l. [DOI] [PubMed] [Google Scholar]
- 36.Zrazhevskiy P, Sena M, Gao XH. Designing Multifunctional Quantum Dots for Bioimaging, Detection, and Drug Delivery. Chem Soc Rev. 2010;39:4326–4354. doi: 10.1039/b915139g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zrazhevskiy P, Gao XH. Multifunctional Quantum Dots for Personalized Medicine. Nano Today. 2009;4:414–428. doi: 10.1016/j.nantod.2009.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]