

Abstract
Adolescent smoking is associated with auditory-cognitive deficits and structural alterations to auditory thalamocortical systems, suggesting that higher auditory function is vulnerable to nicotine exposure during adolescence. Although nicotinic acetylcholine receptors (nAChRs) regulate thalamocortical processing in adults, it is not known whether they regulate processing at earlier ages since their expression pattern changes throughout postnatal development. Here we investigate nicotinic regulation of tone-evoked current source density (CSD) profiles in mouse primary auditory cortex from just after hearing onset until adulthood. At the youngest ages, systemic nicotine did not affect CSD profiles. However, beginning in early adolescence nicotine enhanced characteristic frequency (CF)-evoked responses in layers 2–4 by enhancing thalamocortical, early intracortical, and late intracortical response components. Nicotinic responsiveness developed rapidly and peaked over the course of adolescence, then declined thereafter. Generally, responsiveness in females developed more quickly, peaked earlier, and declined more abruptly and fully than in males. In contrast to the enhancement of CF-evoked responses, nicotine suppressed shorter-latency intracortical responses to spectrally distant (non-CF) stimuli while enhancing longer-latency responses. Intracortical infusion of nAChR antagonists showed that enhancement of CF-evoked intracortical processing involves α4β2*, but not α7, nAChRs, whereas both receptor subtypes regulate non-CF-evoked late intracortical responses. Notably, antagonist effects in females implied regulation by endogenous acetylcholine. Thus, nicotinic regulation of cortical processing varies with age and sex, with peak effects during adolescence that may contribute to the vulnerability of adolescents to smoking.
Introduction
Adolescent smoking increases the risk of long-term dependence (Breslau and Peterson, 1996; Mackesy-Amiti et al., 1997; Chen and Millar, 1998; Adriani et al., 2003), with adolescent girls developing dependence faster after initial exposure (DiFranza et al., 2002). Although psychological and social factors undoubtedly contribute to smoking behavior, biological differences in nicotine sensitivity may underlie gender differences in abuse risk (Johnson et al., 2005). Studies indicate that adolescence is a vulnerable period for adaptation and lasting changes in brain structure and function due to nicotine exposure (Slotkin, 2002), with females being particularly vulnerable (Slotkin et al., 2007). Notably, adolescent smoking is associated with altered structure of subcortical white matter—axons that project to and from temporal, including auditory, cortex—and deficits in auditory-cognitive performance (Jacobsen et al., 2005; Jacobsen et al., 2007a,b). Thus, nicotine exposure during adolescence may interfere with thalamocortical maturation and negatively influences sensory processing and cognitive functions (Heath and Picciotto, 2009).
Nicotine is known to influence sensory-cognitive function in adults (Wesnes and Warburton, 1983; Levin, 2002; Picciotto, 2003; Sacco et al., 2004), including auditory-cognitive functions such as attention, even in nonsmokers (Provost and Woodward, 1991; Le Houezec et al., 1994) (but see Foulds et al., 1996; Levin et al., 1998; Vossel et al., 2008). Mechanisms underlying the enhancement of auditory-cognitive function by nicotine are complex, but likely involve increased cortical responsiveness to auditory stimuli (Harkrider and Champlin, 2001a,b; Harkrider et al., 2001). In adult animal models, nicotine enhances sensitivity to sound by lowering the acoustic threshold via nicotinic acetylcholine receptors (nAChRs) in primary auditory cortex (A1) (Liang et al., 2006). Although nicotinic activation depolarizes auditory thalamic neurons (McCormick and Prince, 1987; Curró Dossi et al., 1991) and enhances the excitability of axons in the auditory thalamocortical pathway (Kawai et al., 2007), it is not clear how these actions regulate cortical information processing, nor whether they occur in younger animals where they might contribute to adolescent vulnerability to nicotine.
Anatomical studies indicate that expression patterns of cortical nAChRs change during development, with different patterns for subtypes of nAChRs (for review, see Metherate, 2004). Receptors containing α7 subunits (α7 nAChRs) are present at birth, peak during the second postnatal week around the onset of hearing, and decline by the end of the third week in rodents (Fuchs, 1989; Broide et al., 1995, 1996), while those containing α4, β2, and other subunits (α4β2* nAChRs) increase during the second and third weeks (Prusky et al., 1988; Kumar and Schliebs, 1992). However, the functional relevance of altered expression patterns is not clear, and given the increased vulnerability of adolescents to smoking, it is possible that the increased nAChR levels underlie altered function. Here, using mice, we have investigated acute effects of systemic nicotine on tone-evoked cortical responses during preadolescent and adolescent periods (P21–P40) and adults (P70–P100). We determined that adolescence is a period of heightened sensitivity for nicotinic modulation of auditory processing in A1, including sex differences that imply earlier and greater sensitivity for females.
Materials and Methods
In vivo electrophysiology.
