

Abstract
Mycobacterium tuberculosis (Mtb) and Yersinia pestis (Yp) produce siderophores with scaffolds of nonribosomal peptide-polyketide origin. Compounds with structural similarities to these siderophores were synthesized and evaluated as antimicrobials against Mtb and Yp under iron-limiting conditions mimicking the iron scarcity these pathogens encounter in the host and under standard iron-rich conditions. Several new antimicrobials were identified, including some with increased potency in the iron-limiting condition. Our study illustrates the possibility of screening compound libraries in both iron-rich and iron-limiting conditions to identify antimicrobials that may selectively target iron scarcity-adapted bacteria and highlights the usefulness of building combinatorial libraries of compounds having scaffolds with structural similarities to siderophores to feed into antimicrobial screening programs.
Keywords: nonribosomal peptide, polyketide, siderophore, Mycobacterium tuberculosis, Yersinia pestis, antimicrobial compound, pyrazoline, hydroxyhydrazine
Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis, and Yersinia pestis (Yp), the etiologic agent of plague, are bacterial pathogens with serious impacts on global public health. Multidrug-resistant (MDR) tuberculosis is an emerging pandemic, whereas the more recent emergence of extensively drug-resistant (XDR) tuberculosis poses a new global threat.1 Plague is a re-emerging disease for which the documented occurrence of MDR Yp strains and self-transferable Yp plasmids conferring antibiotic resistance raises concerns about future Plague control.2 These grim scenarios underscore the need for expanding the anti-tuberculosis and anti-plague drug armamentarium.
Many bacteria utilize secreted, small (<1,000 Da) Fe3+-chelating compounds (Kd <10−25 M) called siderophores to scavenge Fe3+ from their microenvironments and transport it into the cell.3 The Mtb siderophore (mycobactin/carboxymycobactin) and the Yp siderophore (yersiniabactin) are based on substituted scaffolds of nonribosomal peptide-polyketide origin (Figure 1).4 Studies in cellular and animal models of infection have established the relevance of the mycobactin/carboxymycobactin and yersiniabactin siderophore systems in these pathogens.5 The siderophores are believed to facilitate iron scavenging inside the host, where free iron is scarce (10−25-10−15 M) and the pathogens experience and must adapt to iron-limiting conditions.6 These observations suggest that the Mtb and Yp siderophore systems represent potential in vivo conditionally essential target candidates for the development of alternative therapeutics against tuberculosis and plague.7
Figure 1.

Structures of M. tuberculosis and Y. pestis siderophores.
We hypothesize that screening compounds with structural features resembling Mtb and Yp siderophores for growth inhibitory activity against these pathogens may lead to the discovery of novel antimicrobial scaffolds. Such novel antimicrobials could illuminate alternative paths to drug development and/or be useful as small-molecule tools to assist in the elucidation of new target candidates for drug development. Compounds with structural features resembling Mtb and Yp siderophores may impair the siderophore systems (e.g., by inhibiting siderophore biosynthesis or transport) and halt bacterial growth in the host’s iron-limiting environments. Alternatively, these compounds might gain access to the intracellular environment using siderophore transport systems and inhibit essential functions unrelated to iron acquisition. Consistent with these views, we have recently demonstrated potent antimicrobial activity against Mtb and Yp for novel diaryl-carbothioamide-pyrazoline derivatives with structural features resembling the hydroxyphenyl-oxazoline/thiazoline-containing half of Mtb and Yp siderophores.8
In an effort to identify additional novel inhibitors of Mtb and Yp growth, we synthesized and evaluated the antimicrobial activity of new 3,5-diaryl-substituted pyrazoline (DAP) derivatives (cmpds 1–22,9a Table 1). In addition, we synthesized and tested the activity of a group of (2E)-2-benzylidene-N-hydroxyhydrazine carbo(ox/thio/oximid)-amide (BHHC) derivatives (cmpds 23–32,9b Table 2) with hydroxyphenyl-cap functionalities resembling that of the siderophores. The compounds were tested for growth inhibitory activity against Mtb and Yp in iron-limiting media, which mimic the iron-scarcity condition that the pathogens encounter in the host, and in standard iron-rich media.10a We also assessed selected compounds for mode of action (bactericidal or bacteriostatic) in iron-limiting media and for cytotoxicity toward mammalian cells.10b
Table 1.
