

Abstract
Follicular lymphoma (FL) and diffuse large B-cell lymphoma (DLBCL) are the two most common non-Hodgkin lymphomas (NHLs). To identify genes with mutations in B-cell NHL we sequenced tumour and matched normal DNA from 13 DLBCL cases and one FL case. We analysed RNA-seq data from these and another 113 NHLs to identify genes with candidate mutations, and then re-sequenced tumour and matched normal DNA from these cases to confirm 109 genes with multiple somatic mutations. Genes with roles in histone modification were frequent targets of somatic mutation. For example, 32% of DLBCL and 89% of FL cases had somatic mutations in MLL2, which encodes a histone methyltransferase. 11.4% of DLBCL and 13.4% of FL cases had somatic mutations in MEF2B, a calcium-regulated gene that cooperates with CREBBP and EP300 in acetylating histones. Our analysis thus suggests a previously unappreciated disruption of chromatin biology in lymphomagenesis.
Keywords: H3K27, H3K4, HDAC, HAT, cancer genomics, methylation, acetylation, EZH2, driver, cancer sequencing
Introduction
Non-Hodgkin lymphomas (NHLs) are cancers of B, T or natural killer lymphocytes. The two most common types of NHL, follicular lymphoma (FL) and diffuse large B-cell lymphoma (DLBCL), together comprise 60% of new B-cell NHL diagnoses each year in North America1. FL is an indolent and typically incurable disease characterized by clinical and genetic heterogeneity. DLBCL is aggressive and likewise heterogeneous, comprising at least two distinct subtypes that respond differently to standard treatments. Both FL and the germinal centre B-cell (GCB) cell of origin (COO) subtype of DLBCL derive from germinal centre B cells whereas the activated B-cell (ABC) variety, which exhibits a more aggressive clinical course, is thought to originate from B cells that have exited, or are poised to exit, the germinal centre2. Current knowledge of the specific genetic events leading to DLBCL and FL is limited to the presence of a few recurrent genetic abnormalities2. For example, 85-90% of FL and 30-40% of GCB DLBCL cases3,4 harbour t(14;18)(q32;q21), which results in deregulated expression of the BCL2 oncoprotein. Other genetic abnormalities unique to GCB DLBCL include amplification of the c-REL gene and of the miR-17-92 microRNA cluster5. In contrast to GCB cases, 24% of ABC DLBCLs harbour structural alterations or inactivating mutations affecting PRDM1, which is involved in differentiation of GCB cells into antibody-secreting plasma cells6. ABC-specific mutations also affect genes regulating NF-κB signalling7,8,9, with TNFAIP3 (A20) and MYD8810 the most abundantly mutated in 24% and 39% of cases respectively. To enhance our understanding of the genetic architecture of B-cell NHL, we undertook a study to (1) identify somatic mutations and (2) determine the prevalence, expression and focal recurrence of mutations in FL and DLBCL. Using strategies and techniques applied to cancer genome and transcriptome characterization by ourselves and others11,12,13, we sequenced tumour DNA and/or RNA from 117 tumour samples and 10 cell lines (Supplementary Tables S1 and S2) and identified 651 genes (Supplementary Figure S1) with evidence of somatic mutation in B-cell NHL. After validation, we showed that 109 genes were somatically mutated in 2 or more NHL cases. We further characterised the frequency and nature of mutations within MLL2 and MEF2B, which were among the most frequently mutated genes with no previously known role in lymphoma.
Identification of recurrently mutated genes
We sequenced the genomes or exomes of 14 NHL cases, all with matched constitutional DNA sequenced to comparable depths (Supplementary Tables S1 and S2). After screening for single nucleotide variants followed by subtraction of known polymorphisms and visual inspection of the sequence read alignments, we identified 717 nonsynonymous (coding single nucleotide variants; cSNVs) affecting 651 genes (Supplementary Figure S1; Methods). We identified between 20 and 135 cSNVs in each of these genomes. Only 25 of the 651 genes with cSNVs were represented in the cancer gene census (December, 2010 release)14.
We performed RNA sequencing (RNA-seq) on these 14 NHL cases and an expanded set of 113 samples comprising 83 DLBCL, 12 FL and 8 B-cell NHL cases with other histologies and 10 DLBCL-derived cell lines (Supplementary Table S2). We analysed these data to identify novel fusion transcripts (Supplementary Table S3) and cSNVs (Figure 1). We identified 240 genes with at least one cSNV in a genome/exome or an RNA-seq “mutation hot spot” (below), and with cSNVs in at least three cases in total (Supplementary Table S4). We selected cSNVs from each of these 240 genes for re-sequencing to confirm their somatic status. We did not re-sequence genes with previously documented mutations in lymphoma (e.g. CD79B, BCL2). We confirmed the somatic status of 543 cSNVs in 317 genes, with 109 genes having at least two confirmed somatic mutations (Supplementary Table S4 and S5). Of the successfully re-sequenced cSNVs predicted from the genomes, 171 (94.5%) were confirmed somatic, 7 were false calls and 3 were present in the germ line. These 109 recurrently mutated genes were significantly enriched for genes implicated in lymphocyte activation (P=8.3×10-4; e.g. STAT6, BCL10), lymphocyte differentiation (P=3.5×10-3; e.g. CARD11), and regulation of apoptosis (P=1.9×10-3; e.g. BTG1, BTG2). Also significantly enriched were genes linked to transcriptional regulation (P= 5.4×10-4; e.g. TP53) and genes involved in methylation (P=2.2×10-4) and acetylation (P=1.2×10-2), including histone methyltransferase (HMT) and acetyltransferase (HAT) enzymes known previously to be mutated in lymphoma (e.g. EZH213 and CREBBP15; Methods).
Figure 1. Genome-wide visualization of somatic mutation targets in NHL.
