

Abstract
Inhibition of the survival kinase Akt can trigger apoptosis but also has been found to activate autophagy, which may confound tumor attack. In this study, we investigated regulatory mechanisms through which apoptosis and autophagy were modulated in tumor cells subjected to Akt inhibition by MK-2206, the first allosteric small molecule inhibitor of Akt to enter clinical development. In human glioma cells, Akt inhibition by MK-2206 or siRNA-mediated attenuation strongly activated autophagy, whereas silencing of eukaryotic elongation factor-2 (eEF-2) kinase, a protein synthesis regulator, blunted this autophagic response. Suppression of MK-2206-induced autophagy by eEF-2 silencing was accompanied by a promotion of apoptotic cell death. Similarly, siRNA-mediated inhibition of eEF-2 kinase potentiated the efficacy of MK-2206 against glioma cells. Together, these results demonstrated that blunting autophagy and augmenting apoptosis by inhibition of eEF-2 kinase could modulate the sensitivity of glioma cells to Akt inhibition. Our findings suggest that targeting eEF-2 kinase may reinforce the anti-tumor efficacy of Akt inhibitors such as MK-2206.
Keywords: Elongation factor-2 kinase, Akt, Autophagy, Apoptosis, MK-2206, Glioblastoma
Introduction
Akt/protein kinase B, an onco-protein with serine/threonine kinase activity, plays a central role in cell signaling downstream of growth factors. The Akt family consists of Akt1, Akt2 and Akt3, all of which share a similar domain structure (1). Aberrant activation of Akt promotes cell growth, survival and proliferation, and is associated with cancer development and progression (2). Thus, Akt represents an attractive target for therapeutic intervention against cancer. The anticancer action of Akt inhibitors is believed to result from induction of apoptosis triggered by Akt inhibition (3). Recently, it has been found that autophagy, a cellular process that degrades cytoplasmic components via the lysosomal machinery, is also activated when Akt is inhibited (4, 5). Although activation of autophagy (also termed type II programmed cell death) can promote autophagic cell death, this form of intracellular degradative processes has also been appreciated to support cell survival (6, 7). Yet, the exact effects of autophagy on the cytocidal activity of Akt inhibitors, and the functional association of autophagy with apoptosis in response to Akt inhibition, remain largely unknown, but this knowledge should be useful to optimizing the use of Akt inhibitors, which are actively being developed as anticancer agents.
Eukaryotic elongation factor-2 (eEF-2) kinase, also known as calmodulin-dependent protein kinase III, is a unique calcium/calmodulin-dependent enzyme that regulates protein synthesis (8). The only known substrate of this kinase is eEF-2, a 95 kDa protein that promotes ribosomal translocation from the A to the P-site, the reaction that induces movement of mRNA along the ribosome during translation in eukaryotic tissues. eEF-2 kinase phosphorylates eEF-2 on Thr56 and terminates peptide elongation by decreasing the affinity of the elongation factor for the ribosome (9). The implication of eEF-2 kinase in cancer was suggested by the observation that this kinase is up-regulated in various types of neoplasms such as malignant glioma and breast cancer (10–13), and that inhibiting this kinase results in a decreased viability of tumor cells (14). More recently, we have reported that eEF-2 kinase can act as a positive regulator of autophagy under environmental or metabolic stresses, including nutrient deprivation (15), growth factor inhibition (16), and energy stress caused by the glycolytic inhibitor, 2-deoxy-D-glucose (17). As the Akt-mediated signaling participates in the regulation of protein synthesis through mTOR and S6 kinase, a pathway known to control the activity of eEF-2 kinase, in the current study we sought to determine whether eEF-2 kinase was involved in the activation of autophagy caused by Akt inhibition, and whether and how autophagy regulated by this kinase affected the cytocidal efficacy of MK-2206, a small molecule and allosteric inhibitor of Akt (18, 19). We found that inhibiting Akt by either MK-2206 or by RNA interference induced an eEF-2 kinase-dependent, cytoprotective autophagic response, and suppression of eEF-2 kinase enhanced the effectiveness of MK-2206 against glioma cells by promoting the switch from autophagy to apoptotic cell death.
Materials and Methods
Cell lines and culture
The human glioblastoma cell lines, T98G and LN-229, were purchased from and genetically characterized by American Type Culture Collection (ATCC). T98G cells were cultured in Ham’s F-10: DMEM (10:1) medium, and LN-229 cells were cultured in DMEM medium. These media were supplemented with 10% fetal bovine serum, 100 units/mL penicillin, and 100 μg/mL streptomycin. Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2/95% air. All cultures were monitored routinely and found to be free of contamination by mycoplasma or fungi. All cell lines were discarded after three months and new lines propagated from frozen stocks.
