

Summary
The glucocorticoid receptor (GR), like other eukaryotic transcription factors, regulates gene expression by interacting with chromatinized DNA response elements. Photobleaching experiments in living cells indicate that receptors transiently interact with DNA on the time scale of seconds and predict that the response elements may be sparsely occupied on average. Here, we show that the binding of one receptor at the glucocorticoid response element (GRE) does not reduce the steady-state binding of another receptor variant to the same GRE. Mathematical simulations reproduce this non-competitive state using short GR/GRE residency times and relatively long times between DNA binding events. At many genomic sites where GR-binding causes increased chromatin accessibility, concurrent steady-state binding levels for the variant receptor are actually increased, a phenomenon termed assisted loading. Temporally sparse transcription factor-DNA interactions induce local chromatin reorganization, resulting in transient access for binding of secondary regulatory factors.
Keywords: glucocorticoid receptor, dynamics, transcriptional regulation, genome-wide regulation, transcription factors, ligand specificity
Introduction
The access of transcription factors to regulatory elements in their chromosomal context can be strongly limited by local chromatin architecture. Furthermore, many lines of investigation now indicate that these restrictive features of nucleoprotein structure are organized in a cell specific fashion, and contribute significantly to the cell selective activity of a given regulatory site. An emerging paradigm suggests that the local chromatin structure of response elements contributes strongly to their tissue specific action. Thus, the mechanisms that govern site specific activity of distal regulatory sites are central to our understanding of eucaryotic gene regulation.
Characterization of regulatory protein binding sites has been addressed primarily through the technique of chromatin immunoprecipitation, coupled with the use of antibodies specific either to transcription factors (Robertson et al., 2007; Johnson et al., 2007; Jothi et al., 2008; Valouev et al., 2008). This approach has revealed cell selective binding for several transcription factors, including highly selective patterns for nuclear receptors (John et al., 2008; Krum et al., 2008; He et al., 2010). Local transitions in chromatin structure have also been studied on the genome scale by mapping of in vivo DNA accessibility to DNaseI and the delineation of DNaseI hypersensitive sites (DHSs) (Sabo et al., 2006; Hesselberth et al., 2009; John et al., 2011). These approaches can only resolve binding events and chromatin transitions on the time scale of minutes to hours, and lead, in general, to models wherein groups of transcription factors are bound to response elements on long time scales. In contrast, studies of site-specific factor binding in living cells have revealed residence times on chromatin measured in seconds for many site-specific DNA binding proteins (McNally et al., 2000; Hager et al., 2009). The detailed mechanisms by which transcription factors bind to, and reorganize, local nucleosome structures are thus subject to considerable variation in interpretation.
If most proteins are localized on binding elements over long periods, these sites should in fact be saturable, and subject to competitive displacement, as is normally observed for site-specific DNA binding proteins, both on DNA (Cann, 1989) and nucleosomal templates in vitro (Li and Wrange, 1997). In concert with this model, competitive displacement has been argued for transcription factors (Nalley et al., 2006), and saturable DNA response element binding has been assumed in a general quantitative model of eukaryotic transcription activation (Kim and O’Shea, 2008). However, it remains to be determined if competitive behavior and saturable binding are general properties of highly dynamic eukaryotic transcription factors in vivo.
We have directly addressed this question using a well studied sequence-specific DNA binding protein, the glucocorticoid receptor (GR). We have examined competitive binding for this receptor, using a pBox variant of the estrogen receptor with an identical DNA binding specificity to GREs (Mader et al., 1989). Competition was examined both in intact cells, using fluorescently tagged receptors and an amplified array of promoter elements, and at a variety of response elements throughout the murine genome, using chromatin immunoprecipitation. We report that the two proteins fail to manifest significant competition when binding to the same response element. Indeed, at a large subset of binding elements, we find that GR will “assist” binding of the ER pbox protein, even though they bind exactly the same site. We present a model (assisted loading), under which the interaction of multiple transcription factors to a given response element are understood as a highly dynamic process. Under this model, the direct participation of ATP-dependent remodeling complexes is central to a process of continual nucleosome reorganization and cycling.
Results
Lack of factor competition at a hormone response element
We developed a fluorescence microscopy assay to measure the steady-state binding of the GR and ER-pBox to GREs in single cells. When treated with an individual selective steroid hormone, either GR or ER pBox individually associate with a GRE. However in the presence of both Dexamethasone (Dex) and Estradiol (E2), both receptors are concurrently competent to interact with the GRE. The 6644 cell line was engineered to visualize this process in single cells (see methods). The 6644 cell line cultures, which express GFP-labeled GR (GFP-GR) and mCherry-labeled ER pBox (Ch-ER pBox), were processed for RNA FISH to detect transcription from a 200 copy tandem repeat of MMTV LTR promoter-reporter cassettes that is integrated at a single genomic site (termed the MMTV array). The MMTV LTR contains well characterized GREs that are essential for MMTV transcriptional activation (Figure S4A) (Ostrowski et al., 1984; Payvar et al., 1983). Following simultaneous treatment with Dex and E2, both labeled receptors concentrate in the subnuclear region containing the MMTV array, which is clearly identified by the RNA FISH signal (Figure 1A).
Figure 1.

Single cell level analysis of competitive steroid receptor interactions with chromatin. Treatment with Dex or E2 allows DNA-binding of GR or ER pBox, respectively. (A) 6644 cell line cultures were treated for 30 minutes with both Dex and E2, then processed for RNA FISH to detect transcription from the MMTV array. The micrographs display a single nucleus with each fluorescence channel shown separately and merged in the overlay. Computationally defined ROIs for the nucleus and MMTV array FISH signal are outlined in white. (B–D) Based on automatic detection of the MMTV array FISH signal in large numbers of individual cells (n = 500 to 1000 per condition), steady-state receptor interactions with the MMTV array are measured following 30m hormone treatment. (D) The points of the scatter plot show the relative amount of GR and ER pBox interaction with the MMTV array for individual cells within the Dex+E2 treated population, and the red line represents best fit relationship between these parameters. (E) The image analysis algorithm also simultaneously measures MMTV RNA FISH signal. Bars represent the mean of cellular measurements, error bars denote SEM, and markers “a,b,c” indicate homogenous statistical subsets of conditions. Each subset is considered to be statistically different because it consists of measurements with HSD p-values < 0.05 versus the values in another defined subset. (F) Diagrams illustrate the dynamic interaction of GR protein and/or ER pBox protein with the chromatinized GRE located in the B-nucleosome region of the MMTV-LTR promoter. See also Supplemental Figure 1.