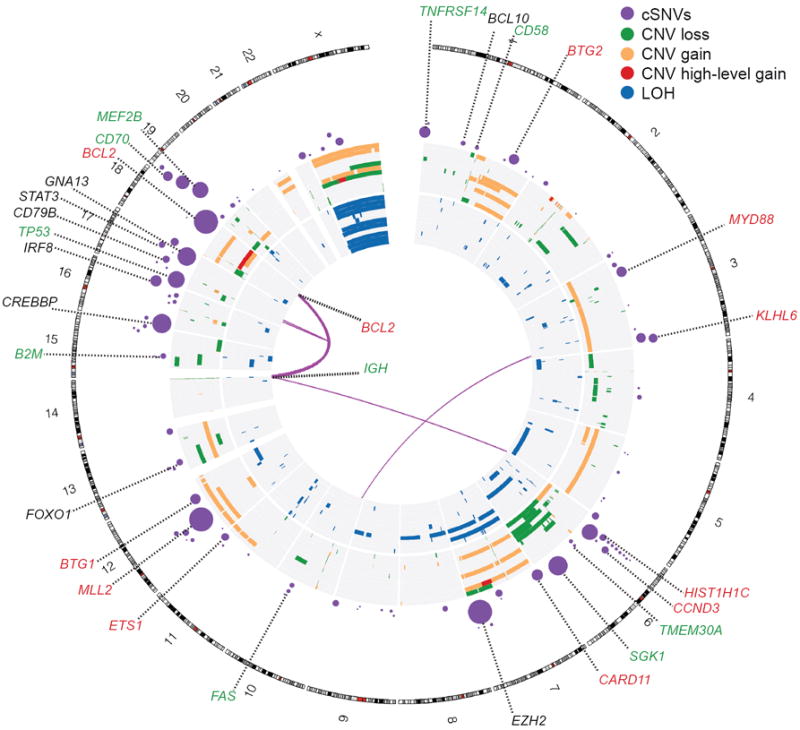
Overview of structural rearrangements and copy number variations (CNVs) in the 11 DLBCL genomes and cSNVs in the 109 recurrently mutated genes identified in our analysis. Inner arcs represent somatic fusion transcripts identified in one of the 11 genomes. The CNVs and LOH detected in each of the 11 DLBCL tumour/normal pairs are displayed on the concentric sets of rings. The inner 11 rings show regions of enhanced homozygosity plotted with blue (interpreted as LOH). The outer 11 rings show somatic CNVs. Purple circles indicate the position of genes with at least two confirmed somatic mutations with circle diameter proportional to the number of cases with cSNVs detected in that gene. Circles representing the genes with significant evidence for positive selection are labelled. Coincidence between recurrently mutated genes and regions of gain/loss are colour-coded in the labels (green=loss, red=gain). For example B2M, which encodes beta-2-microglobulin, is recurrently mutated and is deleted in two cases.
Mutation hot spots can result from mutations at sites under strong selective pressure and we have previously identified such sites using RNA-seq data13. We searched our RNA-seq data for genes with mutation hot spots, and identified 10 genes that were not mutated in the 14 genomes (PIM1, FOXO1, CCND3, TP53, IRF4, BTG2, CD79B, BCL7A, IKZF3 and B2M), of which five (FOXO1, CCND3, BTG2, IKZF3 and B2M) were not previously known targets of point mutation in NHL (Supplementary Table S6; Methods). FOXO1, BCL7A and B2M exhibited hot spots affecting their start codons. The effect of a FOXO1 start codon mutation, which was observed in three cases, was further studied using a cell line in which the initiating ATG was mutated to TTG. Western blots probed with a FOXO1 antibody revealed a band with a reduced molecular weight, indicative of a FOXO1 N-terminal truncation (Supplementary Figure S2) consistent with utilization of the next in-frame ATG for translation initiation. A second hot spot in FOXO1 at T24 was mutated in two cases. T24 is reportedly phosphorylated by AKT subsequent to B-cell receptor (BCR) stimulation16 inducing FOXO1 nuclear export.
We analysed the RNA-seq data to determine whether any of the somatic mutations in the 109 recurrently mutated genes showed evidence for allelic imbalance with expression favouring one allele. Of 380 expressed heterozygous mutant alleles, we observed preferential expression of the mutation for 16.8% (64/380) and preferential expression of the wild-type for 27.8% (106/380; Supplementary Table S7). Seven genes displayed evidence for significant preferential expression of the mutant allele in at least two cases: (BCL2, CARD11, CD79B, EZH2, IRF4, MEF2B and TP53; Methods). In 27 of 43 cases with BCL2 cSNVs, expression favoured the mutant allele, consistent with the previously-described hypothesis that the translocated (and hence, transcriptionally deregulated) allele of BCL2 is targeted by somatic hypermutation17. Examples of mutations at known oncogenic hot spot sites such as F123I in CARD1118 exhibited allelic imbalance favouring the mutant allele in some cases. Similarly, we noted expression favouring two novel hot spot mutations in MEF2B (Y69 and D83) and two sites in EZH2 not previously reported as mutated in lymphoma (A682G and A692V).
We sought to distinguish new cancer-related mutations from passenger mutations using the approach proposed by Greenman et al19. We reasoned that this would reveal genes with strong selection signatures, and mutations in such genes would be good candidate cancer drivers. We identified 26 genes with significant evidence for positive selection (FDR 0.03, Methods), with either selective pressure for acquiring non-synonymous point mutations or truncating/nonsense mutations (Methods; Table 1; Supplementary Table S8). Included were known lymphoma oncogenes (BCL2, CD79B9, CARD1118, MYD8810 and EZH213), all of which exhibited signatures indicative of selection for non-synonymous variants.
Table 1.