Reagents and antibodies
MK-2206 was a gift from Merck & Co. Inc. Bafilomycin A1, acridine orange (AO), 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), 2′,7′-dichlorofluorescein diacetate (DCF-DA) and 3-methyladenosine (3-MA) were purchased from Sigma. Anti–LC-3, anti–α-tubulin, anti–p62, anti–phospho-eEF-2 (Thr56), anti–eEF-2, anti–eEF-2 kinase, anti–Akt, anti–Akt1, anti–Akt2, anti–Akt3, anti–phospho-mTOR, anti–mTOR, anti–phospho-S6 kinase (Ser371), anti–S6 kinase, anti-PARP, anti–cytochrome c, and anti–COX-IV antibodies, were purchased from Cell Signaling Technologies. Anti-HIF-1α antibody was purchased from BD Biosciences. Anti–TSC2 and anti–BNIP3 antibodies were purchased from Santa Cruz. All cell culture media and other reagents were purchased from Invitrogen. Western blot reagents were obtained from Pierce Biotechnology.
siRNA transfection
siRNA duplexes targeting eEF-2 kinase, Akt1, Akt2 and Akt3 were prepared by Dharmacon Research. Non-silencing, scrambled (non-targeting) siRNA was used as a control. Transfection of siRNA was performed according to the manufacturer’s protocol. Briefly, cells in exponential phase of growth were plated in six-well tissue culture plates at 1×105cells per well, grown for 24 h, then transfected with siRNA using Oligofectamine and OPTI-MEM I–reduced serum medium. The concentrations of siRNAs were chosen based on dose-response studies.
Measurement of autophagy
Autophagy was monitored using the following methods as described previously (15, 16): 1) Western blot analysis of LC3; 2) microscopic observation of GFP-LC3 puncta; 3) flow cytometry analysis of AO staining for acidic organelles; 4) electron microscopic examination of double or multi-membrane vacuoles in the cytoplasm.
Western blot analysis
Cells were lysed in M-PER mammalian protein extraction reagent (Thermo Scientific) supplemented with a protease inhibitor cocktail (Roche) at room temperature for 5 minutes followed by centrifugation at 14,000 x g for 10 minutes. Protein Concentrations of the cell lysates were measured using the Bio-Rad DC assay (Bio-Rad). Proteins (20–40 μg) were resolved on SDS-PAGE and transferred to PVDF membrane (Bio-Rad). The blots were incubated with indicated antibodies in 3% BSA/TBST at 4°C for overnight followed by incubation with secondary antibodies at room temperature for 1 h. The protein signals were detected by ECL method.
Apoptosis assays
Apoptosis was determined by: 1) flow cytometric analysis of Annexin V and 7-AAD staining. Briefly, 100 μl Guava Nexin reagent (Millipore) was added to 1 × 105 cells (in 100 μl) and the cells were incubated with the reagent for 20 min at room temperature in the dark. At the end of incubation, the cells were analyzed by a Guava EasyCyte™ Plus FlowCytometry System (Millipore). 2) flow cytometric analysis of activated caspases. The Guava multi-caspases assay protocol (Millipore) was followed. Briefly, 10 μl of the Guava caspase detection reagent (Millipore) was added to 1 × 105 cells (in 100 μl) and the cells were incubated for 1 h at 37 °C in a humidified atmosphere containing 5% CO2/95% air. At the end of incubation, the cells were washed twice with washing buffer, re-suspended in 200 μl of the caspase/7-AAD working solution (Millipore), and then further incubated for 10 min at room temperature. Populations of cells with activated caspases were analyzed on a EasyCyte™ Plus Flow Cytometry System (Millipore).
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential was measured by flow cytometry following JC-1 staining. Briefly, four μl of Guava cell staining solution (including JC-1 and MitoPotential 7-AAD) from Millipore was added to 2 × 105 cells (in 200 μl) and the cells were incubated with the reagent for 30 min at a 37 °C in a humidified atmosphere containing 5% CO2/95% air. At the end of incubation, the cells were analyzed by a Guava EasyCyte™ Plus FlowCytometry System.
Detection of ROS
ROS generation was measured using flow cytometry. Briefly, treated cells were collected and incubated with 10μM of DCF-DA for 30 min at 37 °C in a humidified atmosphere containing 5% CO2/95% air. At the end of incubation, the samples were analyzed on a EasyCyte™ Plus Flow Cytometry System.
Cellular viability assay
Cell viability was measured by MTT assay. Briefly, cells were plated at 5×103 cells per well in 96-well tissue culture plates and incubated at 37 °C in a humidified atmosphere containing 5% CO2/95% air. The formazan product, formed after 4h incubation with MTT, was dissolved in DMSO and read at 570 nm on a Victor3 Multi Label plate reader (PerkinElmer).
Animal study
All animal experiments were performed according to the protocols approved by the Institutional Animal Care and Use Committee at The Pennsylvania State University. Briefly, 4–6 week-old female nude mice were inoculated subcutaneously with LN229 cells (5 × 106 cells/per site) with or without silencing of eEF-2 kinase. At day 7 after inoculation, MK-2206 (120 mg/kg, p.o.) was administered to the tumor-bearing mice. Tumors were harvested 24 h post drug administration for analysis of autophagy and apoptosis. Apoptosis was measured using the terminal deoxynucleotidyl transferase-mediated dUTP nick-end labeling tetramethylrhodamine red apoptosis kit from Roche (Mannheim, Germany), and using Western blot analysis of cleaved caspase 3. Autophagy was detected by Western blot analysis of LC3 II.
Statistical analysis
The difference between the samples with silencing of eEF-2 kinase and the samples without silencing of the enzyme, or the samples with inhibition of autophagy and the samples without inhibition of autophagy, were analyzed using a two-sample t-test.