3.5-Diarvl-substituted pyrazoline (DAP) derivatives (1–22)
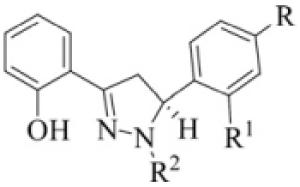 |
|||
|---|---|---|---|
| Compound | R | R’ | R |
| 1 | –H | –OH | –H |
| 2 | –OH | –H | –H |
| 3 | –H | –OH |
|
| 4 | –OH | –H |
|
| 5 | –H | –OH |
|
| 6 | –OH | –H |
|
| 7 | –H | –OH |
|
| 8 | –OH | –H |
|
| 9 | –H | –OH |
|
| 10 | –H | –CH3 |
|
| 11 | –H | –OH |
|
| 12 | –OH | –H |
|
| 13 | –H | –OH |
|
| 14 | –OH | –H |
|
| 15 | –H | –OH |
|
| 16 | –OH | –H |
|
| 17 | –H | –OH |
|
| 18 | –OH | –H |
|
| 19 | –H | –OH |
|
| 20 | –OH | –H |
|
| 21 | –H | –OH |
|
| 22 | –OH | –H |
|
Table 2.
(2E)-2-Benzylidene-N-hydroxyhydrazine carbo(ox/thio/oximid)-amide (BHHC) derivatives (23–32)
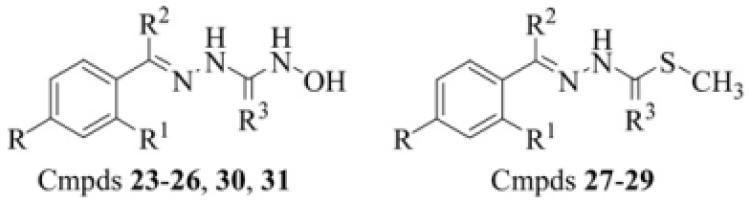 | ||||
|---|---|---|---|---|
| Compound | R | R1 | R2 | R3 |
| 23 | –H | –OH | –H | =O |
| 24 | –OH | –H | –H | =O |
| 25 | –H | –OH | –CH3 | =O |
| 26 | –OH | –H | –CH3 | =O |
| 27 | –H | –OH | –H | =S |
| 28 | –OH | –H | –H | =S |
| 29 | –H | –OH | –CH3 | =S |
| 30 | –H | –OH | –H | –NH |
| 31 | –OH | –H | –H | –NH |
| 32 |
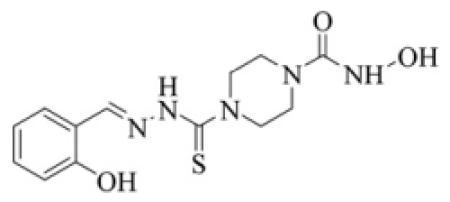
|
|||
Testing against Mtb revealed that 17 compounds (1, 2, 5, 8–15, 24, 27–31) had IC50s and MICs (3–222 μM range, Table 3) within the concentration series tested in the iron-limiting medium, GASTD. Of these 17 compounds, 15 (1, 2, 5, 9-12, 14, 15, 24, 27–31) also had determinable IC50s and MICs (2–132 μM range) in the iron-rich medium, GASTD+Fe. Examination of IC50GASTD+Fe/IC50GASTD and MICGASTD+Fe/MICGASTD ratios revealed that the inhibitors had no noteworthy increased potency in the iron-limiting medium within the concentration series tested. This suggests that interference with iron acquisition, or any other bacterial process differentially required for growth under the iron-limiting condition, is not a property that significantly contributes to the compounds’ antimicrobial activity against Mtb. Interestingly, 14 is 5-fold more potent against Mtb cultured in GASTD+Fe, as judged by MIC values. This phenomenon might suggest that the mechanism(s) of action of 14 against Mtb might be potentiated by an elevated production of cytotoxic hydroxyl radicals originated through increased levels of Fenton reaction in the iron-rich medium. Such a potentiation would be in line with recent findings of Collins and coworkers regarding antibiotic-induced cell death.16 Alternatively, 14 might inhibit an oxidative stress protection function(s) more critically needed in the iron-rich medium. Testing for cytotoxicity at the MIC against Mtb (125 μM) revealed that 14 had no significant cytotoxicity at short cell-cmpd contact time (4 hr), yet cell viability was reduced by 70% relative to untreated controls after prolonged contact time (24 hr) (Supplementary Data, Fig. S1).
Table 3.