Overview of cSNVs and confirmed somatic mutations in most frequently mutated genes
| Cases | Total | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | NS | S | T | NS | S | T | Somatic cSNVs (RNA-seq cohort)* | P (raw) | q | NS SP | T SP | Skew (M, WT, both) *** |
| MLL2† | 16 | 8 | 17 | 17 | 8 | 18 | 10 | 6.85×10-8 | 8.50×10-7 | 0.834 | 14.4 | WT |
| TNFRSF14 G† | 7 | 1 | 7 | 8 | 1 | 7 | 11 | 6.85×10-8 | 8.50×10-7 | 7.52 | 118 | both |
| SGK1 G† | 18 | 6 | 6 | 37 | 10 | 6 | 9 | 6.85×10-8 | 8.50×10-7 | 19.5 | 61.7 | - |
| BCL10† | 2 | 0 | 4 | 3 | 0 | 4 | 4 | 6.85×10-8 | 8.50×10-7 | 3.62 | 112 | WT |
| GNA13 G† | 21 | 1 | 2 | 33 | 1 | 2 | 5 | 6.85×10-8 | 8.50×10-7 | 24.1 | 25.7 | both |
| TP53 G† | 20 | 2 | 1 | 23 | 3 | 1 | 22 | 6.85×10-8 | 8.50×10-7 | 15.6 | 14.1 | both |
| EZH2 G† | 33 | 0 | 0 | 33 | 0 | 0 | 33 | 6.85×10-8 | 8.50×10-7 | 11.4 | 0.00 | both |
| BTG2† | 12 | 6 | 1 | 14 | 6 | 1 | 2 | 6.85×10-8 | 8.50×10-7 | 23.9 | 35.1 | - |
| BCL2 G† | 42 | 45 | 0 | 96 | 105 | 0 | 43 | 9.35×10-8 | 8.50×10-7 | 3.78 | 0.00 | M |
| BCL6†** | 11 | 2 | 0 | 12 | 2 | 0 | 2 | 9.35×10-8 | 8.50×10-7 | 0.175 | 0.00 | M |
| CIITA†** | 5 | 3 | 0 | 6 | 3 | 0 | 2 | 9.35×10-8 | 8.50×10-7 | 0.086 | 0.00 | |
| FAS† | 2 | 0 | 4 | 3 | 0 | 4 | 2 | 1.52×10-7 | 1.17×10-6 | 2.54 | 66.5 | WT |
| BTG1† | 11 | 6 | 2 | 11 | 7 | 2 | 10 | 1.52×10-7 | 1.17×10-6 | 17.5 | 52.5 | both |
| MEF2B G† | 20 | 2 | 0 | 20 | 2 | 0 | 10 | 2.05×10-7 | 1.47×10-6 | 14.2 | 0.00 | M |
| IRF8† | 11 | 5 | 3 | 14 | 5 | 3 | 3 | 4.55×10-7 | 3.03×10-6 | 8.82 | 28.2 | WT |
| TMEM30A† | 1 | 0 | 4 | 1 | 0 | 4 | 4 | 6.06×10-7 | 3.79×10-6 | 0.785 | 65.0 | WT |
| CD58† | 2 | 0 | 3 | 2 | 0 | 3 | 2 | 2.42×10-6 | 1.43×10-5 | 2.29 | 69.2 | - |
| KLHL6† | 10 | 2 | 2 | 12 | 2 | 2 | 4 | 1.00×10-5 | 5.26×10-5 | 5.42 | 16.4 | - |
| MYD88 A† | 13 | 2 | 0 | 14 | 2 | 0 | 9 | 1.00×10-5 | 5.26×10-5 | 12.4 | 0.00 | WT |
| CD70† | 5 | 0 | 1 | 5 | 0 | 2 | 3 | 1.70×10-5 | 8.48×10-5 | 7.08 | 44.0 | - |
| CD79B A† | 7 | 2 | 1 | 9 | 2 | 1 | 5 | 2.00×10-5 | 9.52×10-5 | 10.9 | 18.3 | M |
| CCND3† | 7 | 1 | 2 | 7 | 1 | 2 | 6 | 2.80×10-5 | 1.27×10-4 | 6.55 | 36.3 | WT |
| CREBBP† | 20 | 7 | 4 | 24 | 7 | 4 | 9 | 1.00×10-4 | 4.35×10-4 | 2.72 | 6.04 | both |
| HIST1H1C† | 9 | 0 | 0 | 10 | 0 | 0 | 6 | 1.80×10-4 | 7.50×10-4 | 11.9 | 0.00 | both |
| B2M† | 7 | 0 | 0 | 7 | 0 | 0 | 4 | 3.90×10-4 | 1.56×10-3 | 16.6 | 0.00 | WT |
| ETS1† | 10 | 1 | 0 | 10 | 1 | 0 | 4 | 4.10×10-4 | 1.58×10-3 | 5.76 | 0.00 | WT |
| CARD11† | 14 | 3 | 0 | 14 | 3 | 0 | 3 | 1.90×10-3 | 7.04×10-3 | 3.37 | 0.00 | both |
| FAT2†** | 2 | 1 | 0 | 2 | 1 | 0 | 2 | 6.30×10-3 | 2.25×10-2 | 0.128 | 0.00 | - |
| IRF4†** | 9 | 4 | 0 | 26 | 5 | 0 | 5 | 7.00×10-3 | 2.41×10-2 | 0.569 | 0.00 | both |
| FOXO1† | 8 | 4 | 0 | 10 | 4 | 0 | 4 | 7.60×103 | 2.53×10-2 | 4.02 | 0.00 | - |
| STAT3 | 9 | 0 | 0 | 9 | 0 | 0 | 4 | 2.19×10-2 | 6.08×10-2 | - | - | both |
| RAPGEF1 | 8 | 3 | 0 | 10 | 3 | 0 | 3 | 2.98×10-2 | 7.45×10-2 | - | - | WT |
| ABCA7 | 12 | 3 | 0 | 15 | 3 | 0 | 2 | 7.76×10-2 | 1.67×10-1 | - | - | WT |
| RNF213 | 10 | 8 | 0 | 10 | 8 | 0 | 2 | 7.87×10-2 | 1.67×10-1 | - | - | - |
| MUC16 | 17 | 12 | 0 | 39 | 25 | 0 | 2 | 8.32×10-2 | 1.73×10-1 | - | - | - |
| HDAC7 | 8 | 4 | 0 | 8 | 4 | 0 | 2 | 8.94×10-2 | 1.82×10-1 | - | - | WT |
| PRKDC | 7 | 3 | 0 | 7 | 4 | 0 | 2 | 1.06×10-1 | 2.05×10-1 | - | - | - |
| SAMD9 | 9 | 2 | 0 | 9 | 2 | 0 | 2 | 1.79×10-1 | 3.01×10-1 | - | - | - |
| TAF1 | 10 | 0 | 0 | 10 | 0 | 0 | 2 | 3.03×10-1 | 4.74×10-1 | - | - | - |
| PIM1 | 20 | 19 | 0 | 33 | 34 | 0 | 11 | 3.40×10-1 | 5.23×10-1 | - | - | WT |
| COL4A2 | 8 | 2 | 0 | 8 | 2 | 0 | 2 | 7.64×10-1 | 8.99×10-1 | - | - | - |
| EP300 | 8 | 7 | 1 | 8 | 7 | 1 | 3 | 9.54×10-1 | 1.00 | - | - | WT |
Individual cases with nonsynonymous (NS), synonymous (S) and truncating (T) mutations and total number of mutations of each class is shown separately as some genes contained multiple mutations in the same case. The P values indicated in bold are the upper limit on the P value for that gene determined with the approach described by Greenman et al (see Methods)19, q is the Benjamini-corrected q value, and NS, SP and T SP refer to selective pressure estimates from this model for the acquisition of nonsynonymous or truncating mutations, respectively.
genes significant at an FDR of 0.03. SNVs in BCL2 and previously confirmed hot spot mutations in EZH2 and CD79B are likely somatic in these samples based on published observations of others.
Additional somatic mutations identified in larger cohorts and insertion/deletion mutations are not included in this total.
Selective pressure estimates are both <1 indicating purifying selection rather than positive selection acting on this gene.