Results
Inhibition of Akt activates an eEF-2 kinase-dependent autophagy in glioma cells
Inhibition of Akt can trigger apoptosis, and this is believed to account for the antitumor effect of Akt inhibitors. To better understand how inhibiting Akt produces antitumor actions and the possible influence of autophagy, we examined the effects of the Akt inhibitor, MK-2206 (18), on the level of LC3 II, a hallmark of autophagy. We observed that treatment with MK-2206 caused a robust, concentration-dependent activation of autophagy in human glioma cell lines LN229 and T98G, as evidenced by an increase in the amount of LC3 II (Fig. 1A). LC3 II level was also elevated in the presence of bafilomycin A1, an inhibitor of autophagosome-lysosome fusion and LC3 II degradation, indicating that autophagic flux was enhanced by this Akt inhibitor (Fig. 1B). The stimulatory effect of MK-2206 on autophagy was confirmed by an increase in the GFP-LC3 dots in the cells treated with the drug, as examined by GFP-LC3 puncta formation assay (Fig. 1C), by an increase in the acridine orange staining for acidic vesicular organelles (Fig. 1D), and by an increase in autophagosomes as observed by electron microscopy (Fig. 1E). We have identified eEF-2 kinase as a regulator of autophagy under environmental or metabolic stress (15–17). Here, we asked whether the activity of this kinase also affected autophagy induced by Akt inhibition. As shown in Fig. 2A and Fig. 2B, MK-2206 not only activated eEF-2 kinase in a concentration-dependent manner, as demonstrated by increased phosphorylation of eEF-2 (Fig. 2A), the substrate for the enzyme, but also correspondingly induced autophagy in LN229 and T98G cells (Fig. 2B). To prove the role of eEF-2 kinase in activation of autophagy by MK-2206, we carried out paralleled experiments in the cells with silencing of the expression of eEF-2 kinase. Fig. 2B demonstrates that when eEF-2 kinase was silenced, there was a decrease in the amounts of LC3 II and an increase in the amounts of p62, a selective substrate of macroautophagy, as compared with the cells without silencing of the kinase, indicating that autophagic response to MK-2206 treatment was blunted by inactivation of eEF-2 kinase.
Figure 1. Effect of MK-2206 on autophagy in human glioma cells.

(A) LN229 and T98G cells cultured in medium supplemented with 10% fetal bovine serum were treated with MK2206 for 24h, the level of LC3 was examined by Western blot. (B) LN229 and T98G cells were treated with MK2206 for 24h in the absence or presence of 10 nM of bafilomycin A1, and the level of LC3 was examined by Western blot. Tubulin was used as a loading control. (C) LN229 and T98G cells were transfected with a GFP-LC3 plasmid, followed by treatment with 2.5 or 5 μM MK2206 for 24h. At the end of treatment, the cells were inspected at 60x magnification for numbers of GFP-LC3 puncta. Bars are the quantification of the percentage of cells with 10 or more GFP-LC3 puncta. At least 100 cells were scored in each treatment. * p< 0.05; * *p < 0.01, t-test, MK-2206 vs. vehicle. (D) LN229 and T98G cells were treated with 10 μM of MK-2206 for 24h, and the AO fluorescent intensity in the treated cells was analyzed by flow cytometry. (E) LN229 cells treated with MK-2206 (2.5 μM) or vehicle were harvested by trypsinization, fixed and embedded in spur resin. Ninety nm thin sections were cut and examined at 80 Kv with a JEOL 1200EX transmission electron microscope. Arrows indicate autophagic vacuoles.
Figure 2. Akt inhibition - induced autophagy is eEF-2 kinase - dependent.
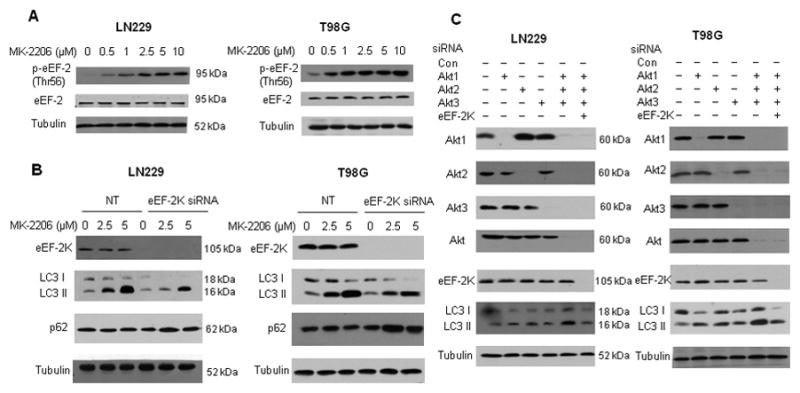
(A) LN229 and T98G cells cultured in medium supplemented with 10% fetal bovine serum were treated with a series of concentrations of MK-2206, and the levels of phospho-eEF-2 and eEF-2 were examined by Western blot. Tubulin was used as a loading control. (B) LN229 and T98 G cells were transfected with a non-targeting RNA or a siRNA targeting eEF-2 kinase, followed by treatment with MK-2206 for 24h. eEF-2 kinase, LC3 and p62 were examined by Western. Tubulin was used as a loading control. (C) LN229 and T98G cells were transfected with a non-targeting RNA, Akt1 siRNA, Akt2 siRNA, Akt3 siRNA, or an eEF-2 kinase siRNA alone, or with combination of these siRNAs. Akt1, Akt2, Akt3, Akt, eEF-2 kinase, and LC3 were detected by Western blot. Tubulin was used as a loading control.