Supplemental Figure 1 displays example fluorescent micrographs of cell nuclei under all hormone treatment conditions. Automated image analysis based on algorithmic identification of the RNA FISH signal indicates that GR interacts at similar steady-state levels following treatment with Dex alone or with both Dex and E2 (Figure 1B). Surprisingly, ER pBox interacts at a greater steady-state level in the presence of Dex and E2 compared to treatment with E2 alone (Figure 1C). We refer to this phenomenon as “assisted loading.” Single cells that exhibit high-levels of GR/MMTV association also show high-levels of ER pBox/MMTV association (Figure 1D), suggesting that the local chromatin state regulates binding of the two receptors without competition. FISH signal was also measured by the automated analysis method, revealing that liganded ER pBox was much weaker as a transactivator compared to liganded GR (Figure 1E). Since total steady-state binding to the GRE was similar for the two receptors under these conditions, this transcriptional difference likely reflects divergent interactions with cofactors. The interaction of ER pBox with the MMTV GREs is extremely transient (Figure S1E, S1F), on the same time scale as those reported previously for GR/MMTV interactions (Becker et al., 2002; McNally et al., 2000). The hyper-dynamic interactions of both receptors may permit both receptor types to alternately bind the promoter without competing with one another (Figure 1F). This alternate transient binding of receptors without measurable steady-state competition requires that the MMTV GREs are unsaturated during hormone-dependent transcriptional activation.
Biochemical characterization of assisted loading
To extend our studies of the GRE saturation state, we employed a modified version of a competitive-ChIP assay that can measure the dynamics of TF binding if the REs are near steady-state saturation (Nalley et al., 2006). These previous studies, which showed competition between transcription factors, were interpreted as evidence for TF/DNA interactions on the time scale of many minutes. ChIP experiments using the 6644 cell line treated with a single ligand show that Dex causes only GR to interact with the consensus GRE located in the B nucleosome of the MMTV LTR promoter, while E2 causes only ER pBox to interact with the same GRE (Figure 2A). Individually, GR and ER pBox bind to the GRE at statistically identical steady-state levels under these conditions. When both hormones are added concurrently, there is no statistical decrease in the steady state binding level of either receptor compared to binding levels in the presence of their single respective hormones (Figure 2A). In fact, as found with the imaging analysis, activation of GR increases the level of ER pBox binding. Repeating these experiments using the 7281 cell line, which was developed for additional over expression of ER-pBox (Figure S2B), also showed no competitive binding behavior (Figure 2B). The lack of competition in these two cell lines again suggests that the GREs are far from saturation.
Figure 2.
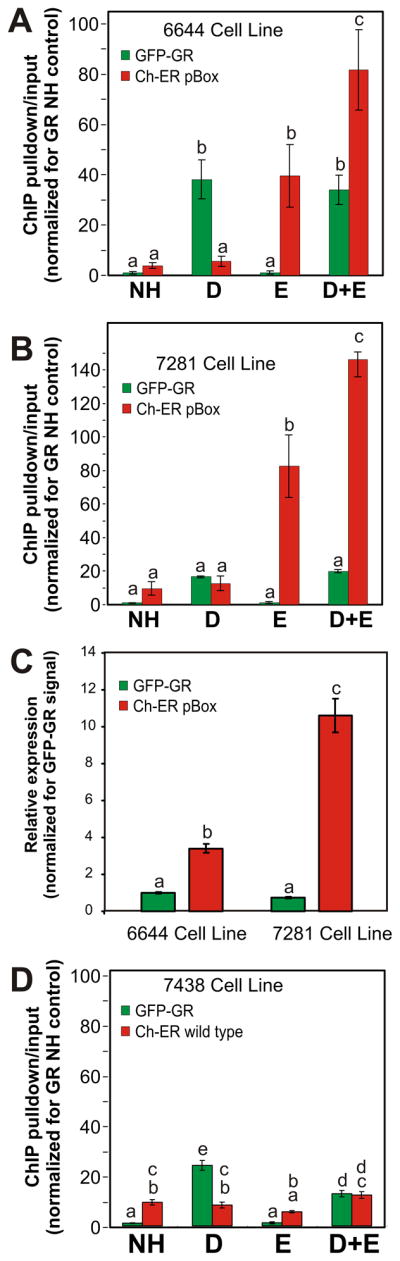
Non-competitive GRE association is observed following increased steroid receptor expression. ChIP assays measure the relative steady-state amount of MMTV B nucleosome GRE chromatin occupied by GR and ER pBox following 30 minute hormone treatment of the (A) 6644 cell line or (B) the 7281 cell line. (C) Parallel quantitative microscopy experiments estimate the relative expression levels of the GFP-GR and Cherry-ER pBox in these two cell lines. (D) ChIP assays measure the relative steady-state amount of MMTV B nucleosome GRE chromatin bound by GR and wild type-ER in the 7438 cell line following 30 minutes of hormone treatment. Bars represent the mean of biological repeats, error bars denote SEM, and markers “a,b,c” indicate homogenous statistical subgroups of conditions. Each subset is considered to be statistically different because it consists of measurements with HSD p-values < 0.05 versus the values in another defined subset. See also Supplemental Figures 1 and 2.
We measured the fluorescence intensity of GFP and Cherry proteins using a single dual tagged fusion protein (data not shown), and employed this information to estimate the relative expression of the GFP-GR and Ch-ER in the 6644 cell line and 7281 cell line based on nuclear fluorescence intensity. Since these two cell lines respectively express over 3× and 10× fold more Ch-ER pBox compared to GFP-GR (Figure 2C), and GFP-GR is expressed at 2–5× fold endogenous GR levels (Figure S2A) it is unlikely that low receptor abundance is the cause of non-competitive behavior. Thus, ER pBox exhibits total steady-state binding that is similar to GR binding levels measured by imaging and biochemical assays, despite expression of ER pBox at much higher levels compared to GR (Figure 1B,C, Figure 2A,C). Combined with the measurements showing that increased expression causes increased steady-state binding of ER pBox to the GRE (compare Figures 2A and 2B), these results indicate that ER-box protein may less efficiently recruit cofactors that are required for efficient binding to GREs in the context of chromatin. Providing further support for this model, ER pBox shows a reduced ability to remodel the MMTV GRE when compared to GR (Figure S4A and B). As receptors bind alternately to the RE over time, GR could recruit cofactors to efficiently bind and open the local chromatin structure. Following the transient binding and release of GR, ER pBox could then interact more efficiently with the more accessible chromatin state at the GRE. Consistent with this model, concurrent treatment with Dex and E2 significantly increased the binding of ER pBox to the MMTV GRE (Figure 1C [p-value = 4.8×10−13], Figure 2A [p-value = 7.6×10−3], 2B [p-value = 6.2×10−5]).
Reduction in GR binding by squelching with wild-type ER
We next repeated the above experiments using the 7438 cell line, which expresses Ch-ER wild-type (wt) protein, GFP-GR, and contains the MMTV array integrated in the same genomic location as 6644 cells. The Ch-ER wt protein differs from Ch-ER pBox protein by only three amino acids that are located in the proximal zinc finger of the DNA binding domain [(Mader et al., 1989) Figure S2C, D]. Because of these three amino acids, the ER wt protein binds to a consensus estrogen RE (ERE) that is distinct from the consensus GRE (Figure S2E). As expected, the Ch-ER wt protein does not interact with the MMTV promoter in an estrogen-dependent manner when measured by ChIP or a steady-state imaging assay (compare Figures 2A,B with Figure 2D; also see Figure S2I). Comparison of fluorescence intensities between the 6644 and 7438 cell lines indicates that expression levels of GR/pBox ER and GR/wt ER are nearly identical in the two cell lines (data not shown). Thus, the levels of steady-state binding we measure for the ER pBox protein are due to specific ER pBox interactions with the GRE, not secondary protein-protein interactions between the two receptor complexes.