“both” indicates we observed separate cases in which skewed expression was seen but where this skew was not consistent for the mutant or wild-type allele. Genes with a superscript of either A or G were found to have mutations significantly enriched in ABC or GCB cases, respectively (P< 0.05, Fisher Exact test).
Evidence for selection of inactivating changes
We expected tumour suppressor genes to exhibit strong selection for the acquisition of nonsense mutations. In our analysis, the eight most significant genes included seven with strong selective pressure for nonsense mutations, including the known tumour suppressor genes TP53 and TNFRSF1420 (Table 1). CREBBP, recently reported as commonly inactivated in DLBCL15, also showed some evidence for acquisition of nonsense mutations and cSNVs (Supplementary Figure S3; Supplementary Table S9). We also observed enrichment for nonsense mutations in BCL10, a positive regulator of NF-κB, in which oncogenic truncated products have been described in lymphomas21. The remaining strongly significant genes (BTG1, GNA13, SGK1 and MLL2) had no reported role in lymphoma. GNA13 was affected by mutations in 22 cases including multiple nonsense mutations. GNA13 encodes the alpha subunit of a heterotrimeric G-protein coupled receptor responsible for modulating RhoA activity22. Some of the mutated residues negatively impact its function23,24, including a T203A mutation, which also exhibited allelic imbalance favouring the mutant allele (Supplementary Table S7). GNA13 protein was reduced or absent on Western blots in cell lines harbouring either a nonsense mutation, a stop codon deletion, a frame shifting deletion, or changes affecting splice sites (Methods; Supplementary Figure S4).
SGK1 encodes a PI3K-regulated kinase with functions including regulation of FOXO transcription factors25, regulation of NF-κB by phosphorylating IkB kinase26, and negative regulation of NOTCH signalling27. SGK1 also resides within a region of chromosome 6 commonly deleted in DLBCL (Figure 1)5. The mechanism by which SGK1 and GNA13 inactivation may contribute to lymphoma is unclear but the strong degree of apparent selection towards their inactivation and their overall high mutation frequency (each mutated in 18 of 106 DLBCL cases) suggests that their loss contributes to B-cell NHL. Certain genes are known to be mutated more commonly in GCB DLBCLs (e.g. TP5328 and EZH213). Here, both SGK1 and GNA13 mutations were found only in GCB cases (P = 1.93×10-3 and 2.28×10-4, Fisher exact test; n=15 and 18, respectively)(Figure 2). Two additional genes (MEF2B and TNFRSF14) with no previously described role in DLBCL showed a similar restriction to GCB cases (Figure 2).
Figure 2. Overview of mutations and potential cooperative interactions in NHL.
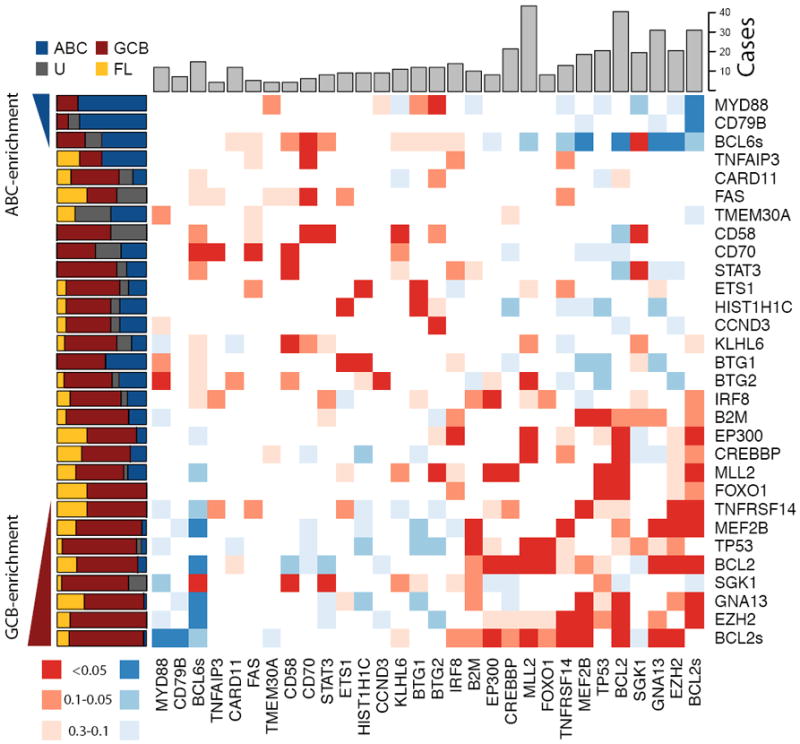
This heat map displays possible trends towards co-occurrence (red) and mutual exclusion (blue) of somatic mutations and structural rearrangements. Colours were assigned by taking the minimum value of a left- and right-tailed Fisher exact test. To capture trends a P-value threshold of 0.3 was used, with the darkest shade of the colour indicating those meeting statistical significance (P <=0.05). The relative frequency of mutations in ABC (blue), GCB (red), unclassifiable (black) DLBCLs and FL (yellow) cases is shown on the left. Genes were arranged with those having significant (P<0.05, Fisher exact test) enrichment for mutations in ABC cases (blue triangle) towards the top (and left) and those with significant enrichment for mutations in GCB cases (red triangle) towards the bottom (and right). The total number of cases in which each gene contained either cSNVs or confirmed somatic mutations is shown at the top. The cluster of blue squares (upper-right) results from the mutual exclusion of the ABC-enriched mutations (e.g. MYD88, CD79B) from the GCB-enriched mutations (e.g. EZH2, GNA13). Presence of structural rearrangements involving the two oncogenes BCL6 and BCL2 (indicated as BCL6s and BCL2s) was determined with FISH techniques utilizing break-apart probes (Methods).
Inactivating MLL2 mutations
MLL2 showed the most significant evidence for selection and the largest number of nonsense SNVs. Our RNA-seq analysis indicated that 26.0% (33/127) of cases carried at least one MLL2 cSNV. To address the possibility that variable RNA-seq coverage of MLL2 failed to capture some mutations, we PCR amplified the entire MLL2 locus (~36kb) in 89 cases (35 primary FLs, 17 DLBCL cell lines, and 37 DLBCLs). 58 of these cases were among the RNA-seq cohort. Illumina amplicon resequencing (Methods) revealed 78 mutations, confirming the RNA-seq mutations in the overlapping cases and identifying 33 additional mutations. We confirmed the somatic status of 46 variants using Sanger sequencing (Supplementary Table S10), and showed that 20 of the 33 additional mutations were insertions or deletions (indels). Three SNVs at splice sites were also detected, as were 10 new cSNVs that had not been detected by RNA-seq.