Animal care and use were approved by the University of California, Irvine, Institutional Animal Care and Use Committee. Mice (age P21∼100, FVB strain) were anesthetized with urethane (0.7 g/kg, i.p.; Sigma) and xylazine (13 mg/kg, i.p.; Phoenix Pharmaceuticals), placed in a sound-attenuating chamber (IAC), and maintained at 36–37°C. Anesthesia was supplemented as necessary with 0.13 g/kg, i.p., urethane and 1.3 mg/kg, i.p., xylazine, via a catheter to avoid movement. A craniotomy was performed, and the right auditory cortex was exposed. We first recorded local field potentials (LFPs) in response to acoustic stimuli (see Acoustic stimulation, below) on the cortical surface to determine the possible location of A1, as described previously (Kawai et al., 2007), with modification. Briefly, the tonotopic characteristic frequency (CF; frequency with the lowest threshold) gradient expected for A1 was identified using a glass pipette (∼1 MΩ at 1 kHz) with tones in 5 kHz steps at near-threshold intensities (−10 to 20 dB SPL), and recording in the anterior–posterior axis using LFPs on the cortical surface. LFPs were filtered and amplified (1 Hz to 10 kHz, AI-401 or AI-405 CyberAmp380), digitized (AxoGraph), and stored on an Apple Macintosh computer. Reversal of tonotopy, indicating the border with an anterior auditory field, was detected in most cases. We then located central A1 in the dorsal–ventral (D–V) axis by determining the site with the shortest latency and largest amplitude surface LFP to 10–70 dB tones recorded along the D–V axis. After the cortical surface mapping, we confirmed tonotopy in A1 by recording multiunit activity in the middle layers (typically 300 μm below the pia) using a tungsten electrode (1∼2 MΩ) at a few sites while constructing tuning curves. CFs determined this way matched within ∼1 kHz those determined by surface LFPs.
We then inserted a 16-channel silicon multiprobe (impedance 2–3 MΩ, 100 μm separation between recording sites) (NeuroNexus) into a selected site in A1 perpendicular to the pial surface to record LFPs throughout the cortical depth for current source density (CSD) analysis (see Data analysis, below). Typically, the top one or two recording sites of the multiprobe were above the pia and visible using a surgical microscope. After multiprobe insertion, we redetermined CF more precisely by analyzing the initial slope of LFPs recorded at a depth of 300–400 μm. CF was determined in 1 kHz steps as the frequency that elicited the lowest threshold, shortest latency LFP. Threshold was determined in 5 dB steps as the lowest intensity to evoke LFPs above baseline (>3 SDs above average >100 ms before tone onset) with consistent onset latencies. At threshold, onset latencies averaged (±SEM) 36 ± 4 ms (n = 11) in adult animals. Following CF determination, we recorded intensity functions (from below threshold to 70 dB SPL in 5–10 dB steps) at CF and two octaves below CF (non-CF).
For nicotine injection experiments, baseline LFPs were recorded by presenting 25 tones per frequency/intensity combination at 0.5 stimuli per second. Four different tones (CF tones at 0, 5, and 60 dB above threshold; non-CF tone at the same intensity as CF high-intensity tone) were presented sequentially and repeated until stable baseline responses were obtained. We estimated the stability of baseline LFPs online by measuring the initial slope (1–2 ms duration from estimated onsets) at a recording channel in the upper layer. At the end of stable baseline recording of at least 20 min., saline was injected intraperitoneally for control recordings for ∼20 min, and then nicotine [(-)-nicotine hydrogen tartrate salt, Sigma; 2 mg/kg, i.e., 0.7 mg/kg free base; all nicotine doses reported as free base) prepared in saline was injected, and recordings resumed immediately and lasted for ∼1 h. In some experiments, nicotine was injected after baseline without saline injection; data were combined since no significant differences were seen.
For cortical injection of nicotinic antagonists, a multiprobe fused to a cannula with the port (tip outer diameter 165 μm, inner diameter 100 μm) located at the eighth channel from the top of a 16-channel multiprobe was inserted into A1 (fluidic multiprobe, NeuroNexus). We attached a 0.5 ml Hamilton syringe to a connector on the fluidic multiprobe. The multiprobe was positioned in A1 such that the fluid port was 400 μm beneath the cortical surface. Following a stable baseline recording, we injected 100 nl of either ACSF (vehicle) or ACSF containing 1 μm dihydro-β-erythroidine (DHβE) or 10 nm methyllycaconitine (MLA) at a rate of 10 nl/min for 10 min, and recordings resumed immediately with the four-tone cycle repeated five times over ∼20 min. We then injected nicotine systemically and continued recordings.
At the end of each experiment, animals were killed with a lethal dose of anesthesia.
Acoustic stimulation.
Acoustic stimuli were digitally synthesized and controlled using MALab (Kaiser Instruments) on a dedicated Macintosh computer, and was delivered through a speaker (ES-1 or FF-1 with ED-1 driver, Tucker-Davis Technologies) positioned ∼3 cm in front of the left ear (open field). For calibration (SPL in decibels relative to 20 μPa) a microphone (model 4939 with Nexus amplifier, Brüel and Kjaer) was positioned in place of the animal at the tip of the left ear bar. Pure tones (100 ms duration, 5 ms rise/fall ramps) were presented to the contralateral ear with a frequency range of 3–40 kHz and an intensity range of −10 to 70 dB SPL.