When cells are simultaneously treated with both hormones, we detect a significant decrease in Dex-dependent GR binding to the GRE (Figure 2D [p-value = 2.4×10−6]) and Dex-dependent activation of the MMTV promoter (Figure S2J [p-value < 1×10−3]). These results are consistent with the previously reported sequestration of cofactors by heterologous steroid receptors (Kraus et al., 1995; Meyer et al., 1989), which suggest the decrease in GR binding is unlikely to involve direct interactions of ER wt with the MMTV promoter. Importantly, the decrease in GR binding after simultaneous treatment with Dex and E2 is a technical control, demonstrating that both imaging and biochemical methods can detect decreases in steady-state receptor/GRE binding (Figure 2D, and Figure S2H).
Our control experiments using Ch-ER wt strongly suggest that protein-protein interactions are an unlikely mechanism in targeting ER pBox to GREs. We sought an independent strategy to confirm this. We performed a series of time-resolved “pulse-chase” treatments using combinations of E2 and corticosterone (Ct), the natural glucocorticoid ligand in rodents. Ct can be rapidly washed out of cells, causing a rapid loss of GR binding to GREs and a sharp decrease in GR-dependent transcription (Stavreva et al., 2009). Following a concurrent 30 min pretreatment with Ct and E2, maintaining dual hormone treatment for an additional 30 min (or 15 min, data not shown) resulted in a significant decrease of GR from GREs at the MMTV array (histograms 2 vs 6, Figure 3A [p-value = 8.9×10−3]) while ER pBox significantly increased association with GREs at the array (histograms 2 vs 6 in Figure 3B [p-value = 7.0×10−9]). The opposing trends of GR and ER pBox binding during the time course further validates chromatin remodeling as the likely mechanism behind assisted loading. The decrease of GR-binding observed from 30 minutes to 60 minutes in presence of Ct plus E2 is also observed in the continued presence of Dex alone, indicating that the decrease in GR binding is not caused by competition from ER pBox. Additional supporting evidence comes from re-ChIP experiments showing that GRE chromatin that is immunoprecipitated with an anti-GR antibody contains undetectable levels of co-associated ER pBox protein (Figure S4C). Further detailed in vitro characterization will be required to resolve the mechanisms by which sequential factor binding leads to increased chromatin accessibility and transcriptional activation.
Figure 3.

Pulse/Chase experiments with sequential hormone treatments. Combinations of corticosterone (cort) or estradiol demonstrate assisted loading of ER pBox protein even when assisted loading is anti-correlated with GR binding levels at later time points. Cells were pre-treated with hormone for 30 minutes, washed with hormone free media, then treated for another 15 or 30 minutes (treatment time line shown in panel C). Following fixation, (A) GFP-GR or (B) Ch-Pbox steady-state binding at the MMTV chromatin array was measured by fluorescence microscopy. Data are shown as the mean of automated measurements from 500–1000 cells per conditions; error bars denote SEM. All manipulations, including hormone addition, cell washing, cell fixation, and microscopic imaging were performed by high-throughput automated methods (see methods section for details).
Assisted loading at multiple endogenous response elements
We have previously characterized global GR binding sites using genome-wide ChIP-seq (John et al., 2011). To explore the saturation-state at these endogenous sites, we performed manual ChIP assays at multiple GREs throughout the genome. Figure 4 presents the findings for a GRE located near the Lcn2 gene and another GRE located near the Slc5a5 gene (Figure 4). As was found in the MMTV experiments, single steroid receptor binding was not strongly reduced under concurrent binding conditions. The slight reduction in GR binding during treatment with Dex+E2 (Figure 4A) was not detected when the experiment was repeated using the 7281 cell line (Data not shown), which expresses additional ER pBox protein, suggesting this trend is due to technical variability in the quantitative ChIP assay. Interestingly, the Lcn2 GRE behaves similarly to the MMTV LTR, where concurrent receptor binding causes increased ER pBox association with the target chromatin (assisted loading). In contrast, assisted loading on the target chromatin does not occur at the GRE in the Slc5a5 locus. This distinct behavior caused us to further investigate the difference between these two genomic loci.
Figure 4.

Steroid receptors interact non-competitively with endogenous GREs. ChIP assays results measure the relative steady-state amount of either (A) Lcn2 gene locus GRE or (B) Slc5a5 gene locus GRE chromatin bound by each receptor following 30 minutes of hormone treatment. Experiments were performed using the 6644 cell line. Genome browser tracks are show the results of genome wide DNase-Seq and GR ChIP-Seq at the region surrounding the (C) Lcn2 gene locus GRE or the (D) Slc5a5 gene locus GRE. The GRE sequences, which were detected by bioinfomatics methods, are shown below the genome browser tracks. The conserved hexameric GRE half-sites are underlined in green. See also Supplemental Figure 3.
We have also performed genome-wide experiments to detect DNaseI hypersensitive (DHS) chromatin regions, which are more accessible to nuclear proteins compared to surrounding regions of chromatin (Li et al., 2007). Interestingly, GR binding sites are highly correlated with DHS regions across the genome (John et al., 2008; John et al., 2011). Some DHS sites, such as the site near the Lcn2 gene (Figure 4C) and the MMTV LTR (data not shown), become accessible only when GR interacts with the colocalized receptor binding site; we refer to this class of DHS elements as de novo sites. Other DHS sites are hypersensitive before Dex treatment (preprogrammed), for example the GRE near the Slc5a5 gene (Figure 4D). These initial results suggests that GR-dependent chromatin remodeling renders the chromatin more accessible during transient GR/GRE interaction and this “open” site is then more frequently bound by ER pBox.
For a given genomic locus, the extent of chromatin accessibility can be represented as the peak of DHS sequence tag density (see Figure S3 for additional examples). A large ratio of DHS peak tag density value in the presence of Dex over the DHS peak tag density value in the absence of Dex indicates GR-dependent reprogramming of the locus via chromatin remodeling, i.e. a de novo event. Considering the examples of GR binding sites shown in Figure 4, the DHS tag density ratio for the Slc5a5 locus is 1 (preprogrammed) and the DHS tag density ratio for Lcn2 locus is 55 (de novo). To generate a single parameter that measures receptor competition or assisted receptor interaction for a given GR binding site, we used the ratio of receptor specific ChIP values in the presence of both hormones over the ChIP values in the presence of the receptor-specific single hormone. If this ChIP ratio is less than 1 it indicates that the specific receptor is being competed off the site, and if the ChIP ratio is greater than 1 it indicates that the specific receptor is assisted in its interactions with the site. We performed this DHS ratio versus ChIP ratio analysis on 15 different genomic GR binding sites in triplicate (see Supplemental Table 1A for loci and values). Data from an additional 26 de novo sites are presented in Supplemental Table 1B.