The somatic mutations were distributed across MLL2 (Figure 3A). 37% (n=29/78) of these were nonsense mutations, 46% (n=36/78) were indels that altered the reading frame, 8% (n=6/78) were point mutations at splice sites and 9% (n=7/78) were non-synonymous amino acid substitutions (Table 2). Four of the somatic splice site mutations had effects on MLL2 transcript length and structure. For example, two heterozygous splice site mutations resulted in the use of a novel splice donor site and an intron retention event.
Figure 3. Summary and effect of somatic mutations affecting MLL2 and MEF2B.
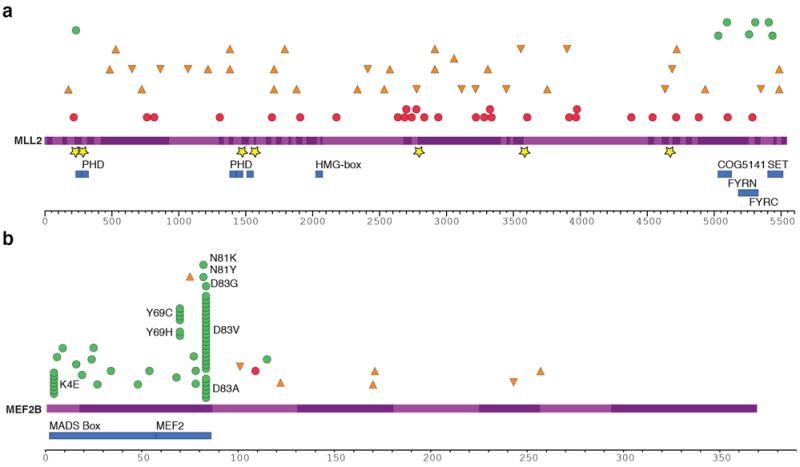
(A) Re-sequencing the MLL2 locus in 89 samples revealed mainly nonsense (red circles) and frameshift-inducing indel mutations (orange triangles). A smaller number of non-synonymous somatic mutations (green circles) and point mutations or deletions affecting splice sites (yellow stars) were also observed. All of the non-synonymous point mutations affected a residue within either the catalytic SET domain, the FYRC domain (“FY-rich C-terminal domain”) or PHD zinc finger domains. The effect of these splice site mutations on MLL2 splicing was also explored (Supplementary Figure S7). (B) The cSNVs and somatic mutations found in MEF2B in all FL and DLBCL cases sequenced are shown with the same symbols. Only the amino acids with variants in at least two patients are labelled. cSNVs were most prevalent in the first two protein coding exons of MEF2B (exons 2 and 3). The crystal structure of MEF2 bound to EP300 supports the idea that two of the mutated sites (L67 and Y69) are important in the interaction between these proteins (Supplementary Figure S8; Supplementary Discussion)50.
Table 2.
Summary of types of MLL2 somatic mutations
| Sample Type | FL | DLBCL | DLBCL cell-line | Centroblast |
|---|---|---|---|---|
| Truncation | 18 | 4 | 7 | 0 |
| Indel with frameshift | 22 | 8 | 6 | 0 |
| Splice site | 4 | 2 | 0 | 0 |
| SNV | 3 | 2 | 2 | 0 |
|
| ||||
| Any mutation (number of cases) | 31 / 35 | 12 / 37 | 10 / 17 | 0 / 8 |
|
| ||||
| Percentage | 89% | 32% | 59% | 0% |
Approximately half of the NHL cases we sequenced had two MLL2 mutations (Supplementary Table S10). We used BAC clone sequencing in eight FL cases to show that in all eight cases the mutations were in trans, affecting both MLL2 alleles. This observation is consistent with the notion that there is a complete, or near-complete, loss of MLL2 in the tumour cells of such patients.
With the exception of two primary FL cases and two DLBCL cell lines (Pfeiffer and SU-DHL-9), the majority of MLL2 mutations appeared to be heterozygous. Analysis of Affymetrix 500k SNP array data from two FL cases with apparent homozygous mutations revealed that both tumours exhibited copy number neutral loss of heterozygosity (LOH) for the region of chromosome 12 containing MLL2 (Methods). Thus, in addition to bi-allelic mutation, LOH is a second, albeit less common mechanism by which MLL2 function is lost.
MLL2 was the most frequently mutated gene in FL, and among the most frequently mutated genes in DLBCL (Figure 2). We confirmed MLL2 mutations in 31 of 35 FL patients (89%), in 12 of 37 DLBCL patients (32%), in 10 of 17 DLBCL cell lines (59%) and in none of the eight normal centroblast samples we sequenced. Our analysis predicted that the majority of the somatic mutations observed in MLL2 were inactivating (91% disrupted the reading frame or were truncating point mutations), suggesting to us that MLL2 is a tumour suppressor of significance in NHL.
Recurrent point mutations in MEF2B
Our selective pressure analysis also revealed genes with stronger pressure for acquisition of amino acid substitutions than for nonsense mutations. One such gene was MEF2B, which had not previously been linked to lymphoma. 20 (15.7%) cases had MEF2B cSNVs and 4 (3.1%) cases had MEF2C cSNVs. All cSNVs detected by RNA-seq affected either the MADS box or MEF2 domains. To determine the frequency and scope of MEF2B mutations, we Sanger-sequenced exons 2 and 3 in 261 primary FL samples; 259 DLBCL primary tumours; 17 cell lines; 35 cases of assorted NHL (IBL, composite FL and PBMCL); and eight non-malignant centroblast samples. We also used a capture strategy (Methods) to sequence the entire MEF2B coding region in the 261 FL samples, revealing six additional variants outside exons 2 and 3. We thus identified 69 cases (34 DLBCL; 12.67% and 35 FL; 15.33%) with MEF2B cSNVs or indels, failing to observe novel variants in other NHL and non-malignant samples. 55 (80%) of the variants affected residues within the MADS box and MEF2 domains encoded by exons 2 and 3 (Supplementary Table S11; Figure 3B). Each patient generally had a single MEF2B variant and we observed relatively few (8 total, 10.7%) truncation-inducing SNVs or indels. Non-synonymous SNVs were by far the most common type of change observed, with 59.4% of detected variants affecting K4, Y69, N81 or D83. In 12 cases MEF2B mutations were shown to be somatic, including representative mutations at each of K4, Y69, N81 and D83 (Supplementary Table S12). We did not detect mutations in ABC cases, indicating that somatic mutations in MEF2B play a role unique to the development of GCB DLBCL and FL (Figure 2).