Antimicrobial activity against M. tuberculosis
| Compound | IC50a (μM) |
Ratio | MIC90b (μM) |
Ratio | Mode of actionc | ||
|---|---|---|---|---|---|---|---|
| GASTD+Fe | GASTD | GASTD+Fe | GASTD | ||||
| DAP series | |||||||
| 1 | 18 | 25 | 1 | 59 | 66 | 1 | BC (2 × MIC) |
| 2 | 37 | 46 | 1 | 125 | 125 | 1 | BC (I × MIC) |
| 3 | >16 | >31 | nd | >16 | >32 | nd | nd |
| 4 | >8 | >8 | nd | >8 | >8 | nd | nd |
| 5 | 64 | 77 | 1 | 104 | 208 | 0.5 | BC (2 × MIC) |
| 6 | >32 | >63 | nd | >63 | >63 | nd | nd |
| 7 | >32 | 54 | nd | >31 | >63 | nd | nd |
| 8 | 40 | 42 | 1 | >31 | 125 | nd | BC (1 × MIC) |
| 9 | 50 | 53 | 1 | 125 | 125 | 1 | BC (2 × MIC) |
| 10 | 7 | 7 | 1 | 16 | 16 | 1 | BC (2 × MIC) |
| 11 | 23 | 44 | 1 | 66 | 66 | 1 | BC (2 × MIC)d |
| 12 | 26 | 31 | 1 | 76 | 125 | 1 | BC (2 × MIC)d |
| 13 | 32 | 37 | 1 | >125 | 125 | >l | BC (2 × MIC) |
| 14 | 22 | 47 | 0.5 | 31 | 125 | 0.2 | BC (2 × MIC) |
| 15 | 8 | 8 | 1 | 16 | 16 | 1 | BC (2 × MIC) |
| 16 | >4 | >8 | nd | >4 | >8 | nd | nd |
| 17 | 98 | 84 | 1 | >500 | >500 | nd | nd |
| 18 | >16 | >63 | nd | >63 | >63 | nd | nd |
| 19 | >16 | >63 | nd | >31 | >63 | nd | nd |
| 20 | >31 | >63 | nd | >31 | >63 | nd | nd |
| 21 | >31 | >63 | nd | >31 | >63 | nd | nd |
| 22 | >31 | >63 | nd | >31 | >63 | nd | nd |
| BHHC series | |||||||
| 23 | >16 | >16 | nd | >16 | >16 | nd | nd |
| 24 | 40 | 97 | 0.4 | 97 | 146 | 1 | BC (2 × MIC) |
| 25 | >8 | >8 | nd | >8 | >8 | nd | nd |
| 26 | >500 | >500 | nd | >500 | >500 | nd | nd |
| 27 | 2 | 3 | 1 | 6 | 7 | 1 | BC (4 × MIC) |
| 28 | 33 | 31 | 1 | 59 | 59 | 1 | BC (2 × MIC) |
| 29 | 4 | 4 | 1 | 9 | 10 | 1 | BC (2 × MIC) |
| 30 | 15 | 11 | 1 | 26 | 222 | 1 | BC (2 × MIC) |
| 31 | 43 | 43 | 1 | 132 | 222 | 1 | BC (2 × MIC) |
| 32 | >8 | 20 | nd | >8 | >31 | nd | nd |
| Isoniazid | 0.09 | 0.07 | 1 | 0.2 | 0.2 | 1 | BC (2 × MIC) |
IC50 values were calculated from sigmoidal curves fitted to triplicate sets of dose–response data.
MIC90 values are means of triplicates. Ratio, GASTD+Fe/GASTD. All values >l, <l, >0.5, and ≤0.5 were rounded to the nearest whole number, to 1, and to one significant digit, respectively.
Mode of action was evaluated in GASTD in duplicate. The concentration at which each compound was tested for mode of action is indicated between parentheses.
Some bactericidal activity detected, yet below the 99% killing criterion set for defining bactericidal mode of action. BS, bacteriostatic; BC, bactericidal; nd, not determined.
Among the active DAP derivatives, 10 and 15 were the most potent against Mtb (IC50 = 7–4 μM, MIC = 16 μM, Table 3). Of these two cmpds, only 10 displayed significant cytotoxicity at the MIC against Mtb (16 μM). Cmpd 10 had a modest impact on cell viability, which was reduced only by 24% after 24 hr of cell-cmpd contact (Supplementary Data, Fig. S1). Encouragingly, these and most other inhibitors in the DAP derivatives series examined for mode of action against Mtb were bactericidal (>99% killing relative to inoculum) at concentrations of 1-2 × MICGASTD (Table 1). This finding is significant since bactericidal activity is a desirable property in any early lead compound evaluated for antibacterial drug development programs. It is worth noting that the only two compounds (11 and 12) defined as bacteriostatic in Table 1 showed significant bactericidal activity, yet below the 99% killing criterion set in this study for defining bactericidal mode of action. Among the compounds of the BHHC derivatives series with defined IC50 and MIC values, 27 and 29 were the most active against Mtb (IC50 = 2–4 μM, MIC = 6–10 μM, Table 3). These two compounds displayed no significant cytotoxicity in mammalian cells at their respective MIC values determined against Mtb (Supplementary Data, Fig. S1). Gratifyingly, 27, 29 and other active cmpds in this series displayed bactericidal mode of action against Mtb at concentrations of 1.7-4 × MICGASTD (Table 3).