At genomic sites with a high DHS ratio there is a significant increase in GR-dependent assisted loading of ER pBox compared to the sites with a low DHS ratio (Figure 5A, C [p-value = 5.0×10−3]). The average DHS ratio differs between the two sorted groups of GR binding sites by approximately 49 fold (Figure 5E). MMTV chromatin structure is also extensively remodeled when GR interacts with the B Nucleosome region (Fletcher et al., 2002). As predicted by our previous experiments, GR is not strongly competed away from the individual GR binding sites regardless of their DHS ratio (Figure 5B) or from statistical groups of sites that were sorted by DHS ratio (Figure 5D). Thus, like the GRE in the MMTV promoter, endogenous GR binding sites are not saturated by steady-state receptor interactions. Based on our results, GR-based competition for REs is unlikely to occur at a significant number of genomic sites.
Figure 5.

GR-dependent chromatin remodeling at genomic GREs increases concurrent binding of ER pBox. Endogenous GRE loci were identified and characterized for DNAse hypersensitivity (DHS) by genomic techniques (see methods). The DHS ratio (+Dex/−Dex) represents the degree to which chromatin accessibility at a specific GRE is regulated by GR binding. The ratio of [ChIP measurements in the presence of both Dex and E2]/[ChIP measurements in the presence of a single hormone] provides a metric for receptor competition and assisted receptor loading. The ChIP ratio is plotted for (A) ER pBox binding and (B) GR binding at 15 genomic GREs with the indicated DHS ratios. Each scatter plot point represent the mean of three independent ChIP experiments. The mean (C) ER pBox ChIP ratio or (D) GR ChIP ratio is shown for groups of Genomic GREs that were sorted based on DHS (+Dex/−Dex) ratio. The mean DHS (+Dex/−Dex) ratio is shown for the same sorted groups. Error bars show the SEM within the sorted groups of GRE measurements, n is the number of different GRE loci in each statistical group. (C–E) ANOVA was used to evaluate each pair of measurements, p-values are displayed. See also Supplemental Figure 4 and Supplemental Table 1.
It is interesting to note that there is considerable variation in the degree of assisted ER-pBox loading at different genomic loci (Figure 5A). We have observed that “de novo” remodeling sites are differentially utilized in divergent differentiated cell lines (John et al., 2011; John et al., 2008). This suggests that there are many sub-types of “de novo” sites with distinct properties. These varying properties could be dictated by many factors including: local chromatin structure, covalent modifications of histones/DNA, combinations of bound DNA-binding factors, or recruited coregulatory proteins.
Assisted loading between heterologous transcription factors
We have shown here that the GR-dependent remodeling of chromatin correlates well with GR-assisted loading of ER pBox onto target chromatin. While we interpret “assisted loading” of ER pBox to be a consequence of GR-mediated chromatin remodeling, it remains a formal possibility that pBox binding may be a reflection of protein-protein interactions between GR and ER pBox, or the recruitment of a factor that would be “left behind” after GR has left the site.
Our ongoing studies indicate that GR-dependent assisted loading is not restricted to the ER pBox protein. For example, we have observed GR assisted loading of endogenous AP-1 (Figure S5) at selected sites in the genome. Just as in the ER pBox case, AP-1 binding is associated with GR-dependent chromatin remodeling at the GRE. In agreement with studies regarding transcription factor dynamics (Hager et al., 2009; Becker et al., 2002; McNally et al., 2000), our current results suggest that GR binds its site transiently and induces a chromatin remodeling event (Nagaich et al., 2004). This in turn makes the remodeled region more accessible to other regulatory proteins (Figure 6A). Through this “hit and run” mechanism, dynamic GR interactions allow for the transmission of regulatory information via chromatin to the machinery that regulates transcription.
Figure 6.

Mathematical modeling indicates that dynamic receptor exchange produces low GRE occupancy and prevents receptor competition. Diagram illustrates the dynamic steps of the “Hit and Run Cycle” over the time scale of seconds. (B) Stair plots illustrate a conceptual diagram for receptor(s) randomly binding to a single GRE over time. As the GRE becomes occupied briefly, this is represented as a “step” transition to the bound state event. If binding events are temporally sparse, the two receptors will not compete with each other for the GRE. If the GR binding event causes a remodeling event (indicated by black brackets), and another transcription factor attempts to bind before the chromatin modification is lost, then this results in assisted loading of the second transcription factor. See Supplemental Figure 6 for detailed quantitative simulation of the model. (C) The steady-state GRE occupancy calculated from averaged sets of 1000 single GRE Monte Carlo simulations. Each set of simulations was performed using the “on” probability indicated on the horizontal axis.
Monte Carlo simulation of steady-state saturation
Mathematical modeling was employed to determine the degree of steady-state saturation that accounts for the observed lack of receptor competition (Figure S6A–C). In this case, simplified stochastic receptor binding to a GRE is modeled by Monte Carlo simulations in which random transitions between bound and unbound state occur at a defined time step of 0.1 seconds. A simplified conceptual representation of stochastic binding by two transcription factors to a single RE illustrates how sequential binding prevents observable competition but promotes assisted loading (Figure 6B). In the illustration, each short-lived “step” represents the transient occupancy of the RE by the transcription factor. Binding and unbinding events are probabilistic in nature. However, some binding events lead to chromatin remodeling (Figure 6B, represented by black brackets), which in turn increase the odds that a second factor will bind the RE.
To quantitatively model the short residence time that was previously measured for the GR at the GRE (Becker et al., 2002; McNally et al., 2000), the “off” or “unbinding” probability of GR is set at 0.1 (10% chance of unbinding at each 0.1 second time step). An “on” or “binding” probability that is a much lower value produces a GRE that has relatively long intervals between individual GR binding events and is largely unoccupied over time (Figure S6D, top stair plot). Under these “on”/”off” probability conditions, the population of GREs are less than 10% saturated at a steady-state level (Figure S6D, top panel mean values). If concurrent binding of ER pBox is added to the model (S6D, bottom stair plot), ER pBox binds in a similar way but only reduces the steady-state occupancy of the GR by 7% (compare Figure S6D top and bottom panel mean values). This small degree of competition is unlikely to be measurable by current experimental methods. In contrast, when the “on” probability is sufficiently increased, the dynamically interacting GR nearly saturates the GRE (Figure S6E top stair plot). Because the GREs are almost constantly occupied under these conditions, addition of concurrent ER pBox binding to the model leads to a large amount of competition between the two receptors (Figure S6E bottom panel). Using this method to simulate ER pBox-dependent competition of GR across a wide range of “on” probabilities reveals that the GREs are likely to be less than 10% occupied during GR activation (Figure 6C).
Discussion
There is emerging evidence that the vast majority of regulatory elements in mammalian cells are distributed widely throughout the genome, not in close proximity to target genes. In addition, binding profiles for the transcription factors acting at these elements are known to be highly cell specific. Thus, mechanisms that control cell selective access to these sites are now understood to be a critical feature of transcriptional regulation in higher eucaryotes. The key issue is how access to these sites is initiated and maintained during differentiation or in response to external stimuli.