Discussion
In our study of genome, transcriptome and exome sequences from 127 B-cell NHL cases, we identified 109 genes with clear evidence of somatic mutation in multiple individuals. Significant selection appears to act on at least 26 of these for the acquisition of either nonsense or missense mutations. To the best of our knowledge, the majority of these genes had not previously been associated with any cancer type. We observed an enrichment of somatic mutations affecting genes involved in transcriptional regulation and, more specifically, chromatin modification.
MLL2 emerged from our analysis as a major tumour suppressor locus in NHL. It is one of six human H3K4-specific methyltransferases in the MLL family, all of which share homology with the Drosophila trithorax gene29. Trimethylated H3K4 (H3K4me3) is an epigenetic mark associated with the promoters of actively transcribed genes. By laying down this mark, MLLs are responsible for the transcriptional regulation of developmental genes including the homeobox (Hox) gene family30 which collectively control segment specificity and cell fate in the developing embryo31,32. Each MLL family member is thought to target different subsets of Hox genes33 and in addition, MLL2 is known to regulate the transcription of a diverse set of genes34. Recently, MLL2 mutations were reported in a small-cell lung cancer cell line35 and in renal carcinoma36 but the frequency of nonsense mutations affecting MLL2 in these cancers was not established in these reports. Parsons and colleagues recently reported inactivating mutations in MLL2 or MLL3 in 16% of medulloblastoma patients37 further implicating MLL2 as a cancer gene.
Our data link MLL2 somatic mutations to B-cell NHL. The reported mutations are likely to be inactivating and in eight of the cases with multiple mutations, we confirmed that both alleles were affected, presumably resulting in essentially complete loss of MLL2 function. The high prevalence of MLL2 mutations in FL (89%) equals the frequency of the t(14;18)(q32;q21) translocation, which is considered the most prevalent genetic abnormality in FL3. In DLBCL tumour samples and cell lines, MLL2 mutation frequencies were 32% and 59% respectively, also exceeding the prevalence of the most frequent cytogenetic abnormalities, such as the various translocations involving 3q27, which occur in 25-30% of DLBCLs and are enriched in ABC cases38. Importantly, we found MLL2 mutated in both DLBCL subtypes (Figure 2). Our analyses thus indicate that MLL2 acts as a central tumour suppressor in FL and both DLBCL subtypes.
The MEF2 gene family encodes four related transcription factors that recruit histone-modifying enzymes including histone deacetylases (HDACs) and HATs in a calcium-regulated manner. Although truncating variants were detected in our analysis of MEF2 gene family members, our analysis suggests that, in contrast to MLL2, MEF2 family members tend to selectively acquire non-synonymous amino acid substitutions. In the case of MEF2B, 59.4% of all the cSNVs were found at four sites within the protein (K4, Y69, N81 and D83), and all four of these sites were confirmed to be targets of somatic mutation. 39% of the MEF2B alterations affect D83, resulting in replacement of the charged aspartate with any of alanine, glycine or valine. Although we cannot yet predict the consequences of these substitutions on protein function, it seems likely that their effect would impact the ability of MEF2B to facilitate gene expression and thus play a role in promoting the malignant transformation of germinal centre B cells to lymphoma (Supplementary Discussion).
MEF2B mutations can be linked to CREBBP and EP300 mutations, and to recurrent Y641 mutations in EZH213. One target of CREBBP/EP300 HAT activity is H3K27, which is methylated by EZH2 to repress transcription. There is evidence that the action of EZH2 antagonizes that of CREBBP/EP30039. One function of MEF2 is to recruit either HDACs or CREBBP/EP300 to target genes40, and it has been suggested that HDACs compete with CREBBP/EP300 for the same binding site on MEF241. Under normal Ca2+ levels, MEF2 is bound by type IIa HDACs, which maintain the tails of histone proteins in a deacetylated repressive chromatin state42. Increased cytoplasmic Ca2+ levels induce the nuclear export of HDACs, enabling the recruitment of HATs such as CREBBP/EP300, facilitating transcription at MEF2 target genes. Mutation of CREBBP, EP300 or MEF2B may impact expression of MEF2 target genes owing to reduced acetylation of nucleosomes near these genes (Supplementary Figure S5; Supplementary Discussion). In light of the recent finding that heterozygous EZH2 Y641 mutations enhance overall H3K27 trimethylation activity of PCR243,44, it is possible that mutation of both MLL2 and EZH2 could cooperate in reducing the expression of some of the same target genes. Our data imply that (1) post-transcriptional modification of histones is of key importance in germinal centre B cells and (2) deregulated histone modification due to these mutations likely results in reduced acetylation and enhanced methylation and acts as a core driver event in the development of NHL (Supplementary Figure S5).
Methods Summary
All samples analysed contained at least 50% tumour cells. Genomes, exomes and transcriptomes were sequenced using a combination of Illumina GAIIx and HiSeq 2000 instruments to read lengths of between 36 and 100 nucleotides. Exome capture was performed using the Agilent SureSelect Target Enrichment System Protocol (Version 1.0, September 2009). Alignment was accomplished using BWA45 and variants were identified using SNVmix46. Variants were manually reviewed in IGV and were confirmed (where applicable) by PCR followed by either Sanger sequencing or Illumina re-sequencing. Structural rearrangements in genomes and transcriptomes were identified using ABySS47. Gene expression values used for subtype assignment were calculated as RPKM values48 and subtypes were assigned using an adaptation of the method developed for data from Affymetrix expression arrays49 trained with samples previously classified by this standard approach.