Data analysis.
We averaged LFP responses to each set of 25 stimuli and derived CSD profiles as previously reported (Müller-Preuss and Mitzdorf, 1984; Kaur et al., 2005) using a custom program in AxoGraph X. We analyzed CSD traces in layer 4 or layers 2/3 with stable baseline and consistent sound-evoked responses for baseline (control), post-antagonist injection (if any) and postnicotine responses. The onset latency of each CSD trace was defined as the first point of consecutive data points of at least 3 ms duration that were above a threshold amplitude, defined as 3× SDs of 7–10 ms baseline before response. We analyzed three temporal phases of CF tone-evoked current sinks and two phases of non-CF tone-evoked sinks by measuring the current sink area over a defined time period (see Results). Data were grouped by sex and age. Current sink magnitudes were normalized to average baseline values, and the normalized data were averaged across animals.
Color CSD contours were constructed by averaging CSD traces from tone onset to the end of the tone (i.e., 100 ms) and normalizing amplitudes to the maximum positive (sink) and negative (source) values using DeltaGraph (Red Rock Software). To define the laminar locations, we assumed that the earliest onset sink in the middle layers was located at the border between layer 3 and layer 4, and the other layers arranged according to the laminar pattern determined in adult mice by Anderson et al. (2009): layers 1–4 had a width of 125 μm each, and layers 5 and 6 had a width of 250 μm each. Thus, the laminar boundaries depicted in figures are estimates only.
Statistical analysis.
Statistical comparisons were performed using Microsoft Excel. Changes in current sink amplitude and onset latency before and after nicotine injection (prenicotine vs postnicotine) were compared using paired t tests (two-tailed). Age dependence of nicotine effects was examined using one-way ANOVA (α = 0.05) among all five age groups (unless indicated otherwise) with post hoc t tests for pairs of age groups. Sex differences were examined using t tests at each age. All mean data are reported ±SEM, unless indicated otherwise.
Results
To determine how nicotinic regulation of A1 varies across development, we measured tone-evoked CSD profiles at regular intervals before and after administration of nicotine in 44 mice ranging in age from P21 (∼1 week after hearing onset) to adult. In each animal, we inserted a 16-channel silicon multiprobe into A1 to record LFPs evoked by CF and non-CF (two octaves below CF) stimuli, and constructed CSD profiles off-line. To examine periadolescent trends, separately for male and female mice, we grouped data into five age ranges that, for convenience, we refer to as preadolescence (P21–P25, 3 males, 4 females), early adolescence (P26–P30, 5 males, 5 females), mid-adolescence (P31–P35, 3 males, 4 females), late adolescence (P36–P40, 4 males, 3 females), and adult (P70–P100, 7 males, 6 females). The age range for adolescence is based on that previously established for the rat (P28–P45) (Spear, 2000). We also examined sex differences within each of these age groups.
Tone-evoked cortical responses in A1 of adolescent mice
CSD profiles indicate the location of current sinks (i.e., putative sites of synaptic inputs). In young mice (P21–P40), as in adults (Kawai et al., 2007), CF tones generated one or two large current sinks in the upper layers (Fig. 1A). The earliest onset current sink in the middle layers occurred near the border of layers 3 and 4 (referred to as the “layer 3/4 sink”) (Fig. 1A, green CSD trace). This current sink or the one immediately above it (Fig. 1A, red CSD trace), was the largest amplitude current sink in each animal (the “CF main sink”). We also observed a small amplitude sink with a short duration in the lower layers (the “layer 5/6 sink) (Fig. 1A, blue CSD trace). Onset latencies of the layer 5/6 sink were shorter than those of the layer 3/4 sink in most animals (43 of 44 animals; p < 0.05, paired t test), suggesting that thalamocortical synaptic inputs first contact the lower layers of A1 before reaching layers 3/4. This CSD profile in mouse A1 is in general agreement with previous findings in other species, where the lemniscal thalamocortical input from the ventral division of medial geniculate nucleus (MGv) to layers 3/4 is rapidly amplified intracortically to generate large current sinks, while thalamocortical collateral inputs to layer 5/6 generate smaller current sinks (for monkey, see Müller-Preuss and Mitzdorf, 1984; Steinschneider et al., 1992; for rat, see Barth and Di, 1990; Kaur et al., 2005; Sakata and Harris, 2009; for gerbil, see Happel et al., 2010).
Figure 1.