Testing against Yp revealed that 14 cmpds (1, 2, 7–9, 11, 13, 14, 19, 27–29, 31, 32; Table 1) reached IC50s (0.04–181 μM range, Table 4) within the concentration series tested in the iron-limiting medium, PMHD. Nine of these compounds (1, 11, 13, 14, 27–29, 31, 32) also reached MICs (0.2–388 μM range) in PMHD. Only 29 and 30 had determinable activity in the iron-rich medium, PMHD+Fe (29: IC50 = 156 μM, MIC = 233 μM; 30: IC50 = 305 μM). Interestingly, examination of the IC50PMHD+Fe/IC50PMHD ratios revealed that a number of inhibitors (7, 11, 13, 14, 19, 27, 29, 31, 32) with increased potency (>3-fold) against Yp cultured under iron scarcity. Cmpd 32, with >100-fold and >20-fold higher potency in PMHD based on IC50 and MIC values, respectively, stood out in this group. The higher potency of these compounds in the iron-limiting medium raises the possibility that interference with an iron acquisition function, or other function more critically required for growth under the iron-limiting condition, is a property that significantly contributes to the compounds’ antimicrobial activity against Yp. One of the possible mechanisms of action of these compounds could be related to iron-binding properties. An iron-binding ability strong enough to outcompete the powerful iron chelating capacity of the bacterial siderophore could lead to sequestration of the traces of iron in the iron-limiting medium, thus reducing further iron bioavailability and producing a stronger antimicrobial activity under the iron-scarcity condition. Alternatively, it is possible that these compounds gain intracellular access using the iron-scarcity-unregulated yersiniabactin’s uptake system,5e therefore reducing IC50 and MIC values in PMHD.
Table 4.
Antimicrobial activity against Y. pestis
| Compound | IC50a (μM) |
Ratio | MIC90b (μM) |
Ratio | Mode of actionc | ||
|---|---|---|---|---|---|---|---|
| PMHD+Fe | PMHD | PMHD+Fe | PMHD | ||||
| DAP series | |||||||
| 1 | >8 | 9 | nd | >8 | 31 | nd | BC (1 × MIC) |
| 2 | >31 | 20 | >2 | >31 | >31 | nd | nd |
| 3 | >31 | >31 | nd | >31 | >31 | nd | nd |
| 4 | >16 | >16 | nd | >16 | >16 | nd | nd |
| 5 | >250 | >250 | nd | >250 | >250 | nd | ml |
| 6 | >31 | >31 | nd | >31 | >31 | nd | nd |
| 7 | >31 | 8 | >4 | >3l | >31 | nd | nd |
| 8 | >31 | 13 | >2 | >31 | >31 | nd | nd |
| 9 | >63 | 181 | nd | >63 | >250 | nd | nd |
| 10 | >31 | >62 | nd | >31 | >63 | nd | nd |
| 11 | >63 | 7 | >9 | >63 | 42 | >l | BC (1 × MIC) |
| 12 | >63 | >125 | nd | >63 | >125 | nd | nd |
| 13 | >16 | 4 | >4 | >16 | 16 | >1 | BC (2 × MIC) |
| 14 | >63 | 5 | >13 | >63 | 21 | >3 | BC (2 × MIC) |
| 15 | >16 | >16 | nd | >16 | >16 | nd | nd |
| 16 | >8 | >8 | nd | >8 | >8 | nd | nd |
| 17 | >63 | >31 | nd | >63 | >31 | nd | nd |
| 18 | >16 | >31 | nd | >16 | >3l | nd | nd |
| 19 | >31 | 8 | >4 | >31 | >31 | nd | nd |
| 20 | >31 | >16 | nd | >31 | >16 | nd | nd |
| 21 | >16 | >31 | nd | >16 | >31 | nd | nd |
| 22 | >16 | >31 | nd | >16 | >31 | nd | nd |
| BHHC series | |||||||
| 23 | >4 | >16 | nd | >4 | >16 | nd | nd |
| 24 | >250 | >250 | nd | >250 | >250 | nd | nd |
| 25 | >2 | >4 | nd | >2 | >4 | nd | nd |
| 26 | >500 | >500 | nd | >500 | >500 | nd | nd |
| 27 | >59 | 4 | >15 | >59 | 15 | >4 | BC (2 × MIC) |
| 28 | >233 | 137 | >2 | >233 | 388 | nd | BC (1 × MIC) |
| 29 | 156 | 61 | >3 | 233 | 116 | 2 | BC (2 × MIC) |
| 30 | 305 | 70 | 4 | >500 | 250 | >2 | BC (2 × MIC) |
| 31 | >500 | >500 | nd | >500 | >500 | nd | nd |
| 32 | >4 | 0.04 | >100 | >4 | 0.2 | >20 | BC (2 × MIC) |
| Streptomycin | 1 | 1 | 1 | 2 | 1 | 2 | BC (2 × MIC) |
IC50 values were calculated from sigmoidal curves fitted to triplicate sets of dose response data.