Supplementary Material
Acknowledgments
This study was funded in part by funding from the National Cancer Institute Office of Cancer Genomics (Contract No. HHSN261200800001E.), the Terry Fox Foundation (grant #019001, Biology of Cancer: Insights from Genomic Analyses of Lymphoid Neoplasms), Genome Canada/Genome BC Grant Competition III (Project Title: High Resolution Analysis of Follicular Lymphoma Genomes) to J.M.C., R.D.G. and M.A.M. We acknowledge support from NIH grants P50CA130805-01 “SPORE in Lymphoma, Tissue Resource Core (PI Fisher)” and 1U01CA114778 “Molecular Signatures to Improve Diagnosis and Outcome in Lymphoma (PI Chan)”. A.J.M. is a Career Development Program Fellow of the Leukemia and Lymphoma Society. N.A.J. was a research fellow of the Terry Fox Foundation (award NCIC 019005) and the Michael Smith Foundation for Health Research (ST-PDF-01793). M.A.M. is a Terry Fox Young Investigator and a Michael Smith Senior Research Scholar. R.D.M is a Vanier Scholar (CIHR) and holds a MSFHR senior graduate studentship. M.M.L acknowledges support from a Postdoctoral Fellowship from the Spanish Ministry of Education, under the “Programa Nacional de Movilidad de Recursos Humanos del Plan Nacional de I-D+i 2008-2011”. D.W.S was supported by the Terry Fox Foundation Strategic Health Research Training Program in Cancer Research at Canadian Institutes of Health Research (Grant No. TGT-53912). JJS receives acknowledges funding from The Canadian Cancer Society and the Canadian Institutes of Health Research. RG is supported by a UBC Four Year Fellowship. IMM acknowledges the Canadian Foundation for Innovation for a Leaders Opportunity Fund. The laboratory work for this study was undertaken at the Genome Sciences Centre, British Columbia Cancer Research Centre and the Centre for Translational and Applied Genomics, a program of the Provincial Health Services Authority Laboratories. The authors would like to thank Dr. Chris Greenman for supplying his software and also gratefully acknowledge Dr. Daniela Gerhard and Dr. Samuel Aparicio for helpful discussions and guidance. Special thanks to Cecelia Suragh, Robyn Roscoe, Armelle Troussard and Adrienne Drobnies for expert project management assistance, and to the Library Construction, Sequencing and Bioinformatics Teams at the Genome Sciences Centre. The content of this publication does not necessarily reflect the views of policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. All raw sequence data can be accessed using the DCC portal (http://cgap.nci.nih.gov/Data_Access), study accession phs000235.v2.p1.
Footnotes
Author Contributions: MAM, RDG, DEH, MH and JMC conceived of the study and led the design of the experiments. RDM performed the analysis of sequence data, identified mutations and, with MML, AJM and MAM, produced Figures and wrote the manuscript. MML, AJM, DLT, SC, SC, DS, HM, JS, MM, TZ, AD, KT, YB, MF, JTW and TMS designed and performed experiments to amplify, discover, and validate mutations. RG, MG and IMM contributed to analyses and reviewed the manuscript. NAJ, MB, BW and BM prepared the samples, performed sample sorting and COO analysis and contributed to the text. ABW and JJS collected and prepared constitutional DNA samples. KLM, RC, SL, MF and SJ generated de novo assemblies and identified mutations. MK, SR, MG, OY and EYZ wrote software and contributed to Figures. RC performed copy number analysis and produced Figure and SBN performed confirmatory FISH experiments. YZ and AT produced the sequencing libraries. IB, RH, SJMJ, RM, JS, MH contributed to the development of experimental and analytical protocols. LR provided materials and reviewed the manuscript.
The authors declare no competing financial interests.
The SRA accession for the submission is SRP001599 which is linked the dbGAP study accession phs000235.v2.p1.
References
- 1.Anderson JR, Armitage JO, Weisenburger DD. Epidemiology of the non-Hodgkin”s lymphomas: distributions of the major subtypes differ by geographic locations. Non-Hodgkin”s Lymphoma Classification Project. Ann Oncol. 1998;9:717–720. doi: 10.1023/a:1008265532487. [DOI] [PubMed] [Google Scholar]
- 2.Lenz G, Staudt LM. Aggressive lymphomas. N Engl J Med. 2010;362:1417–1429. doi: 10.1056/NEJMra0807082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Horsman DE, et al. Follicular lymphoma lacking the t(14;18)(q32;q21): identification of two disease subtypes. Br J Haematol. 2003;120:424–433. doi: 10.1046/j.1365-2141.2003.04086.x. [DOI] [PubMed] [Google Scholar]
- 4.Iqbal J, et al. BCL2 translocation defines a unique tumor subset within the germinal center B-cell-like diffuse large B-cell lymphoma. Am J Pathol. 2004;165:159–166. doi: 10.1016/s0002-9440(10)63284-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lenz G, et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc Natl Acad Sci USA. 2008;105:13520–13525. doi: 10.1073/pnas.0804295105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pasqualucci L, et al. Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. J Exp Med. 2006;203:311–317. doi: 10.1084/jem.20052204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kato M, et al. Frequent inactivation of A20 in B-cell lymphomas. Nature. 2009;459:712–716. doi: 10.1038/nature07969. [DOI] [PubMed] [Google Scholar]
- 8.Compagno M, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature. 2009;459:717–721. doi: 10.1038/nature07968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Davis RE, et al. Chronic active B-cell-receptor signalling in diffuse large B-cell lymphoma. Nature. 2010;463:88–92. doi: 10.1038/nature08638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ngo VN, et al. Oncogenically active MYD88 mutations in human lymphoma. Nature. 2011;470:115–119. doi: 10.1038/nature09671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mardis ER, et al. Recurring mutations found by sequencing an acute myeloid leukemia genome. N Engl J Med. 2009;361:1058–1066. doi: 10.1056/NEJMoa0903840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Shah SP, et al. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature. 2009;461:809–813. doi: 10.1038/nature08489. [DOI] [PubMed] [Google Scholar]
- 13.Morin RD, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42:181–185. doi: 10.1038/ng.518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Futreal PA, et al. A census of human cancer genes. Nat Rev Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pasqualucci L, et al. Inactivating mutations of acetyltransferase genes in B-cell lymphoma. Nature. 2011;471:189–195. doi: 10.1038/nature09730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yusuf I, Zhu X, Kharas MG, Chen J, Fruman DA. Optimal B-cell proliferation requires phosphoinositide 3-kinase-dependent inactivation of FOXO transcription factors. Blood. 2004;104:784–787. doi: 10.1182/blood-2003-09-3071. [DOI] [PubMed] [Google Scholar]