Tone-evoked CSD profiles in adolescent mouse A1. A, B, LFPs and derived CSD profiles evoked in response to CF (A; 22 kHz) and non-CF (B; 5.5 kHz; both 60 dB SPL) tones in a P32 female mouse. In CF-evoked CSD traces (top, middle column), the layer 3/4 sink (green trace) had the earliest onset in the upper layers, and the CF main sink (red trace) occurred 100 μm more superficially. Note that the onset of the layer 5/6 sink (blue trace) is earlier than that of the layer 3/4 sink. The non-CF main sink (bottom, middle column, orange trace) was 100 μm below the depth of the CF main sink and had a longer onset latency (vertical dotted lines are aligned to the onset of the CF-evoked layer 3/4 sink). Response traces are from recording sites separated by 100 μm, and trace duration also indicates tone duration (100 ms). Calibration: LFP, 0.2 mV; CSD, 10 mV/mm2. CSD color contours (right) were constructed by normalizing CSD traces to maximum and minimum values; thick horizontal lines indicate five response phases: CF-evoked input (1), early intracortical (2), and late intracortical (3) phases, and non-CF-evoked early (4) and late (5) phases. Numbers on far left indicate cortical layer. Laminar proportions are based on adult values and are estimates only (see Materials and Methods for details). WM, White matter.
We also recorded responses to a spectrally distant (non-CF) tone two octaves below CF at the same intensity. Non-CF stimuli evoked the largest amplitude current sink (the “non-CF main sink”) at the same cortical depth as CF tones did in most animals (26 of 30 preadolescent and adolescent animals; 10 of 13 adult animals; see Nicotinic regulation of non-CF tone evoked current sinks, below), or at an adjacent recording site 100 μm above or below the CF main sink (Fig. 1B). Compared with the CF-evoked sinks, the non-CF-evoked sinks exhibited significantly longer onset latencies with low magnitude, remained elevated longer and spread more widely across the upper cortical layers (i.e., multiple recording sites responding with similar sink magnitude) (Fig. 1B). Direct thalamocortical inputs likely do not contribute to the non-CF main sink, which more likely reflects intracortical synaptic activity (Metherate and Cruikshank, 1999; Kaur et al., 2005; Happel et al., 2010).
Cortical processing matures early in adolescence
To determine whether CSD profiles change across periadolescent development, we analyzed the magnitude of CF-evoked current sinks. CSD traces in the upper layers were divided into three phases—Input, Early Intracortical, and Late Intracortical (Fig. 1A, right, 1–3)—and areas under the traces for each phase were quantified. The input phase is the first 3 ms of the layer 3/4 sink, capturing the linearly rising component of the sink before its peak. This phase reflects presumed thalamocortical inputs. The early intracortical phase (Fig. 1A, phase 2) reflects the sink magnitude 3∼20 ms after onset of the CF main sink, which includes its peak response. In addition, we often observed a late component of current sinks with a secondary peak several tens of milliseconds later; this Late Intracortical phase (Fig. 1A, phase 3) reflects sink magnitude 30∼80 ms after onset of the CF main sink. Comparison of the three phases showed no difference among the five age groups [Fig. 2A (see figure legends for statistics)], suggesting that the magnitude of tone-evoked thalamocortical and intracortical synaptic activities does not change beyond the third week in mouse A1.
Figure 2.

A, B, Periadolescent development of tone-evoked current sink magnitudes (A) and onset latencies (B). A, Magnitudes of CF tone-evoked current sinks for input (black), early intracortical (white), and late intracortical (gray) phases for five age groups, normalized to magnitudes at P21–P25. Current sink areas for P21–P25 were 51.8, 1671, and 3865 mV · ms/mm2 for input, early intracortical, and late intracortical phases, respectively. No significant differences were observed among age groups (p = 0.71, 0.97, and 0.93 for input, early intracortical, late intracortical phases, respectively; five-age, one-way ANOVA). B, Onset latencies of CF tone-evoked layer 3/4 sink (black), layer 5/6 sink (white), and the non-CF main sink (gray). Asterisks indicate differences between groups (p < 0.05, two-tailed t test).
We also analyzed the onset timing of tone-evoked responses. The onset latencies of the layer 3/4 sink, the layer 5/6 sink and the non-CF main sink in preadolescence were longer than those in the adult (Fig. 2B). However, by early adolescence these same latencies had shortened to adult-like values. Consistent with this, the latency difference between the response onset in layer 3/4 and that in layer 5/6, which likely reflects conduction times in thalamocortical afferents, significantly decreased from preadolescence (4.7 ± 0.9 ms, n = 7) to adult-like values in early adolescence (2.3 ± 0.5 ms, n = 10; preadolescence vs early adolescence p < 0.05, t tests) (compare with 2.8 ± 0.5 ms, n = 7, in mid-adolescence; 2.4 ± 0.3 ms, n = 7, in late adolescence; and 2.0 ± 0.4 ms, n = 13, in adults; all values are significantly different from preadolescence). Finally, the difference between the onset latencies of the CF-evoked layer 3/4 sink and the non-CF main sink showed a tendency to decrease from preadolescence (11.2 ± 1.2 ms) to adult-like values in early adolescence (8.4 ± 2.1 ms; compare with 8.4 ± 2.8 ms in mid-adolescence; 8.7 ± 2.0 ms in late adolescence), but this trend did not reach significance (p > 0.05, t tests). The latency difference in preadolescence was, however, significantly different from that in adults (6.9 ± 1.4 ms, n = 13; p < 0.05, t tests), suggesting again that intracortical processing is immature in preadolescence. Overall, these data indicate that some basic aspects of synaptic connectivity in the auditory pathway mature by early adolescence.