To corroborate the involvement of eEF-2 kinase in the Akt inhibition-activated autophagy and to assess whether the effect of inhibiting Akt on autophagy was isozyme - selective, we silenced the expression of Akt1, Akt2 and Akt3, respectively, using the siRNAs specific for each of these Akt isozymes. We found that respective silencing of Akt1, Akt2 or Akt3 expression all caused an increase in the amount of LC3 II (Fig. 2C), indicating that inhibition of each of these isozymes contributes to induction of autophagy. Depletion of eEF-2 kinase by siRNA also blunted autophagy induced by knockdown of all these Akt isozymes (Fig. 2C), providing further support for a role of this enzyme in mediating autophagy activated by Akt inhibition.
The TSC2-mTOR-S6 kinase-eEF-2 kinase pathway is involved in activation of autophagy by MK-2206
To explore the pathways that mediate the effect of eEF-2 kinase on Akt inhibition-induced autophagy, we knocked down TSC2, a tumor suppressor protein that is negatively regulated by Akt and suppresses mTOR activity (20). As shown in Fig. 3, knockdown of TSC2 led to a reduction in the amount of LC3 II in the cells treated with MK-2206, in comparison to the non-targeting controls (Fig. 3A). Concomitantly, knockdown of TSC2 also resulted in a decreased activity of eEF-2 kinase (decreased phospho-eEF-2), an increased activity of S6 kinase (increased phospho-S6 kinase) and mTOR (increased phospho-mTOR) (Fig. 3B). These results suggest that induction of autophagy by Akt inhibition is mediated via the TSC2-mTOR-S6 kinase-eEF-2 kinase pathway.
Figure 3. TSC2/mTOR/S6 kinase/eEF-2 kinase pathway is involved in the activation of autophagy by Akt inhibition.

LN229 and T98G cells were transfected with a non-targeting RNA or a TSC2 siRNA, followed by treatment with MK-2206 for 24h in fully supplemented medium. The levels of TSC2 and LC3 (A), phospho-mTOR (Ser2448), mTOR, phospho-S6 kinase (Ser371), S6 kinase, phospho-eEF-2 (Thr56) and eEF-2 (B) were examined by Western blot. Tubulin was used as a loading control.
Silencing of eEF-2 kinase augments apoptosis in glioma cells subjected to Akt inhibition
As the pro-survival or pro-death role of autophagy in stressed cells is often associated with alteration of apoptosis, we next queried whether there was a functional relationship between apoptosis and the eEF-2 kinase-mediated autophagy when Akt was inhibited. We compared the degree of apoptosis in the MK-2206-treated glioma cells with or without silencing of eEF-2 kinase. As demonstrated in Fig. 4A and Fig. 4B, MK-2206 caused a concentration-dependent increase in apoptosis in the treated cells, as manifested by increases in Annexin V staining (Fig. 4A) and in activated pan-caspases (Fig. 4B); notably, silencing of eEF-2 kinase strengthened the apoptogenic effect of MK-2206 in the glioma cells (Fig. 4A and Fig. 4B)
Figure 4. Silencing of eEF-2 kinase expression augments the MK-2206-induced apoptosis in tumor cells.

(A and B) LN229 and T98G cells were transfected with a non-targeting RNA or a siRNA targeting eEF-2 kinase, followed by treatment with MK-2206 for 24h in fully supplemented medium. Apoptosis was determined by flow cytometric analyses of Annexin staining (A) and activated pan-caspases (B). Each bar represents the mean ± SE of three experiments. *p< 0.05, **p< 0.01 (C) LN229 cells cultured in medium supplemented with 10% fetal bovine serum were treated with 2.5 μM of MK-2206 under normoxia or hypoxia (1% O2) for 24 h, and HIF-1α, LC3, phospho-eEF-2, eEF-2, and PARP were examined by Western blot. Tubulin expression was used as a loading control. (D) LN229 and T98G cells were transfected with a non-targeting RNA or an eEF-2 kinase - targeted siRNA, followed by treatment with MK-2206 (2.5 μM) for 24h under hypoxia (1% O2). HIF-1α, eEF-2 kinase, LC3 and BNIP3 were examined by Western blot. Tubulin was used as a loading control. (E) LN229 and T98G cells were transfected with a non-targeting RNA or an eEF-2 kinase - targeted siRNA, following by treatment with MK-2206 for 24h under normoxia or hypoxia (1% O2). Apoptosis was determined by flow cytometric analyses of Annexin staining. Each bar represents the mean ± SE of three experiments. *p< 0.05, **p< 0.01. (F) Mice bearing LN229 tumor xenografts were given MK-2206 (120 mg/kg). Twenty-four hours later, tumors were removed and analyzed for autophagy and apoptosis. Autophagy was determined by Western blot of LC3 II; apoptosis was measured by Western blot of cleaved caspase 3 and TUNEL assay. Results shown are representative of four tumor specimens from each treatment group. * p < 0.015
Hypoxic microenvironments of solid tumors are believed to be linked to the malignant features, including poor response to therapies. Thus, we next assessed whether suppression of eEF-2 kinase-mediated autophagy could also sensitize hypoxic glioma cells to the apoptogenic effect of MK-2206. As shown in Fig. 4C, under hypoxic condition, MK-2206 induced a stronger autophagic response and higher eEF-2 kinase activity than those under normoxic condition; knockdown of eEF-2 kinase not only blunted autophagy activated by MK-2206 and hypoxia (Fig. 4D), but also further amplified apoptosis in the MK-2206-treated glioma cells subjected to hypoxia (Fig. 4E). To test whether these findings are relevant to in vivo circumstance, we inoculated mice with LN229 cells either with or without knockdown of eEF-2 kinase, and then treated the tumor-bearing mice with MK-2206 (120 mg/kg, p.o.). We observed that following MK-2206 treatment, the tumor xenografts with knockdown of eEF-2 kinase had a lower level of the autophagy marker, LC3 II, but a higher level of the apoptosis marker, cleaved caspase-3, than xenografts without eEF-2 kinase knockdown (Fig. 4F, upper panel). TUNEL assay also demonstrated an increase of apoptosis in tumor specimens with knockdown of eEF-2 kinase (Fig. 4F, lower panel). These in vivo tests were performed using a non-orthotopic xenograft model, and it remains to be shown that these findings are relevant to glioma in an orthotopic setting.