The local nucleosome structure at distal enhancers is frequently characterized by high sensitivity to nucleolytic attack (so-called hypersensitive sites). Motifs for multiple transcription factors appear to be clustered in enhancer regions, suggesting that one or more of these proteins could serve as “pioneer factors” for initiation of a locally modulated nucleoprotein state (Cirillo et al., 2002). Indeed, specific factors have been implicated in the development of selective enhancer activity (Green and Carroll, 2007) for proteins such as the estrogen receptor.
Essentially all of the information available for protein distributions, and chromatin sates, at enhancer elements has been obtained with techniques (ChIP and DHS), that are by their nature insensitive to dynamic processes. Furthermore, these methodologies provide population signals that are averaged across many cells, potentially masking processes that may be quite complex on the molecular time scale (Hager et al., 2009). We have addressed this issue directly for the glucocorticoid receptor. Previous studies (John et al., 2008) have shown that GR can act both as a pioneer factor, opening local chromatin sites, and as a partner factor, binding at sites with nucleosomes already in transition under the direction of other factors. The studies presented here were initiated to address a simple tenet; if GR action at a given response element moves the enhancer structure from one static state (inaccessible DNA; closed chromatin configuration) to an altered state with a modified nucleosome structure and DNA bound receptor, then increasing concentrations of receptor protein in the cell should compete for this binding state on a simple mass action law. Such a result has in fact been observed for GR binding to nucleosomal structures in vitro (Li and Wrange, 1997).
In contrast to the expected results, we find that the ER-pBox protein, which binds the identical GRE recognition element, will not compete for binding to GRE sites in the MMTV promoter in living cells. Furthermore, under the condition where ER-pBox is activated concurrently and allowed to bind to sites in the promoter array, GR not only will not compete for ER-pBox binding, but actually increases the level of ER-pBox protein associated with the array. These findings are most directly interpreted under the “hit-and-run” model for transcripiton factor action (Hager et al., 2002; McNally et al., 2000). Since the receptor is only briefly resident on its binding site in living cells, the condition of competition under mass action (observed in vitro) cannot hold under the physiologically relevant conditions in living cells. According to our computational binding model, this result is only possible if less than 10% of the chromatin sites are steady-state occupied by receptor over time (see Figure 6). Because both transcription factors dynamically interact with REs on the time scale of seconds, we propose that the lack of saturation is due to the balance of association and dissociation rates (association ≪ dissociation). This low steady-state site occupancy has been predicted by quantitative studies of diverse TFs which act combinatorially to direct drosophila development (Segal et al., 2008). Low occupancy of binding sites due to dynamic and probabilistic TF-chromatin interactions is also consistent with stochastic transcription observed at the single cell level (Kaufmann and van, 2007; Darzacq et al., 2007; Voss et al., 2009; Kim and O’Shea, 2008).
Taken together, these diverse studies argue against a mechanism of highly ordered and sequential binding of transcription factors in a long-lived multi-protein complex, which would be predicted to nearly saturate binding sites in chromatin. Our model does not exclude the near saturation of REs by extremely high intranuclear concentrations of transcription factors, but it is not clear that such levels are ever achieved. We do not observe competition between steroid receptors at levels as high as 10- fold over endogenous concentrations, and we predict that most REs are unsaturated during physiological transcriptional activation. The observation that TF binding sites are largely unsaturated in vivo should be incorporated into current quantitative models that utilize steady-state biochemical data to model transcriptional regulation (Kim and O’Shea, 2008; Granek and Clarke, 2005; Wasson and Hartemink, 2009).
We propose that a critical feature of GR action is the recruitment of remodeling activity that opens the local nucleosome structure. Earlier studies in an in vitro reconstituted chromatin remodeling system (Nagaich et al., 2004) showed that GR recruits the Swi/Snf complex to remodel MMTV nucleosome B, but the receptor itself is ejected from the template. During this process, sites are transiently available for binding by the ER-pBox protein (Figure 6A). We further propose that the lifetime of the “remodeled state” is significantly longer than the actual resident time for receptor on DNA. Thus, GR will assist loading of the ER-pBox protein because of the recruited remodeling activity. A corollary of this proposed mechanism is that ER-pBox is ineffective in recruitment of the relevant remodeling system for the MMTV GRE. Indeed, we have recently described a remarkable site specificity for recruitment of remodeling systems by GR (John et al., 2008). This model is also consistent with work from the Kornberg group (Boeger et al., 2008) on the dynamics of nucleosome exchange at the Pho5 promoter in yeast. These investigators argue that the change in occupancy of regulatory nucleosomes results from a shift in the relative rate of assembly and disassembly, rather than from a static displacement of nucleosome components.
A specific prediction of the assisted loading model is that GR enhancement of ER-pBox binding should correlate directly with its pioneering role in chromatin opening. We therefore examined the effect of GR action on ER-pBox binding at specific binding elements throughout the genome. For those elements where chromatin is already in transition (“pre-programmed”) prior to GR activation, there is no assisted loading effect on ER-pBox binding (Figure 5). In these cases, the remodeled state is already in effect under the direction of unknown factors; thus further recruitment of GR to these sites has little effect. However, for those cases where GR itself initiates the remodeled state (“de novo” sites), there is a dramatic effect on co-binding by ER-pBox. These results are in direct support of the assisted loading model.
The findings discussed here represent a specific case of what is likely a general mechanism. Mobilities for a significant number of transcription factors at site-specific response elements have now been studied, and the vast majority of these studies report rapid exchange characteristics. Furthermore, multiple factor binding motifs have been reported at many enhancer elements, and emerging studies indicate that many collaborating factors can function at a given enhancer element. We suggest that the mechanisms underlying the complex actions of multiple binding proteins in initiating and maintaining the activity of cell-specific enhancer elements are likely to be highly dynamic, as described here for elements involving the glucocorticoid receptor.
Methods
Plasmid construction and generation of inducible cell lines
The coding sequence of human estrogen receptor with the three amino acid pBox mutations (from pHE82, (Mader et al., 1989) fused to the GFP coding sequence in the Clonetech EGFP-C1 plasmid backbone was a gift of Paul Yen. Using BspE I and Mfe I restriction enzymes, the ER pBox coding sequence was cloned in frame with mCherry in a shuttle vector, which was described previously (pCh-C1, (Voss et al., 2009). Using Age I and Mfe I restriction enzymes, the mCh-ER pBox coding sequence was then transferred to a tet-inducible retroviral vector (pRev-TRE link, (Voss et al., 2009), producing the pRev-TRE-Link ER pBox vector. The nucleotides containing the pBox mutations were replaced with corresponding sequence from the GFP-ER wt plasmid by restricting with Not I and Mfe I to generate the prev-TRE-Link ERwt plasmid.
Retroviral particles were produced in phoenix cells by standard methods. Mouse mammary carcinoma 3617 cells, which contain the integrated MMTV-reporter gene array and a tet-inducible GFP-GR expression vector (Walker et al., 1999) were infected with retroviral particles encoding tet-regulated Ch-ER pBox or Ch-ER wt to produce the 6644 cell line or 7438 cell line, respectively. Cells with integrated retrovirus were selected with Hygromycin for at least 14 days prior to the start cell line characterization. For maintenance, 6644, and 7438 cells were cultured with DMEM supplemented with 10% fetal calf serum and 5 μg/ml tetracyclin to repress expression of the tet-regulated fusion proteins (Walker et al., 1999). In the presence of tetracycline, there was minimal GFP and Cherry signal detectable by fluorescence microscopy in any of the cell lines (data not shown).