MIC90 values are means of triplicates. Ratio, PMHD+Fe/PMHD. All values >1, <l, >0.5, and ≤0.5 were rounded to the nearest whole number, to 1 and to one significant digit, respectively.
Mode of action was evaluated in PMHD in duplicate The concentration at which each compound was tested for mode of action is indicated between parentheses. nd, not determined; US, bacteriostatic; BC, bactericidal.
Compounds 13 (IC50PMHD = 4 μM, MICPMHD = 16 μM) and 14 (IC50PMHD = 5 μM, MICPMHD = 21 μM) were the most potent among the active DAP derivatives with detectable activity against Yp (Table 4). These two compounds displayed no significant cytotoxicity when they were evaluated at their respective MIC values determined against Yp (Supplementary Data, Fig. S1). Testing of these and two other active DAP derivatives (1, 11) for mode of action against Yp revealed that the four compounds were bacteriostatic at concentrations of 1-2 × MICPMHD (Table 4). Among the BHHC derivatives with defined IC50 and MIC values in at least one condition (iron limiting or iron rich), 32 stood out due to its remarkable potency (IC50PMHD = 0.04 μM, MICPMHD = 0.2 μM) against Yp cultured under iron scarcity. Encouragingly, 32 displayed no significant cytotoxicity when evaluated in cytotoxicity assays at the MIC determined against Yp (Supplementary Data, Fig. S1). Notably, the activity of 32 was significantly higher than that of streptomycin (IC50 and MIC ~1 μM), a bactericidal drug used to treat plague and included herein as an anti-Yp activity reference. Five compounds (27-29, 31, 32) of the BHHC derivatives series were tested for mode of action against Yp in PMHD at concentrations of 1-2 × MICPMHD. Under the conditions tested, 29 displayed bactericidal activity, whereas 27, 28, 31 and 32 were bacteriostatic.
In sum, 20 of 32 compounds synthesized and evaluated herein have detectable antimicrobial against Mtb and/or Yp in at least one condition, iron scarcity or iron sufficient. To our knowledge, these are novel scaffolds not previously shown to have antimicrobial properties. Most active compounds identified herein have comparable potency in the low and high iron conditions. This finding suggests that their pharmacological targets are likely to be essential bacterial functions required under both these conditions. In line with our aforementioned hypothesis, however, some of our compounds have higher potency under the iron-limiting condition. Under this condition, bacteria depend on siderophores for efficient iron scavenging and engage an adaptive response to tailor their physiology to iron scarcity, thus exposing novel potential in vivo conditional target candidates.7a Some of these antimicrobials may impair siderophore system functioning as discussed above, a property that would result in bacteriostatic activity conditional to environmental iron scarcity (e.g., as seen with 27 and 32 against Yp). Overall, our study illustrates the possibility of screening compound libraries in both iron-sufficient and iron-limiting conditions to identify antimicrobials that may selectively target iron scarcity-adapted bacteria and highlights the usefulness of building combinatorial libraries of compounds having scaffolds with structural similarities to siderophores to feed antimicrobial screening programs.
Supplementary Material
Acknowledgment
This work was supported by NIH Grant 1R01AI075092-01A1 (LENQ). LENQ acknowledges the endowment support from Carol and Larry Zicklin. We are grateful to the Sophisticated Analytical Instrument Facility (CDRI, Lucknow, India) for providing spectral data.