Mitochondrial ROS is a mediator of the MK-2206-stimulated autophagy and apoptosis
To understand the mechanism governing the functional connection between autophagy and apoptosis in the MK-2206-treated tumor cells and the role of eEF-2 kinase in these cellular processes, we determined the effects of MK-2206 and eEF-2 kinase on mitochondrial integrity and reactive oxygen species (ROS) production, as mitochondria and ROS play an important role in the regulation of both apoptosis and autophagy (21, 22). As shown in Fig. 5A and Fig. 5B, treatment of LN229 or T98G cells with MK-2206 impaired the mitochondrial integrity, as evidenced by a decline in mitochondrial membrane potential (Δψm) (Fig. 5A) and an increase in release of cytochrome c into cytosol (Fig. 5B). Knockdown of eEF-2 kinase aggravated the MK-2206-induced loss of mitochondrial integrity (Fig. 5A and Fig. 5B). MK-2206 also stimulated the generation of ROS (Fig. 5C and Fig. 5D), and knockdown of eEF-2 kinase further increased the production of ROS (Fig. 5D). The over-production of ROS appeared to be, at least partially, responsible for activation of both autophagy and apoptosis, as co-treatment with MK-2206 and the ROS scavenger, N-acetyl cystein (NAC), weakened the stimulatory effect of MK-2206 on both autophagy (Fig. 5E) and apoptosis (Fig. 5F). Because mitochondria are a source of ROS production, in order to further demonstrate the importance of the mitochondria in the MK-2206-induced autophagy and apoptosis and the role of the eEF-2 kinase-regulated autophagy in mitigating cellular stress, we compared the level of the mitochondrial marker cytochrome oxidase subunit IV (COX IV), a specific marker for mitochondria, in the MK-2206-treated cells with or without silencing of eEF-2 kinase. Fig. 5G shows that silencing of eEF-2 kinase, which can suppress autophagy (Fig. 2 and Fig. 4), rescued the decreases of COX IV in cells treated with MK-2206, suggesting that suppression of autophagy slowed down the degradation of the damaged mitochondria. The results shown in Fig. 5H demonstrated an increase of the amount of LC 3 II mainly in the mitochondrial fraction of the MK-2206 treated cells, verifying the mitochondrial autophagy that was activated by MK-2206.
Figure 5. Mitochondrial ROS level is associated with the effects of MK-2206 and eEF-2 kinase silencing on autophagy and apoptosis.

(A, B) LN229 and T98 G cells were transfected with a non-targeting RNA or a siRNA targeting eEF-2 kinase, followed by treatment with MK-2206 for 24 h in fully supplemented medium. Mitochondrial membrane potential was measured by JC-1 staining by flow cytometry (A). The level of cytochrome c in cytosol was examined by Western blot. Tubulin was used as a loading control (B). (C, D) LN229 and T98G cells with or without silencing of eEF-2 kinase were treated with the indicated concentrations of MK-2206 in fully supplemented medium, and the level of ROS was measured by staining with DCF-DA and analyzing by flow cytometry. Each bar represents the mean ± SE of three experiments. *p< 0.05, **p< 0.01 (E, F) LN229 and T98G cells were treated with MK-2206 for 24h in the absence or presence of 2mM NAC. The level of LC3 was examined by Western blot. Tubulin was used as a loading control (E). Apoptosis was determined by flow cytometric analyses of Annexin staining (F). Each bar represents the mean ± SE of three experiments. *p< 0.05, **p< 0.01 (G) LN229 and T98G cells with or without silencing of eEF-2 kinase were treated with the indicated concentrations of MK-2206, and the amount of COX-IV was determined by Western blot. (H) LN229 and T98G cells were treated with MK2206 for 24h. The level of LC3 in mitochondrial or cytosol fraction was examined by Western blot. Tubulin was used as a loading control.