RNA fluorescence in situ hybridization (FISH) and epifluorescence microscopy
18 hours prior to RNA FISH experiments, 6644, and 7438 cells were plated on number 1 German 22 mm x 22 mm square coverslips in DMEM supplemented with 10% charcoal-stripped calf serum. Tetracycline was omitted from the medium to induce expression of the fluorescent fusion proteins. Cells on coverslips were treated with no homone medium, 100 nM Dex, 100 nM E2, or a combination of 100 nM Dex and 100 nM E2 for 0.5 hours then immediately processed for RNA FISH to detect the MMTV controlled reporter gene as previously described (Voss et al., 2009). 3D fluorescent image stacks of randomly selected fields were acquired using a Deltavision digital deconvolution microscope exactly as previously described (Voss et al., 2009).
Automated image analysis
Digitally deconvolved RNA FISH/Fluorescent fusion protein images were subjected to two different automated analysis algorithms. Both of these automated methods were developed and implemented using Matlab software and image processing and statistics toolboxes. The first algorithm, which depends on the RNA FISH signal, was executed exactly as previously described (Voss et al., 2009). Briefly, the algorithm measures the maximal GFP or RFP signal that colocalizes with the RNA FISH signal region of interest and also measures the nucleoplasmic GFP or RFP mean intensity. The ratio of the FP intensity at the RNA FISH signal over the nucleoplasmic signal, which we term the loading ratio, is a measure of steady-state GFP-GR, Ch-ER pBox, or Ch-ER wt enrichment at the MMTV array in each single cell. The second algorithm measures GFP-GR, Ch-ER pBox, or Ch-ER enrichment at the MMTV array independently of the RNA FISH signal ROI. The FP-images are first processed with both linear and anisotropic filters to reduce fine scale noise. Then edge detection based methods are used to segment all subnuclear structures into ROIs. Measurements of intensity, area, etc are taken for all subnuclear ROIs. A manually selected test set containing approximately 100× GFP-GR “strong array ROIs” was established based on subnuclear morphology of the GFP-GR signal in each nucleus. Unambiguous selection of manual “strong array ROIs” was aided by also confirming colocalization with the RNA FISH signal in each nucleus. Statistical analysis of the “strong array ROIs” and all other “non-array” ROIs in the test set identifies specific measurement parameters which can identify only the “strong arrays”. These specific parameter values are then applied to the experimental measurements of all subnuclear ROIs. Strength of FP-receptor/MMTV array interactions in the cell population are reported as the fraction of cells with detectable FP-Receptor/MMTV array binding.
High-throughput microscopy methods for time course experiments
Cultures of 6644 cells were seeded in 96-well thin glass bottom imaging plates (Matrical Biosciences) and grown for 18 hours - tetracycline to induce expression of FP-fusion proteins. A separate imaging plate was prepared for each time point. Using the multi-dispense head of a Janus automated liquid handling robot (Perkin Elmer Health Sciences), all hormone pre-treatment media were added simultaneously to the specific wells of the imaging plate. The plates were then incubated for 30 minutes at 37C with 5% CO2. A BioTek EL 406 automated plate washer then rapidly washed the plates 6× (total time less than 2 min) with hormone free media. The Janus then again simultaneously added all indicated treatment media to the specific wells of the plate. The plates were returned to the incubator for the indicated time, and were then fixed with 4% formaldehyde/PBS from an automated Thermo MultiDrop dispenser. To ensure that the washout was complete, RNA FISH was performed to detect transcript from the MMTV chromatin array reporter gene (data not shown). Following staining of the nuclei with Dapi, the wells of the plates were imaged using a 40× water immersion objective (resultant image pixel size = 320 nm) and an Opera high-throughput spinning-disk confocal microscopy system (Perkin Elmer Health Sciences). Multiple image fields in each micro-well were blindly selected and z-stack images of the entire cell volume were captured (9 optical sections at 1 micron spacing). Images were automatically analyzed using custom Matlab algorithms that identify the GFP-GR or Ch-ER pBox proteins concentrating at the MMTV array structure as described in the preceding section.
Chromatin immunoprecipitation (ChIP) assays
Chromatin immunoprecipitations were performed essentially as described in standard protocols (Upstate), with some optimization (John et al, 2008). Briefly, cells were treated for 30 min with 10% CSS medium supplemented with either vehicle, 100 nM dexamethasone, 100 nM Estradiol, or a combination of the two hormones. Incubation with 1% formaldehyde for 10 minutes at 37 °C fixed the cells. Subsequent, addition of 150 mM glycine quenched the cross-linking reaction for 10 min. Input material for each immunoprecipitation sample contained 100 ug of soluble, sonicated chromatin. Cross-linked Protein-DNA complexes were precipitated with an anti-GR antibody cocktail (5 μg of PA1-511A antibody, ABR, 5 ug of MA1-510 antibody, ABR and 1 ug of sc-1004, Santa Cruz) or an anti-ER antibody cocktail (1 ug of sc-543, Santa Cruz and 2 ug of ms-315-P1ABX, Labvision/Thermo Scientific). DNA isolates from immunoprecipitates were used as templates for real-time quantitative PCR amplification. All ChIP experiments were performed at least three times.
Meta-Analysis of genome-wide DNaseI hypersensitivity data and genome-wide ChIP-Seq data
We utilized genome-wide profiles of GR ChIP and DNaseI hypersensitivity in the parental cell line 3134 that we characterized elsewhere (John et al., 2011). Briefly, two biological replicates of DHS samples from cells either unstimulated or stimulated with Dex for 1 hour were subjected to deep sequencing by Solexa/Illumina Genome Analyzer. After confirming replicate reproducibility, sequence tags from duplicates were pooled to generate tag density profiles for Dex-treated and untreated samples. For GR ChIP-seq, triplicate samples were sequenced and the highest quality/resolution dataset was chosen to generate tag density profile. DHS sites were identified as hotspots of DNaseI sensitivity by an algorithm that accounts for local variabilities due to mappability and other biases (John et al., 2011). GR ChIP hotspots were similarly identified after DNA input adjustment, and more narrowly defined 150 bp-width peaks within hotspots were obtained by a specific algorithm (John et al., 2011). Two subsets of GR ChIP peaks were retrieved based on whether the chromatin accessibility was dramatically increased or unchanged after Dex treatment, as measured by DHS tag density. We selected a small number of GR binding sites by requiring a reproducibly strong GR tag density signal and a high scoring GRE sequence (position p value < 0.0001 from a genome scan by MAST, using a de novo discovered GRE position weight matrix from a MEME analysis on GR ChIP peaks [see (John et al., 2011)]. Combining this information allowed us to identify two distinct classes of GR binding sites. The first class exhibits a high level of DNase accessibility before GR binding and during GR binding. The second class of sites is resistant to DNAse cleavage before GR binding but becomes much more accessible after GR binding. There were relatively few robust examples of the first class, and top five sites on the list were selected for manual ChIP analysis. For the second class, the top 10 sites were selected for manual ChIP analysis.