Footnotes
Supplementary Data Supplementary data associated with this article can be found, in the online version, at doi:
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1. (a).Dye C. Global epidemiology of tuberculosis. Lancet. 2006;367:938. doi: 10.1016/S0140-6736(06)68384-0. [DOI] [PubMed] [Google Scholar]; (b) Gandhi NR, Moll A, Sturm AW, Pawinski R, Govender T, Lalloo U, Zeller K, Andrews J, Friedland G. Extensively drug-resistant tuberculosis as a cause of death in patients co-infected with tuberculosis and HIV in a rural area of South Africa. Lancet. 2006;368:1575. doi: 10.1016/S0140-6736(06)69573-1. [DOI] [PubMed] [Google Scholar]; (c) Zignol M, Hosseini MS, Wright A, Weezenbeek CL, Nunn P, Watt CJ, Williams BG, Dye C. Global incidence of multidrug-resistant tuberculosis. J. Infect. Dis. 2006;194:479. doi: 10.1086/505877. [DOI] [PubMed] [Google Scholar]
- 2. (a).Galimand M, Guiyoule A, Gerbaud G, Rasoamanana B, Chanteau S, Carniel E, Courvalin P. Multidrug resistance in Yersinia pestis mediated by a transferable plasmid. N. Engl. J. Med. 1997;337:677. doi: 10.1056/NEJM199709043371004. [DOI] [PubMed] [Google Scholar]; (b) Guiyoule A, Gerbaud G, Buchrieser C, Galimand M, Rahalison L, Chanteau S, Courvalin P, Carniel E. Transferable plasmid-mediated resistance to streptomycin in a clinical isolate of Yersinia pestis. Emerg. Infect. Dis. 2001;7:43. doi: 10.3201/eid0701.010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. (a).Ratledge C, Dover LG. Iron metabolism in pathogenic bacteria. Annu. Rev. Microbiol. 2000;54:881. doi: 10.1146/annurev.micro.54.1.881. [DOI] [PubMed] [Google Scholar]; (b) Drechsel H, Winkelmann G. Iron chelation and siderophores. In: Winkelmann G, Carrano CJ, editors. Transition metals in microbial metabolism. Harwood Accademic; Amsterdam: 1997. p. 1. [Google Scholar]; (c) Neilands JB. Siderophores: structure and function of microbial iron transport compounds. J. Biol. Chem. 1995;270:26723. doi: 10.1074/jbc.270.45.26723. [DOI] [PubMed] [Google Scholar]; (d) Wooldridge KG, Williams PH. Iron uptake mechanisms of pathogenic bacteria. FEMS Microbiol. Rev. 1993;12:325. doi: 10.1111/j.1574-6976.1993.tb00026.x. [DOI] [PubMed] [Google Scholar]; (e) Wandersman C, Delepelaire P. Bacterial iron sources: from siderophores to hemophores. Annu. Rev. Microbiol. 2004;58:611. doi: 10.1146/annurev.micro.58.030603.123811. [DOI] [PubMed] [Google Scholar]
- 4.Quadri LEN. Assembly of aryl-capped siderophores by modular peptide synthetases and polyketide synthases. Mol. Microbiol. 2000;37:1. doi: 10.1046/j.1365-2958.2000.01941.x. [DOI] [PubMed] [Google Scholar]
- 5. (a).De Voss JJ, Rutter K, Schroeder BG, Su H, Zhu Y, Barry CE., III The salicylate-derived mycobactin siderophores of Mycobacterium tuberculosis are essential for growth in macrophages. Proc. Natl. Acad. Sci. USA. 2000;97:1252. doi: 10.1073/pnas.97.3.1252. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Rodriguez GM, Smith I. Identification of an ABC transporter required for iron acquisition and virulence in Mycobacterium tuberculosis. J. Bacteriol. 2006;188:424. doi: 10.1128/JB.188.2.424-430.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Bearden SW, Fetherston JD, Perry RD. Genetic organization of the yersiniabactin biosynthetic region and construction of avirulent mutants in Yersinia pestis. Infect. Immun. 1997;65:1659. doi: 10.1128/iai.65.5.1659-1668.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Fetherston JD, Bertolino VJ, Perry RD. YbtP and YbtQ: two ABC transporters required for iron uptake in Yersinia pestis. Mol. Microbiol. 1999;32:289. doi: 10.1046/j.1365-2958.1999.01348.x. [DOI] [PubMed] [Google Scholar]; (e) Fetherston JD, Kirillina O, Bobrov AG, Paulley JT, Perry RD. The yersiniabactin transport system is critical for the pathogenesis of bubonic and pneumonic plague. Infect. Immun. 2010;78:2045. doi: 10.1128/IAI.01236-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6 (a).Jurado RL. Iron, infections, and anemia of inflammation. Clin. Infect. Dis. 1997;25:888. doi: 10.1086/515549. [DOI] [PubMed] [Google Scholar]; (b) Ward CG, Bullen JJ, Rogers HJ. Iron and infection: new developments and their implications. J. Trauma. 1996;41:356. doi: 10.1097/00005373-199608000-00030. [DOI] [PubMed] [Google Scholar]