Suppression of autophagy via inhibiting eEF-2 kinase enhances the cytocidal effect of MK-2206 in human glioma cells
To evaluate whether or not the eEF-2 kinase-mediated autophagy has an impact on the antitumor action of Akt inhibitors, we determined the effects of inhibiting eEF-2 kinase and autophagy on viability of the glioma cells treated with MK-2206. We found that the cytotoxicity of MK-2206 against T98G and LN229 cells was significantly greater when eEF-2 kinase was silenced, as compared to that in the cells transfected with a non-targeting RNA (Fig. 6A). Fig. 6B shows that enhanced sensitivity to MK-2206 was also achieved in glioma cells co-treated with 0.25 μM of a small molecule inhibitor of eEF-2 kinase, NH125 (14). To ascertain that the reinforcing effect of inhibiting eEF-2 kinase on cytocidal activity of MK-2206 was attributed to suppression of autophagy, we tested MK-2206 in the glioma cells in which autophagy was suppressed by either silencing of beclin 1, a key autophagy regulator, or by use of 3-methyladenine (3-MA), a chemical inhibitor of autophagy. We observed that the cytocidal activity of MK-2206 was also markedly increased in the cells subjected to silencing of beclin 1 expression (Fig. 6C) or with the treatment of 1 mM of 3-MA (Fig. 6D), suggesting that inhibition of autophagy can indeed sensitize tumor cells to the Akt inhibitor, MK-2206.
Figure 6. Suppression of autophagy increases the cytocidal activity of MK-2206 in glioma cells.

(A) LN229 and T98G cells were transfected with a non-targeting RNA or a siRNA targeting eEF-2 kinase, followed by treatment with a series concentration of MK-2206 for 48 h in fully supplemented medium. (B) LN229 and T98G cells cultured in medium supplemented with 10% fetal bovine serum were treated with a series concentration of MK-2206 for 48 h in the presence or absence of 0.25 μM of NH125. (C) LN229 and T98G cells were transfected with a non-targeting siRNA or a beclin 1-targeted siRNA, followed by treatment with a series concentration of MK-2206 for 48h. (D) LN229 and T98G cells were treated with a series concentration of MK-2206 for 48 h in the presence or absence of 1 mM of 3-MA. At the end of treatment, cell viability was measured by MTT assay. Results shown were mean ± SD of quadruplicate determinations from one of three identical experiments; *p< 0.05, **p< 0.01.
Discussion
What exact roles autophagy plays in response of tumor cells to cancer therapies, what signaling pathways are involved in the regulation of autophagy, and how to exploit autophagy in devising novel strategies for cancer therapy, are the questions being actively and extensively investigated. Inhibiting Akt, a serine/threonine kinase mediating mitogenic and anti-apoptotic responses that result from activation of multiple signaling cascades, has been shown to induce autophagy (4, 5) as well as to promote apoptotic cell death and suppress proliferation and growth of tumor cells (23); yet, whether and how autophagy affects the efficacy of Akt inhibitors, a promising and new class of cancer therapeutic agents, remains poorly understood. The results of our current study informs that autophagy induced by Akt inhibition can indeed affect the cytotoxicity of Akt inhibitors, and that manipulating autophagic activity can modulate sensitivity of tumor cells to the cytocidal effect of Akt inhibitors. We show that, like other Akt inhibitors (5), the novel allosteric Akt inhibitor, MK-2206, could induce autophagy in tumor cells (Fig. 1, Fig. 2B, Fig. 3A and Fig. 4C). Moreover, we demonstrate in this study that autophagy induced by Akt-inhibition was cytoprotective and could mitigate the cytotoxicity of the Akt inhibitor, as blunting autophagic response by either genetic or pharmacologic approaches could enhance the cytocidal efficacy of MK-2206 (Fig. 6) as well as the apoptotic cell death induced by the compound (Fig. 4). Furthermore, we revealed that eEF-2 kinase, a unique calcium/calmodulin-dependent enzyme that regulates protein synthesis and is overexpressed in malignant gliomas and other malignancies, played an essential role in regulating autophagy that is activated by Akt inhibition (Fig. 2), and this regulatory role appeared to be mediated through the TSC2/mTOR/S6 kinase pathway (Fig. 3). We have previously reported that eEF-2 kinase serves as a central component of the mammalian macroautophagy pathway that is activated in response to environmental or metabolic stress (15–17). The present study further underscores the importance of this kinase as a regulator of autophagy. However, the precise molecular mechanism by which eEF-2 kinase controls autophagy is yet to be defined.