Computer modeling of RE occupancy
A kinetic Monte Carlo model (Burghaus et al., 2006) was implemented in the Matlab computing environment to simulate the occupancy of REs in the presence of a single RE targeted TF, or two separate RE targeted TFs. The GRE in this simplified model is considered to be in one of two states, unoccupied or occupied by a transcription factor. At each discrete time step, any GRE in an unoccupied state randomly transitions into an occupied state based on an association or binding probability that is defined by the model. In this scheme, any GRE in the occupied state randomly changes to the unbound state based on the dissociation or unbinding probability per time step. The experimentally measured dynamic interactions are temporally over-sampled in the simulation by setting the discrete time step to 100 msec. For each RE the occupancy state was tracked for 1000× time steps (Figure S6B). Graphical examination revealed that the RE reached steady state by 400× time steps for all tested association/dissociation probabilities (data not shown). The subsequent 600× time steps were averaged to calculate the steady-state mean occupancy and other mean parameters over a simulated 60 second time interval. To model a population of REs in many cells the kinetic simulation was run 1000× (Figure S6C). When both TFs were simultaneously binding to the RE in the model, the two TFs were given equal probability (0.5 probability each) to bind first to any unoccupied RE. Modeling this equal binding probability characteristic (which takes differences in expression levels into account) was consistent with the similar steady-state GRE occupancy observed for GR and ER pBox binding in imaging and ChIP experiments in 6644 cells (Figure 1, Figure 2, Figure 4).
Western blots
Cultures of 3617 cells were grown without tetracycline, trypsinized, counted, and lysed in SDS-PAGE sample buffer at 95C for 5 min. The final concentration of cells in the lysis buffer was 10,000 cells per ul. The lysates (10 ul) were separated by electrophoresis on a 3–8% TAE Novex gel. Following transfer to nitrocellulose membranes, the Western blots were stained with Poneau S stain and scanned to evaluate the consistency of protein loading, electrophoresis, and transfer. The primary anti-GR antibody (PA1-511A,1 Affinity Bioreagents/Thermo Scientific) or anti-ER antibody (ms-315-P1ABX, Labvision/Thermo Scientific) was diluted 1:1000 in TBST, 5% milk and incubated with the membranes overnight at 4C. Following washing, the membranes were probed with HRP conjugated secondary antibodies. After further washing, the membranes were visualized with Super Signal Pico detection reagent (Pierce) and images were captured on x-ray film.
Statistical analysis
Data were obtained from at least three biological repeats. Statistical analysis of measurements from whole cultures (biochemical assays) or individual cells (microscopy assays) was performed using SPSS16 software. In brief, means and standard errors were calculated, and then significant differences were determined by analysis of variance (ANOVA) combined with the Tukey s HSD multiple comparison post hoc test (alpha set at 0.05 to define homogenous subsets of conditions within a given assay). Therefore, each subset is considered to be statistically different because it consists of measurements with HSD p-values < 0.05 versus the values in another defined subset.
Supplementary Material
Acknowledgments
The authors thank Anindya Indrawan for general technical support of these experiments. Tatiana Karpova, manager of the NCI Fluorescence Imaging Core Facility), provided expert assistance for epifluorescence microscopy experiments. The resources of the NCI High-Throughput Imaging Facility (HiTIF) were also essential for the rapid completion of this study. This research was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research, and by the D.O.D. s U.S. Congressionally Directed Medical Research Programs Grant W81XWH-06-1-0776.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Becker M, Baumann CT, John S, Walker D, Vigneron M, McNally JG, Hager GL. Dynamic behavior of transcription factors on a natural promoter in living cells. EMBO Rep. 2002;3:1188–1194. doi: 10.1093/embo-reports/kvf244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boeger H, Griesenbeck J, Kornberg RD. Nucleosome retention and the stochastic nature of promoter chromatin remodeling for transcription. Cell. 2008;133:716–726. doi: 10.1016/j.cell.2008.02.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burghaus U, Stephan J, Vattuone L, Rogowska JM. A Practical Guide to Kinetic Monte Carlo Simulations and Classical Molecular Dynamics Simulations: An Example Book. Hauppauge NY: Nova Science Publishers; 2006. pp. 1–194. [Google Scholar]
- Cann JR. Phenomenological theory of gel electrophoresis of protein-nucleic acid complexes. J Biol Chem. 1989;264:17032–17040. [PubMed] [Google Scholar]
- Cirillo LA, Lin FR, Cuesta I, Friedman D, Jarnik M, Zaret KS. Opening of compacted chromatin by early developmental transcription factors HNF3 (FoxA) and GATA-4. Mol Cell. 2002;9:279–289. doi: 10.1016/s1097-2765(02)00459-8. [DOI] [PubMed] [Google Scholar]
- Darzacq X, Shav-Tal Y, de Turris V, Brody Y, Shenoy SM, Phair RD, Singer RH. In vivo dynamics of RNA polymerase II transcription. Nat Struct Mol Biol. 2007;14:796–806. doi: 10.1038/nsmb1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingber DE, Mancini MA. Estrogen-receptor-alpha exchange and chromatin dynamics are ligand- and domain-dependent. J Cell Sci. 2006;119:4101–4116. doi: 10.1242/jcs.03161. [DOI] [PubMed] [Google Scholar]
- Fletcher TM, Xiao N, Mautino G, Baumann CT, Wolford RG, Warren BS, Hager GL. ATP-dependent mobilization of the glucocorticoid receptor during chromatin remodeling. Mol Cell Biol. 2002;22:3255–3263. doi: 10.1128/MCB.22.10.3255-3263.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granek JA, Clarke ND. Explicit equilibrium modeling of transcription-factor binding and gene regulation. Genome Biol. 2005;6:R87. doi: 10.1186/gb-2005-6-10-r87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green KA, Carroll JS. Oestrogen-receptor-mediated transcription and the influence of co-factors and chromatin state. Nat Rev Cancer. 2007;7:713–722. doi: 10.1038/nrc2211. [DOI] [PubMed] [Google Scholar]
- Hager GL, Elbi CC, Becker M. Protein dynamics in the nuclear compartment. Curr Opin Genet Dev. 2002;12:137–141. doi: 10.1016/s0959-437x(02)00278-2. [DOI] [PubMed] [Google Scholar]