Nicotine regulation of tone-evoked CSD profiles in A1 varies with age and sex
We next examined the effects of acute, systemic nicotine (0.7 mg/kg, i.p.) on tone-evoked CSD profiles. This nicotine dose had been determined in preliminary experiments to be low but effective, since lower doses (0.17 and 0.35 mg/kg) did not alter tone-evoked responses. Figure 3 shows examples of CSD profiles for CF-evoked responses in young mice. Qualitatively, nicotine had little effect on a preadolescent female (Fig. 3A), but strongly enhanced responses in an early-adolescent female (Fig. 3B; response amplitude color scale normalized to peak value in nicotine). To quantify nicotine effects, we analyzed current sink areas for the three phases of CF-evoked responses (Fig. 1A, 1–3), measured at regular intervals (∼4 min) before and after systemic nicotine. Figure 4A shows the time course for the input phase of the layer 3/4 sink (presumed thalamocortical input), separately for male and female animals in each of the five age groups. No nicotine enhancement was seen in preadolescents of either sex. In all other age groups, nicotine enhanced the Input phase, an effect that for most groups peaked within ∼12 min and declined over ∼30–60 min. In early-adolescent females, the nicotine enhancement was especially strong, peaked after the initial 12 min period, and lasted ∼50 min. The effect of nicotine on input responses for each sex and age group is summarized in Figure 4A (bottom), which depicts current sink magnitude averaged over 12 min time windows (Fig. 4A, top, vertical lines) up to ∼48 min postnicotine. Nicotine enhanced Input phase beginning in early adolescence for both sexes (Fig. 4, asterisks) (p < 0.05, prenicotine vs postnicotine, paired t tests). Enhancement in females peaked during early adolescence [for 12–24 min: F(2,11) = 4.27, p = 0.042, three age groups (preadolescence, early adolescence, and adult), ANOVA; for 24–36 min: F(4,17) = 4.01, p = 0.018, ANOVA]. Post hoc t tests show enhancement for early adolescence and adults compared with preadolescence. The peak enhancement in females differed from the nicotine effect in males of the same age for postnicotine intervals of 12–48 min (p < 0.05, t tests). Nicotine enhancement in males first became apparent in early adolescence, and increased in magnitude during late adolescence (P36–P40; 0–12 min: F(4,17) = 3.64, p = 0.026, ANOVA). Post hoc t tests show significant enhancement for all adolescent age groups and adults over preadolescence.
Figure 3.
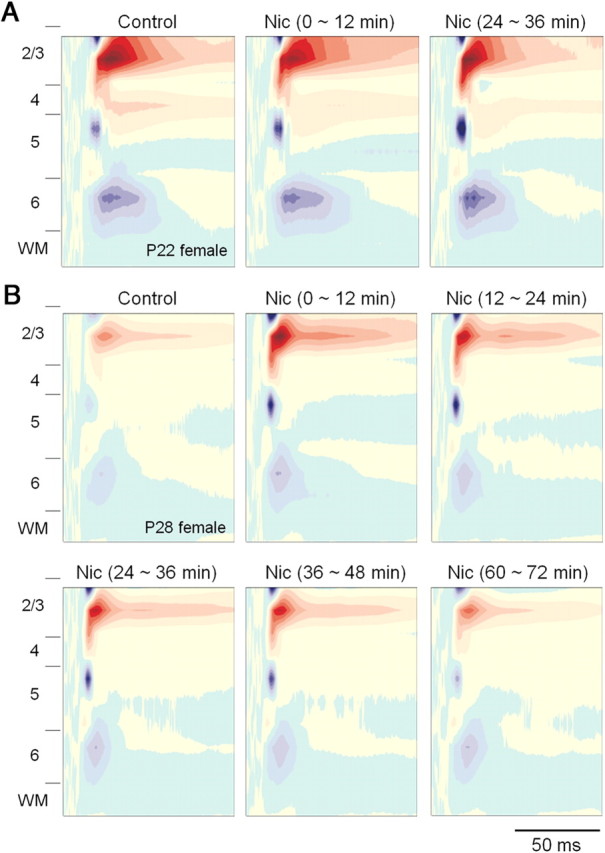
Effects of systemic nicotine on tone-evoked CSD profiles become apparent during adolescence. A, B, Nicotine had little effect on CSD profile in a preadolescent female (P22; CF stimulus, 21 kHz, 65 dB SPL) (A), but enhanced CSD profile in an early-adolescent female (P28; CF stimulus, 20 kHz, 65 dB SPL) (B). Each CSD profile is an average of three from within a 12 min time period: for example, before (Control), 0∼12 min after (compare with Fig. 4A, shaded area), 24∼36 min after nicotine (Nic), with profiles for each animal normalized to maximum source and sink values among all conditions.
Figure 4.