As reported previously (3–5) and demonstrated in this study (Fig. 1, Fig. 2 and Fig. 4), inhibition of Akt can induce both autophagy and apoptosis; nevertheless, how apoptosis and autophagy affect cellular sensitivity to Akt inhibitors remains unclear. Our observation indicates that the Akt inhibition-activated autophagy plays a protective role in tumor cells, as suppression of autophagy can enhance the cytocidal activity of the Akt inhibitor, MK-2206. Then, is there a functional association between autophagy and apoptosis in cells subjected to Akt inhibition, and how is the interplay between these two cellular processes controlled? To answer these questions, we examined the effects of MK-2206 and eEF-2 kinase activity on level of ROS, a critical regulator of both apoptosis and autophagy, and on integrity of mitochondria, which are both source and target of ROS. We found that MK-2206 increased ROS level in a concentration-dependent manner (Fig. 5C, upper panels) and enhanced mitochondrial permeability (Fig. 5A and Fig. 5B) in the treated tumor cells. Thus, loss of mitochondrial integrity and over-production of ROS may account for activation of apoptosis; also, excess of ROS and activation of the TSC2/mTOR/S6 kinase/eEF-2 kinase pathway (Fig. 3B) both may contribute to induction of autophagy, which appears to favor cell survival. Noteworthily, when autophagy was blunted by inhibition of eEF-2 kinase, degradation of the damaged mitochondria was retarded (Fig. 5G), and this might further increase the accumulation of ROS. Indeed, MK-2206 activated a mitochondrial autophagy, as an up-regulation of the autophagy marker, LC3 II, was only detected in the mitochondrial fractions of the treated cells (Fig. 5H). On the other hand, inhibiting eEF-2 kinase causes a decline in cellular ATP content due to the up-regulation of protein synthesis (16, 17), and depletion of the cellular ATP can also impair mitochondrial function and facilitate ROS generation, which consequently, can elicit more apoptotic cell death. This may explain the augmentation of MK-2206 cytotoxicity by inhibiting eEF-2 kinase, which was observed in this study. It is likely that ROS serves as a mediator in these cellular processes, as NAC, a ROS scavenger, could block the autophagy (Fig. 5E) and apoptosis (Fig. 5F) activated by MK-2206, although not completely, probably due to the dual effects of ROS on autophagy and apoptosis. Based on the results of the present study, we believe that eEF-2 kinase could act as an essential regulator controlling the switch between autophagy and apoptosis in cells experiencing cellular insults such as Akt inhibition, and that a previously unidentified and important pathway that regulates the cross-talk between autophagy and apoptosis and modulates cellular sensitivity to Akt inhibitors, as is depicted in Fig. 7, might emerge.
Figure 7.

Proposed role for eEF-2 kinase in the cross-talk between autophagy and apoptosis.
Because of the central role of Akt in oncogenic signaling and in tumor progression, Akt is considered one of the major targets for development of anticancer drugs. In fact, several inhibitors of Akt are being tested in clinic as promising cancer therapeutic agents (24–26); nevertheless, these compounds seem to have limited activity as a single agent. Strategies for reinforcing the activity of the Akt inhibitors would make this type of drugs more valuable in treatment of cancer. Our findings that suppression of the eEF-2 kinase-mediated autophagy can promote apoptotic cell death and augment the cytotoxicity of MK-2206, even under hypoxic condition, may provide a rationale for designing new and more effective therapeutic intervention for treatment of malignant tumors such as gliomas, i.e., combining an Akt inhibitor with an eEF-2 kinase inhibitor to shuttle cells into more reliable and permanent pathways of cell death. Evaluation of this strategy in pre-clinical glioma and other tumor models is underway. Considering that MK-2206 is now under clinical development as a new anticancer agent, and that we have already identified an inhibitor of eEF-2 kinase (14), the results reported here may have translational potentials and a direct impact on cancer therapy.
In summary, the results reported here demonstrate that suppressing autophagy and augmenting apoptosis via inhibition of eEF-2 kinase can modulate sensitivity of tumor cells to Akt inhibition, and suggest that eEF-2 kinase may be utilized as an effective target for reinforcing the anti-tumor efficacy of Akt inhibitors such as MK-2206.
Footnotes
Supported by grants from the US Public Health Service R01CA135038, and from Merck & Co. Inc.
References
- 1.Hanada M, Feng J, Hemmings BA. Structure, regulation and function of PKB/AKT--a major therapeutic target. Biochim Biophys Acta. 2004;1697:3–16. doi: 10.1016/j.bbapap.2003.11.009. [DOI] [PubMed] [Google Scholar]
- 2.Altomare DA, Testa JR. Perturbations of the AKT signaling pathway in human cancer. Oncogene. 2005;24:7455–64. doi: 10.1038/sj.onc.1209085. [DOI] [PubMed] [Google Scholar]
- 3.Garcia-Echeverria C, Sellers WR. Drug discovery approaches targeting the PI3K/Akt pathway in cancer. Oncogene. 2008;27:5511–26. doi: 10.1038/onc.2008.246. [DOI] [PubMed] [Google Scholar]