- Hager GL, McNally JG, Misteli T. Transcription dynamics. Mol Cell. 2009;35:741–753. doi: 10.1016/j.molcel.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He HH, Meyer CA, Shin H, Bailey ST, Wei G, Wang Q, Zhang Y, Xu K, Ni M, Lupien M, Mieczkowski P, Lieb JD, Zhao K, Brown M, Liu XS. Nucleosome dynamics define transcriptional enhancers. Nat Genet. 2010;42:343–347. doi: 10.1038/ng.545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesselberth JR, Zhang Z, Sabo PJ, Chen X, Sandstrom R, Reynolds AP, Thurman RE, Neph S, Kuehn MS, Noble WS, Fields S, Stamatoyannopoulos JA. Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat Methods. 2009;6:283–289. doi: 10.1038/nmeth.1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S, Sabo PJ, Johnson TA, Sung MH, Biddie SC, Lightman SL, Voss TC, Davis SR, Meltzer PS, Stamatoyannopoulos JA, Hager GL. Interaction of the glucocorticoid receptor with the global chromatin landscape. Mol Cell. 2008;29:611–624. doi: 10.1016/j.molcel.2008.02.010. [DOI] [PubMed] [Google Scholar]
- John S, Thurman RE, Sabo PJ, Sung MH, Biddie SC, Johnson TA, Hager GL, Stamatoyannopoulos JA. Chromatin accessibility dictates de novo regulatory factor binding. Nat Genet. 2011;43:264–268. doi: 10.1038/ng.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. doi: 10.1126/science.1141319. [DOI] [PubMed] [Google Scholar]
- Jothi R, Cuddapah S, Barski A, Cui K, Zhao K. Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucleic Acids Res. 2008;36:5221–5231. doi: 10.1093/nar/gkn488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufmann BB, van Oudenaarden A. Stochastic gene expression: from single molecules to the proteome. Curr Opin Genet Dev. 2007;17:107–112. doi: 10.1016/j.gde.2007.02.007. [DOI] [PubMed] [Google Scholar]
- Kim HD, O’Shea EK. A quantitative model of transcription factor-activated gene expression. Nat Struct Mol Biol. 2008;15:1192–1198. doi: 10.1038/nsmb.1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraus WL, McInerney EM, Katzenellenbogen BS. Ligand-dependent, transcriptionally productive association of the amino- and carboxyl-terminal regions of a steroid hormone nuclear receptor. Proc Natl Acad Sci USA. 1995;92:12314–12318. doi: 10.1073/pnas.92.26.12314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krum SA, Miranda-Carboni GA, Lupien M, Eeckhoute J, Carroll JS, Brown M. Unique ERalpha cistromes control cell type-specific gene regulation. Mol Endocrinol. 2008;22:2393–2406. doi: 10.1210/me.2008-0100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- Li Q, Wrange O. Assays for transcription factors access to nucleosomal DNA. Methods. 1997;12:96–104. doi: 10.1006/meth.1997.0451. [DOI] [PubMed] [Google Scholar]
- Mader S, Kumar V, de Verneuil H, Chambon P. Three amino acids of the oestrogen receptor are essential to its ability to distinguish an oestrogen from a glucocorticoid- responsive element. Nature. 1989;338:271–274. doi: 10.1038/338271a0. [DOI] [PubMed] [Google Scholar]
- McNally JG, Mueller WG, Walker D, Wolford RG, Hager GL. The glucocorticoid receptor: Rapid exchange with regulatory sites in living cells. Science. 2000;287:1262–1265. doi: 10.1126/science.287.5456.1262. [DOI] [PubMed] [Google Scholar]
- Meyer ME, Gronemeyer H, Turcotte B, Bocquel MT, Tasset D, Chambon P. Steroid hormone receptors compete for factors that mediate their enhancer function. Cell. 1989;57:433–442. doi: 10.1016/0092-8674(89)90918-5. [DOI] [PubMed] [Google Scholar]
- Nagaich AK, Walker DA, Wolford RG, Hager GL. Rapid periodic binding and displacement of the glucocorticoid receptor during chromatin remodeling. Mol Cell. 2004;14:163–174. doi: 10.1016/s1097-2765(04)00178-9. [DOI] [PubMed] [Google Scholar]
- Nalley K, Johnston SA, Kodadek T. Proteolytic turnover of the Gal4 transcription factor is not required for function in vivo. Nature. 2006;442:1054–1057. doi: 10.1038/nature05067. [DOI] [PubMed] [Google Scholar]
- Ostrowski MC, Huang AL, Kessel M, Wolford RG, Hager GL. Modulation of enhancer activity by the hormone responsive regulatory element from mouse mammary tumor virus. EMBO J. 1984;3:1891–1899. doi: 10.1002/j.1460-2075.1984.tb02064.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payvar F, DeFranco DB, Firestone GL, Edgar B, Wrange O, Okret S, Gustafsson JA, Yamamoto KR. Sequence-specific binding of glucocorticoid receptor to MTV DNA at sites within and upstream of the transcribed region. Cell. 1983;35(2 Pt 1):381–392. doi: 10.1016/0092-8674(83)90171-x. [DOI] [PubMed] [Google Scholar]
- Robertson G, Hirst M, Bainbridge M, Bilenky M, Zhao Y, Zeng T, Euskirchen G, Bernier B, Varhol R, Delaney A, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat Methods. 2007;4:651–657. doi: 10.1038/nmeth1068. [DOI] [PubMed] [Google Scholar]
- Sabo PJ, Kuehn MS, Thurman R, Johnson BE, Johnson EM, Cao H, Yu M, Rosenzweig E, Goldy J, Haydock A, et al. Genome-scale mapping of DNase I sensitivity in vivo using tiling DNA microarrays. Nat Methods. 2006;3:511–518. doi: 10.1038/nmeth890. [DOI] [PubMed] [Google Scholar]
- Segal E, Raveh-Sadka T, Schroeder M, Unnerstall U, Gaul U. Predicting expression patterns from regulatory sequence in Drosophila segmentation. Nature. 2008;451:535–540. doi: 10.1038/nature06496. [DOI] [PubMed] [Google Scholar]
- Stavreva DA, Wiench M, John S, Conway-Campbell BL, McKenna MA, Pooley JR, Johnson TA, Voss TC, Lightman SL, Hager GL. Ultradian hormone stimulation induces glucocorticoid receptor-mediated pulses of gene transcription. Nat Cell Biol. 2009;11:1093–1102. doi: 10.1038/ncb1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valouev A, Johnson DS, Sundquist A, Medina C, Anton E, Batzoglou S, Myers RM, Sidow A. Genome-wide analysis of transcription factor binding sites based on ChIP-Seq data. Nat Methods. 2008;5:829–834. doi: 10.1038/nmeth.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Werven FJ, van Teeffelen HA, Holstege FC, Timmers HT. Distinct promoter dynamics of the basal transcription factor TBP across the yeast genome. Nat Struct Mol Biol. 2009;16:1043–1048. doi: 10.1038/nsmb.1674. [DOI] [PubMed] [Google Scholar]
- Voss TC, Schiltz RL, Sung MH, Johnson TA, John S, Hager GL. Combinatorial probabilistic chromatin interactions produce transcriptional heterogeneity. J Cell Sci. 2009;122:345–356. doi: 10.1242/jcs.035865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker D, Htun H, Hager GL. Using inducible vectors to study intracellular trafficking of GFP-tagged steroid/nuclear receptors in living cells. Methods (Companion to Methods in Enzymology) 1999;19:386–393. doi: 10.1006/meth.1999.0874. [DOI] [PubMed] [Google Scholar]
- Wasson T, Hartemink AJ. An ensemble model of competitive multi-factor binding of the genome. Genome Res. 2009;19:2101–2112. doi: 10.1101/gr.093450.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.