- 17.Saito M, et al. BCL6 suppression of BCL2 via Miz1 and its disruption in diffuse large B cell lymphoma. Proc Natl Acad Sci USA. 2009;106:11294–11299. doi: 10.1073/pnas.0903854106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lenz G, et al. Oncogenic CARD11 mutations in human diffuse large B cell lymphoma. Science. 2008;319:1676–1679. doi: 10.1126/science.1153629. [DOI] [PubMed] [Google Scholar]
- 19.Greenman C, Wooster R, Futreal PA, Stratton MR, Easton DF. Statistical analysis of pathogenicity of somatic mutations in cancer. Genetics. 2006;173:2187–2198. doi: 10.1534/genetics.105.044677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cheung KJ, et al. Acquired TNFRSF14 mutations in follicular lymphoma are associated with worse prognosis. Cancer Res. 2010;70:9166–9174. doi: 10.1158/0008-5472.CAN-10-2460. [DOI] [PubMed] [Google Scholar]
- 21.Du MQ, et al. BCL10 gene mutation in lymphoma. Blood. 2000;95:3885–3890. [PubMed] [Google Scholar]
- 22.Kreutz B, Hajicek N, Yau DM, Nakamura S, Kozasa T. Distinct regions of Galpha13 participate in its regulatory interactions with RGS homology domain-containing RhoGEFs. Cell Signal. 2007;19:1681–1689. doi: 10.1016/j.cellsig.2007.03.004. [DOI] [PubMed] [Google Scholar]
- 23.Bhattacharyya R, Wedegaertner P. Galpha 13 requires palmitoylation for plasma membrane localization, Rho-dependent signaling, and promotion of p115-RhoGEF membrane binding. J Biol Chem. 2000;275:14992–14999. doi: 10.1074/jbc.M000415200. [DOI] [PubMed] [Google Scholar]
- 24.Manganello JM, Huang J, Kozasa T, Voyno-Yasenetskaya TA, Le Breton GC. Protein kinase A-mediated phosphorylation of the Galpha13 switch I region alters the Galphabetagamma13-G protein-coupled receptor complex and inhibits Rho activation. J Biol Chem. 2003;278:124–130. doi: 10.1074/jbc.M209219200. [DOI] [PubMed] [Google Scholar]
- 25.Brunet A, et al. Protein Kinase SGK Mediates Survival Signals by Phosphorylating the Forkhead Transcription Factor FKHRL1 (FOXO3a) Mol Cell Biol. 2001;21:952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tai DJC, Su C, Ma Y, Lee EHY. SGK1 phosphorylation of IkappaB Kinase alpha and p300 Up-regulates NF-kappaB activity and increases N-Methyl-D-aspartate receptor NR2A and NR2B expression. J Biol Chem. 2009;284:4073–4089. doi: 10.1074/jbc.M805055200. [DOI] [PubMed] [Google Scholar]
- 27.Mo J, et al. Serum- and glucocorticoid-inducible kinase 1 (SGK1) controls Notch1 signaling by downregulation of protein stability through Fbw7 ubiquitin ligase. J Cell Sci. 2011;124:100–112. doi: 10.1242/jcs.073924. [DOI] [PubMed] [Google Scholar]
- 28.Young KH, et al. Structural profiles of TP53 gene mutations predict clinical outcome in diffuse large B-cell lymphoma: an international collaborative study. Blood. 2008;112:3088–3098. doi: 10.1182/blood-2008-01-129783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shilatifard A. Molecular implementation and physiological roles for histone H3 lysine 4 (H3K4) methylation. Current Opinion in Cell Biology. 2008;20:341–348. doi: 10.1016/j.ceb.2008.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Milne T, et al. MLL Targets SET Domain Methyltransferase Activity to Hox Gene Promoters. Mol Cell. 2002;10:1107–1117. doi: 10.1016/s1097-2765(02)00741-4. [DOI] [PubMed] [Google Scholar]
- 31.Krumlauf R. Hox genes in vertebrate development. Cell. 1994;78:191–201. doi: 10.1016/0092-8674(94)90290-9. [DOI] [PubMed] [Google Scholar]
- 32.Canaani E, et al. ALL-1//MLL1, a homologue of Drosophila TRITHORAX, modifies chromatin and is directly involved in infant acute leukaemia. Br J Cancer. 2004;90:756–760. doi: 10.1038/sj.bjc.6601639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wiedemann L, et al. Global Analysis of H3K4 Methylation Defines MLL Family Member Targets and Points to a Role for MLL1-Mediated H3K4 Methylation in the Regulation of Transcriptional Initiation by RNA Polymerase II. Mol Cell Biol. 2009;29:6074–6085. doi: 10.1128/MCB.00924-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Issaeva I, et al. Knockdown of ALR (MLL2) Reveals ALR Target Genes and Leads to Alterations in Cell Adhesion and Growth. Mol Cell Biol. 2007;27:1889–1903. doi: 10.1128/MCB.01506-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pleasance ED, et al. A small-cell lung cancer genome with complex signatures of tobacco exposure. Nature. 2010;463:184–190. doi: 10.1038/nature08629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dalgliesh GL, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–363. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Parsons DW, et al. The Genetic Landscape of the Childhood Cancer Medulloblastoma. Science. 2011;331:435–439. doi: 10.1126/science.1198056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Iqbal J, et al. Distinctive patterns of BCL6 molecular alterations and their functional consequences in different subgroups of diffuse large B-cell lymphoma. Leukemia. 2007;21:2332–2343. doi: 10.1038/sj.leu.2404856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pasini D, et al. Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res. 2010 doi: 10.1093/nar/gkq244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Giordano A, Avantaggiati M. p300 and CBP: partners for life and death. J Cell Physiol. 1999;181:218–230. doi: 10.1002/(SICI)1097-4652(199911)181:2<218::AID-JCP4>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 41.Han A, He J, Wu Y, Liu JO, Chen L. Mechanism of recruitment of class II histone deacetylases by myocyte enhancer factor-2. J Mol Biol. 2005;345:91–102. doi: 10.1016/j.jmb.2004.10.033. [DOI] [PubMed] [Google Scholar]
- 42.Youn H, Liu J. Cabin1 represses MEF2-dependent Nur77 expression and T cell apoptosis by controlling association of histone deacetylases and acetylases with MEF2. Immunity. 2000;13:85–94. doi: 10.1016/s1074-7613(00)00010-8. [DOI] [PubMed] [Google Scholar]
- 43.Yap DB, et al. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 2011;117:2451–2459. doi: 10.1182/blood-2010-11-321208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sneeringer CJ, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci USA. 2010;107:20980–20985. doi: 10.1073/pnas.1012525107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Goya R, et al. SNVMix: predicting single nucleotide variants from next-generation sequencing of tumors. Bioinformatics. 2010;26:730–736. doi: 10.1093/bioinformatics/btq040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Robertson G, et al. De novo assembly and analysis of RNA-seq data. Nat Meth. 2010;7:909–912. doi: 10.1038/nmeth.1517. [DOI] [PubMed] [Google Scholar]
- 48.Mortazavi A, Williams BA, Mccue K, Schaeffer L, Wold B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat Meth. 2008;5:621–628. doi: 10.1038/nmeth.1226. [DOI] [PubMed] [Google Scholar]
- 49.Wright G, et al. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci USA. 2003;100:9991–9996. doi: 10.1073/pnas.1732008100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.He J, et al. Structure of p300 bound to MEF2 on DNA reveals a mechanism of enhanceosome assembly. Nucleic Acids Res. 2011 doi: 10.1093/nar/gkr030. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.