- 7. (a).Quadri LEN. Strategic paradigm shifts in the antimicrobial drug discovery process of the 21st century. Infect. Disord. Drug Targets. 2007;7:230. doi: 10.2174/187152607782110040. [DOI] [PubMed] [Google Scholar]; (b) Ferreras JA, Ryu JS, Di Lello F, Tan DS, Quadri LE. Small-molecule inhibition of siderophore biosynthesis in Mycobacterium tuberculosis and Yersinia pestis. Nat. Chem. Biol. 2005;1:29. doi: 10.1038/nchembio706. [DOI] [PubMed] [Google Scholar]
- 8.Stirrett KL, Ferreras JA, Jayaprakash V, Sinha BN, Ren T, Quadri LE. Small molecules with structural similarities to siderophores as novel antimicrobials against Mycobacterium tuberculosis and Yersinia pestis. Bioorg. Med. Chem. Lett. 2008;18:2662. doi: 10.1016/j.bmcl.2008.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. (a).Cmpds 1–14 were synthesized from appropriate 2′-hydroxy chalcone derivatives as outlined in Scheme 1 (Supplementary Data). Cmpds 9 and 10 were prepared as we reported earlier.11 1N-acetyl pyrazolines (3 and 4) were obtained by refluxing appropriate 2′-hydroxy chalcones with hydrazine hydrate (80%) in acetic acid.12 Similarly, 5 and 6 as well as 11 and 12 were obtained by refluxing appropriate 2′-hydroxy chalcones with semicarbazide hydrochloride/aminoguanidine bicarbonate in methanol for 4–6 hr. Cmpds 7 and 8 were synthesized from 1 and 2, respectively. Ethyl chloroformate was added to a solution of pyrazolines (1 and 2) in methanol with constant stirring at room temperature. Triethylamine was added to neutralize the acid liberated. Hydroxylamine in methanol was then added with stirring at room temperature for 2 hr to obtain 7 and 8. Cmpds 13 and 14 were synthesized from 9 and 9a, respectively. A slight excess of methyl iodide was added to 9 and 9a dropwise with stirring (<10°C, 1h). Hydroxylamine in methanol was then added and stirring continued (22°C, 30 min) to obtain 13 and 14. Cmpds 15–22 were synthesized from appropriate 2′-hydroxy chalcone derivatives as outlined in Scheme 2 (Supplementary Data). Cmpds 15 and 16 were prepared in similar manner to 5 and 6 and by replacing semicarbazide hydrochloride with isoniazide. Cmpds 17–22 were prepared from 1 and 2 as we reported earlier.13 Reaction of benzoyl chloride with 1 and 2 in pyridine provided 17 and 18, respectively. Similarly, reaction of sulphonyl chlorides with 1 and 2 in THF provided 19–22. Cmpds 23–32 were synthesized as outlined in Scheme 3 (Supplementary Data). (b) Cmpds 23–26 were prepared as reported.14 N-hydroxy semicarbazides were prepared by reaction of hydroxylamine, phenylchloroformate and hydrazine hydrate (80%) in moist ether. The N-hydroxy semicarbazides were then condensed with appropriate aldehyde/ketone to yield 23–26. Methyl hydrazinecarbodithioate was prepared as reported15 and condensed with appropriate aldehyde/ketone to produce 27–29. Cmpds 30 and 31 were prepared by a strategy similar to that used for 13 and 14. A slight excess of methyl iodide followed by hydroxylamine to thiosemicarbazones of appropriate aldehydes provided 30 and 31. Cmpds 32 was synthesized as we described earlier.15a Reaction of 27 with N-hydroxypiperidine-4-carboxamide provided 32. The intermediates were characterized by elemental analysis and FT-IR spectra. Final cmpds were characterized through 1H-NMR and FAB-MS spectral data. Synthetic procedures, physicochemical and spectral characteristics of 1–32 are presented in Supplementary Data.
- 10. (a).Antimicrobial activity was determined in dose–response experiments using 96-well-plate-based, twofold-microdilution assays as reported.7b,8,17 Virulent Mtb H37Rv was grown in iron-limiting GASTD medium and GASTD supplemented with 100 μM FeCl3 (GASTD+Fe).7b,17 Avirulent Yp KIM6-2082.1+ was grown in iron-limiting PMHD medium and PMHD supplemented with 100 μM FeCl3 (PMHD+Fe).7b,17 Cultures were started (OD600 = 0.001) from deferrated culture stocks prepared as reported.7b Growth was assessed as OD600 after incubation at 37 °C (Mtb: 10 days, stationary condition; Yp: 26 hr, 200 rpm) using a Spectra Max Plus reader (Molecular Dynamics). Compounds were evaluated at up to the highest concentration permitted by their solubility, with a 500 μM upper testing limit, and added as DMSO solutions. DMSO was kept at 0.5% in treated and DMSO-control cultures. IC50s were calculated from sigmoidal curves fitted to triplicate dose–response data using KaleidaGraph (Synergy Software) as reported.7b MIC90s were calculated as the lowest