- 4.Degtyarev M, De Maziere A, Orr C, et al. Akt inhibition promotes autophagy and sensitizes PTEN-null tumors to lysosomotropic agents. J Cell Biol. 2008;183:101–16. doi: 10.1083/jcb.200801099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fujiwara K, Iwado E, Mills GB, Sawaya R, Kondo S, Kondo Y. Akt inhibitor shows anticancer and radiosensitizing effects in malignant glioma cells by inducing autophagy. Int J Oncol. 2007;31:753–60. [PubMed] [Google Scholar]
- 6.Kundu M, Thompson CB. Macroautophagy versus mitochondrial autophagy: a question of fate? Cell Death Differ. 2005;12 (Suppl 2):1484–9. doi: 10.1038/sj.cdd.4401780. [DOI] [PubMed] [Google Scholar]
- 7.Eskelinen EL. Doctor Jekyll and Mister Hyde: autophagy can promote both cell survival and cell death. Cell Death Differ. 2005;12 (Suppl 2):1468–72. doi: 10.1038/sj.cdd.4401721. [DOI] [PubMed] [Google Scholar]
- 8.Ryazanov AG, Shestakova EA, Natapov PG. Phosphorylation of elongation factor 2 by EF-2 kinase affects rate of translation. Nature. 1988;334:170–3. doi: 10.1038/334170a0. [DOI] [PubMed] [Google Scholar]
- 9.Ryazanov AG. Ca2+/calmodulin-dependent phosphorylation of elongation factor 2. FEBS Lett. 1987;214:331–4. doi: 10.1016/0014-5793(87)80081-9. [DOI] [PubMed] [Google Scholar]
- 10.Bagaglio DM, Cheng EH, Gorelick FS, Mitsui K, Nairn AC, Hait WN. Phosphorylation of elongation factor 2 in normal and malignant rat glial cells. Cancer Res. 1993;53:2260–4. [PubMed] [Google Scholar]
- 11.Bagaglio DM, Hait WN. Role of calmodulin-dependent phosphorylation of elongation factor 2 in the proliferation of rat glial cells. Cell Growth Differ. 1994;5:1403–8. [PubMed] [Google Scholar]
- 12.Cheng EH, Gorelick FS, Czernik AJ, Bagaglio DM, Hait WN. Calmodulin-dependent protein kinases in rat glioblastoma. Cell Growth Differ. 1995;6:615–21. [PubMed] [Google Scholar]
- 13.Parmer TG, Ward MD, Yurkow EJ, Vyas VH, Kearney TJ, Hait WN. Activity and regulation by growth factors of calmodulin-dependent protein kinase III (elongation factor 2-kinase) in human breast cancer. Br J Cancer. 1999;79:59–64. doi: 10.1038/sj.bjc.6690012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Arora S, Yang JM, Kinzy TG, et al. Identification and characterization of an inhibitor of eukaryotic elongation factor 2 kinase against human cancer cell lines. Cancer Res. 2003;63:6894–9. [PubMed] [Google Scholar]
- 15.Wu H, Yang JM, Jin S, Zhang H, Hait WN. Elongation factor-2 kinase regulates autophagy in human glioblastoma cells. Cancer Res. 2006;66:3015–23. doi: 10.1158/0008-5472.CAN-05-1554. [DOI] [PubMed] [Google Scholar]
- 16.Cheng Y, Li H, Ren X, Niu T, Hait WN, Yang J. Cytoprotective effect of the elongation factor-2 kinase-mediated autophagy in breast cancer cells subjected to growth factor inhibition. PLoS One. 5:e9715. doi: 10.1371/journal.pone.0009715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wu H, Zhu H, Liu DX, et al. Silencing of elongation factor-2 kinase potentiates the effect of 2-deoxy-D-glucose against human glioma cells through blunting of autophagy. Cancer Res. 2009;69:2453–60. doi: 10.1158/0008-5472.CAN-08-2872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bilodeau MT, Balitza AE, Hoffman JM, et al. Allosteric inhibitors of Akt1 and Akt2: a naphthyridinone with efficacy in an A2780 tumor xenograft model. Bioorg Med Chem Lett. 2008;18:3178–82. doi: 10.1016/j.bmcl.2008.04.074. [DOI] [PubMed] [Google Scholar]
- 19.Hirai H, Sootome H, Nakatsuru Y, et al. MK-2206, an allosteric Akt inhibitor, enhances antitumor efficacy by standard chemotherapeutic agents or molecular targeted drugs in vitro and in vivo. Mol Cancer Ther. 9:1956–67. doi: 10.1158/1535-7163.MCT-09-1012. [DOI] [PubMed] [Google Scholar]
- 20.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol. 2002;4:648–57. doi: 10.1038/ncb839. [DOI] [PubMed] [Google Scholar]
- 21.Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J. 2009;28:3015–26. doi: 10.1038/emboj.2009.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wong CH, Iskandar KB, Yadav SK, Hirpara JL, Loh T, Pervaiz S. Simultaneous induction of non-canonical autophagy and apoptosis in cancer cells by ROS-dependent ERK and JNK activation. PLoS One. 5:e9996. doi: 10.1371/journal.pone.0009996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Thompson JE, Thompson CB. Putting the rap on Akt. J Clin Oncol. 2004;22:4217–26. doi: 10.1200/JCO.2004.01.103. [DOI] [PubMed] [Google Scholar]
- 24.Ghobrial IM, Roccaro A, Hong F, et al. Clinical and translational studies of a phase II trial of the novel oral Akt inhibitor perifosine in relapsed or relapsed/refractory Waldenstrom’s macroglobulinemia. Clin Cancer Res. 16:1033–41. doi: 10.1158/1078-0432.CCR-09-1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oh Y, Herbst RS, Burris H, et al. Enzastaurin, an oral serine/threonine kinase inhibitor, as second- or third-line therapy of non-small-cell lung cancer. J Clin Oncol. 2008;26:1135–41. doi: 10.1200/JCO.2007.14.3685. [DOI] [PubMed] [Google Scholar]
- 26.Garrett CR, Coppola D, Wenham RM, et al. Phase I pharmacokinetic and pharmacodynamic study of triciribine phosphate monohydrate, a small-molecule inhibitor of AKT phosphorylation, in adult subjects with solid tumors containing activated AKT. Invest New Drugs. doi: 10.1007/s10637-010-9479-2. [DOI] [PMC free article] [PubMed] [Google Scholar]