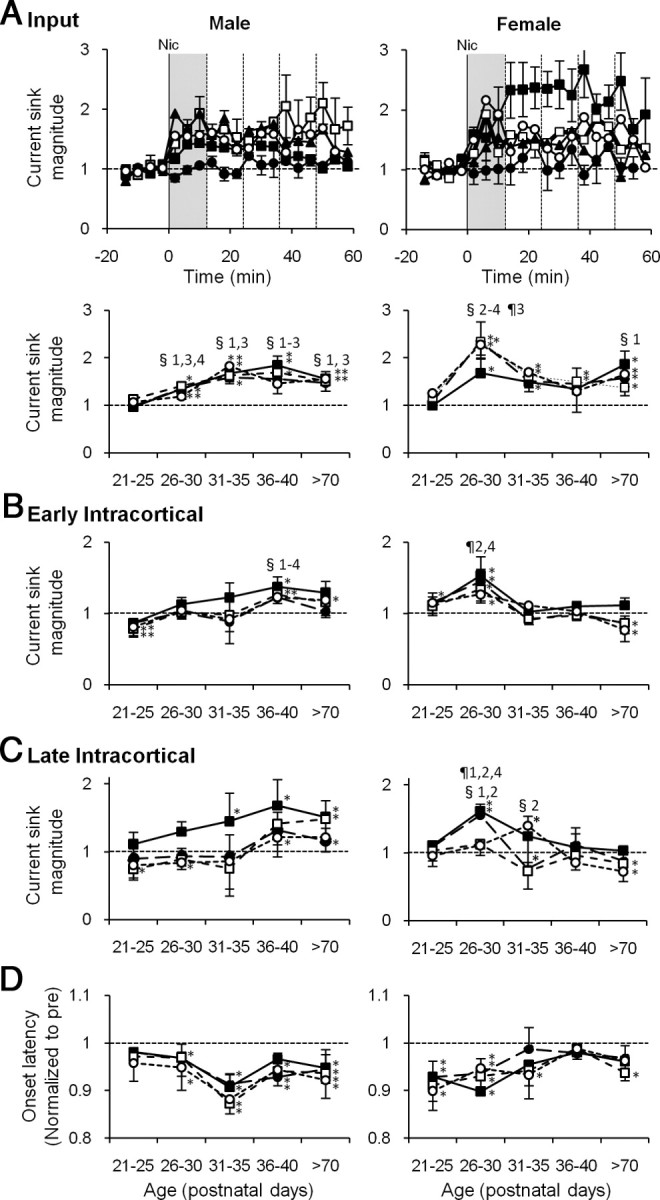
Nicotinic effects on CF-evoked CSD profiles peak during adolescence, earlier for females than males. A, Time course of nicotine's effect on the input phase of layer 3/4 sink. Current sink areas are normalized to prenicotine baseline in males (top left) and females (top right) for each age group: P21–P25 (•); P26–P30 (■); P31–P35 (□); P36–P40 (▴); >P70 (○). Vertical solid lines at 0 min indicate the time of nicotine injection. Error bars for some data omitted for clarity. Bottom, Time course of nicotine effect at each age, plotted for 12 min postnicotine intervals (vertical dotted lines at top): 0∼12 min (■); 12∼24 min (•); 24∼36 min (□); 36∼48 min (○). B, C, Time course of nicotine effect at each age for early intracortical (B) and late intracortical (C) phases of the CF main sinks. D, Time course at each age of nicotine effect on onset latencies of CF tone-evoked layer 3/4 sinks averaged; symbols are as given above, and latencies were normalized to prenicotine baseline. Error bars (±SEM) are indicated upward for black symbols and downward for white symbols for clarity. *Prenicotine versus postnicotine, p < 0.05, paired t test; §significant differences compared with P21–P25; ¶significant differences compared with >P70 (p < 0.05, one-way ANOVA). The numbers by the symbols indicate the group time points with 1 for 0∼12 min, 2 for 12∼24 min, 3 for 24∼36 min, and 4 for 36∼48 min.
For those age groups and time windows with significant enhancement of the Input phase, we also analyzed nicotine effects on the layer 5/6 current sink (i.e., presumed thalamocortical collateral; sink area measured over 3 ms from sink onset). Unexpectedly, nicotine did not enhance the layer 5/6 sink during the 0–12 min postnicotine interval for any age or either sex, and for later postnicotine intervals (up to 48 min) enhancement occurred in only 25% of intervals (7 of 28, 12 min interval: 12–48 min in P26–P30 males; 24–48 min in P36–P40 males; and 24–48 min in >P70 females). These data indicate that there is little correspondence between nicotine effects in layer 3/4 and in layer 5/6.