concentration tested that inhibited growth by ≥90% relative to DMSO controls. Compounds were tested at up to the maximum multiple of the MIC permitted by their solubility. (b) After bacterial cultures were treated with compounds and incubated as noted above, the mode of action was evaluated by enumerating CFU/mL after plating serial 10-fold dilutions of duplicate Mtb and Yp cultures on plates of Middlebrook 7H1118 and plates of TBA17, respectively. Plates were incubated at 37 °C for 30 days for Mtb and at 30 °C for 3 days for Yp before colony counting. Cytotoxicity was assessed against HeLa cells in a 96-well plate platform using a standard ATPLite™ reagent-based cytotoxicity assay (Perkin-Elmer) as previously reported.8
- 11. (a).Azarifar D, Shaabanzadeh M. Synthesis and characterization of new 3,5-dinaphthyl substituted 2-pyrazolines and study of their antimicrobial activity. Molecules. 2002;7:885. [Google Scholar]; (b) Gökhan N, Yeşilada A, Uçar G, Erol K, Bilgin AA. 1-N-substituted thiocarbamoyl-3-phenyl-5-thienyl-2-pyrazolines: Synthesis and evaluation as MAO inhibitors. Arch. Pharm. Pharm. Med. Chem. 2003;336:362. doi: 10.1002/ardp.200300732. [DOI] [PubMed] [Google Scholar]; (c) Rao YK, Fang SH, Tzeng YM. Differential effects of synthesized 2′-oxygenated chalcone derivatives: modulation of human cell cycle phase distribution. Bioorg. Med. Chem. 2004;12:2679. doi: 10.1016/j.bmc.2004.03.014. [DOI] [PubMed] [Google Scholar]
- 12.Tai AW, Lien EJ, Moore EC, Chun Y, Roberts JD. Studies of N-hydroxy-N’-aminoguanidine derivatives by nitrogen-15 nuclear magnetic resonance spectroscopy and as ribonucleotide reductase inhibitors. J. Med. Chem. 1983;6:1326. doi: 10.1021/jm00363a021. [DOI] [PubMed] [Google Scholar]
- 13.Sharma TC, Saksena V, Reddy NJ. Oxidation of some hydroxy aryl pyrazolines with manganese dioxide. Acta Chim. Acad. Sci. Hungar. 1977;93:415. [Google Scholar]
- 14.Klayman DL, Bartosevich JF, Griffin TS, Mason CJ, Scovill JP. 2-Acetylpyridine thiosemicarbazones. 1. A new class of potential antimalarial agents. J. Med. Chem. 1979;22:855. doi: 10.1021/jm00193a020. [DOI] [PubMed] [Google Scholar]
- 15. (a).Bhadaliya C, Bunha M, Jagrat M, Sinha BN, Saiko P, Graser G, Szekeres T, Raman G, Rajendran P, Moorthy D, Basu A, Venkatesan J. Design, synthesis and anticancer activity of piperazine hydroxamates and their histone deacetylase (HDAC) inhibitory activity. Bioorg. Med. Chem. Lett. 2010;20:3906. doi: 10.1016/j.bmcl.2010.05.020. [DOI] [PubMed] [Google Scholar]; (b) Ren S, Wang R, Komatsu K, Bonaz-Krause P, Zyrianov Y, McKenna CE, Csipke C, Tokes ZA, Lien EJ. Synthesis, biological evaluation, and quantitative structure-activity relationship analysis of new Schiff bases of hydroxysemicarbazide as potential antitumor agents. J. Med. Chem. 2002;45:410. doi: 10.1021/jm010252q. [DOI] [PubMed] [Google Scholar]
- 16. (a).Dwyer DJ, Kohanski MA, Hayete B, Collins JJ. Gyrase inhibitors induce an oxidative damage cellular death pathway in Escherichia coli. Mol. Syst. Biol. 2007;3:91. doi: 10.1038/msb4100135. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kohanski MA, Dwyer DJ, Collins JJ. How antibiotics kill bacteria: from targets to networks. Nat. Rev. Microbiol. 2010;8:423. doi: 10.1038/nrmicro2333. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ. A common mechanism of cellular death induced by bactericidal antibiotics. Cell. 2007;130:797. doi: 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- 17.Stirrett KL, Ferreras JA, Rossi SM, Moy RL, Fonseca FV, Quadri LE. A multicopy suppressor screening approach as a means to identify antibiotic resistance determinant candidates in Yersinia pestis. BMC Microbiol. 2008;8:122. doi: 10.1186/1471-2180-8-122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. (a).Chavadi SS, Edupuganti UR, Vergnolle O, Fatima I, Singh SM, Soll CE, Quadri LE. Inactivation of tesA reduces cell-wall lipid production and increases drug susceptibility in mycobacteria. J. Biol. Chem. 2011 doi: 10.1074/jbc.M111.247601. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ferreras JA, Stirrett KL, Lu X, Ryu JS, Soll CE, Tan DS, Quadri LE. Mycobacterial phenolic glycolipid virulence factor biosynthesis: mechanism and small-molecule inhibition of polyketide chain initiation. Chem. Biol. 2008;15:51. doi: 10.1016/j.chembiol.2007.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.