The effects of nicotine on the Intracortical phases of CF-evoked responses also peaked during adolescence and showed striking sex differences. For the Early Intracortical phase (Fig. 4B), enhancement in females peaked in early adolescence and disappeared in older adolescents and adults (P26–P30: 12–24 min, F(4,17) = 3.93, p = 0.019, ANOVA; 12–24 and 36–48 min, p < 0.05, post hoc t tests). In males, after a slight nicotinic suppression in preadolescence, enhancement occurred only in late adolescents and adults (prenicotine vs postnicotine, paired t tests). late adolescence was the only age group that differed from preadolescent males (post hoc t tests). The late-adolescent enhancement also differed significantly from that in females of the same age (for 12–24 and 24–36 min ranges, p < 0.05, t tests). For the Late Intracortical phase (Fig. 4C), females again showed heightened nicotinic enhancement in early adolescence, which declined at later ages (for 12–24 min, F(4,17) = 6.84, p = 0.002, all-age ANOVA), whereas males showed nicotinic enhancement in mid-adolescence and thereafter (prenicotine vs postnicotine, paired t tests). The early-adolescent enhancement in females differed significantly from that in males of the same age (for 12–24 and 36–48 min ranges, p < 0.05, t tests).
Thus, nicotinic enhancement of CF-evoked current sinks begins, and peaks, during adolescence. Notable sex differences include the peak in nicotinic enhancement that occurs ∼10 d earlier in females than in males, and the diminished intracortical effects in females at any age after early adolescence.
Nicotine effect on thalamocortical latency
Nicotine generally decreased the onset latency of the CF-evoked current sink in layer 3/4, and this effect occurred earlier in development than effects on sink magnitude (above). Preadolescent females, but not males, exhibited nicotine-induced reduction of onset latency (Fig. 4D), even though current sink magnitude was not affected (Fig. 4A). Peak effects of ∼10% reduction in onset latency occurred during early adolescence for females and mid-adolescence for males, with lesser but significant effects persisting into adulthood. The nicotine-induced decrease in onset latency persisted for ∼30 min or more, postinjection (data not shown). Thus, again, nicotine effects peaked during adolescence and occurred earlier for females than for males. Nicotinic effects on sink onset latency, but not magnitude, during preadolescence implies regulatory mechanisms with different development time courses and, possibly, different locations (e.g., subcortical and cortical, respectively).
Nicotinic regulation of non-CF tone-evoked current sinks
Nicotinic regulation of non-CF-evoked CSD profiles differed from that of CF-evoked responses. Figure 5A shows an example: in contrast to the uniform enhancement of CF-evoked response phases (as described above), nicotine reduced the Early component of the non-CF main sink and enhanced the Late component. Reduction of the Early component was observed in ∼70% of animals across ages and in both sexes (males: 10 of 15 periadolescents, 5 of 7 adults; females: 11 of 15 periadolescents, 5 of 6 adults). For quantitative analysis, we divided the non-CF main sink into an early phase (0–20 ms after sink onset) (Figs. 1B, 5A, labeled 4) and a Late phase (from 30 ms after sink onset to 80–100 ms) (Figs. 1B, 5A, labeled 5). The time course of nicotine's effect on the Early phase is shown in Figure 5B; quantification of the first three time points after nicotine (Fig. 5B, shaded areas) indicated that nicotine either suppressed or did not affect the Early phase in all age groups and both sexes (Fig. 5B,C). No enhancement of the Early phase was seen at any age. The most consistent and long-lasting suppressive effects of nicotine were observed in adults of both sexes.
Figure 5.
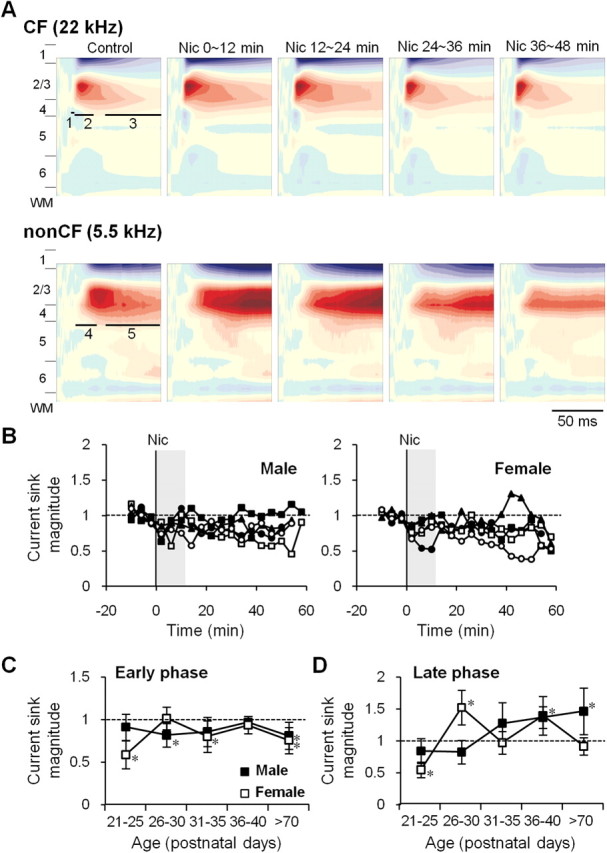
Nicotine has a different effect on non-CF-evoked CSD profiles than on CF-evoked profiles. A, An example of nicotine's differential effects on CSD profiles evoked by CF vs non-CF stimuli (P30, female). Nicotine enhanced all three phases of the CF main sink, but for the non-CF main sink it reduced the early phase (labeled 4) while it enhanced the late phase (labeled 5). CSD profiles are averages of three time points within 12 min intervals before (Control), and 0∼12, 12∼24, 24∼36, and 36∼48 min after nicotine injection (Nic). Maximum and minimum values used for normalization were 50.7 and −51.9 mV/mm2 for CF, and 11.2 and −17.5 mV/mm2 for non-CF, respectively. B, Effect of nicotine on the early phase of non-CF main current sinks. Time course of effects for males (B, left) and females (B, right) for each age group: P21–P25 (•); P26–P30 (■); P31–P35 (□); P36–P40 (▴); >P70 (○). Error bars omitted for clarity. C, Average nicotine effect on early phase (first 12 min after nicotine, i.e., shaded area in B) for males (■) and females (□). D, Effect of nicotine on late phase; asterisk for data at P36–P40 applies to males only. Error bars: ±SEM. *Prenicotine vs postnicotine, p < 0.05, paired t test.
Analysis of nicotine effects on the Late phase of the non-CF main sink (Fig. 5D) shows a developmental pattern that more closely resembles regulation of CF-evoked responses (compare Figs. 4, 5). Nicotinic enhancement of the Late phase peaked in early-adolescent females and late-adolescent males. Thus, CF and non-CF tones may recruit partially overlapping intracortical neural circuits, and nicotine may affect late-intracortical processing for each in a similar manner.
Intracortical injection of nAChR antagonists, DHβE, or MLA
To determine whether the effects of systemic nicotine were due to nAChRs located in A1—and if so, which nAChR subtypes—we injected nicotinic antagonists directly into the cortex via a fluid port assembly fused to the 16-channel multiprobe. The fluid port opening was positioned between recording channels, approximately in layer 4. Experiments were performed at ages of peak nicotine responsiveness for each sex (i.e., in late-adolescent males and early-adolescent females). Following baseline recordings of CF-evoked responses, 100 nl of ACSF or drug was infused slowly (10 nl/min over 10 min) (Fig. 6, vertical shading), and recordings resumed for ∼20 min before delivery of systemic nicotine (Fig. 6, solid vertical lines). Cortical infusion of DHβE (1 μm at port), an antagonist at α4β2* nAChRs (Mulle and Changeux, 1990; Whiting et al., 1991; Alkondon and Albuquerque, 1993; Wong et al., 1995; Buisson et al., 1996), blocked nicotine enhancement of the CF-evoked Early (Fig. 6B) and Late (Fig. 6C) Intracortical phases, but did not block enhancement of the Input phase (Fig. 6A), in both sexes. Notably, in females only, DHβE alone (before nicotine administration) reduced the magnitude of the intracortical sinks (Fig. 6B,C), but not the Input sink, implying sex-specific actions of endogenous transmitter on intracortical processes. DHβE also blocked nicotinic enhancement of the non-CF-evoked Late phase [data not shown; normalized nicotine effect after ACSF: 1.56 ± 0.54 in males (n = 6), 1.27 ± 0.18 in females (n = 7), paired t tests comparing prenicotine vs postnicotine, p < 0.05; nicotine effect after DHβE: 1.14 ± 0.13 in males (n = 5), 0.90 ± 0.11 in females (n = 6); paired t tests, p > 0.05]. Thus, cortical α4β2* nAChRs mediate enhancement of CF- and non-CF-evoked intracortical, but not subcortical, processes in both sexes, with females exhibiting evidence for similar regulation by endogenous ACh.
Figure 6.

Effects of intracortical DHβE injection on nicotinic regulation of CF tone-evoked current sinks. A–C, Time course of effects on Input (A), Early Intracortical (B), and Late Intracortical (C) current sinks are shown for males (P36–P40; left column) and females (P26–P30; middle column). ACSF (□) or DHβE (■; 1 μm at fluid port) was injected intracortically over 10 min (shaded area). Nicotine was injected systemically at the vertical lines (0 min). Right column, Mean current sink magnitude (average of 5 time points) before (□) and after (■) nicotine in the presence of ACSF (males, n = 6; females, n = 8) or DHβE (males, n = 5; females, n = 7) for males (left side) and females (right side). Error bars: ± SEM. *p < 0.05, before versus after nicotine, paired t test; §p < 0.05, baseline control versus DHβE, paired t test.
Cortical infusion of MLA (10 nm at port), a specific antagonist at α7 nAChRs (Alkondon et al., 1992; Drasdo et al., 1992), had no effect on nicotinic enhancement of any CF-evoked current sink: Input, or Early or Late Intracortical (Fig. 7). However, MLA did prevent nicotinic enhancement of the non-CF-evoked Late phase (data not shown; normalized nicotine effect after MLA: 0.83 ± 0.07 in males (n = 7), 1.00 ± 0.13 in females (n = 5); p > 0.05, paired t tests comparing prenicotine vs postnicotine). Thus, α7 nAChRs and α4β2* nAChRs both regulate the non-CF-evoked Late current sink.
Figure 7.

Effects of intracortical MLA injection on nicotinic regulation of CF tone-evoked current sinks. A–C, Time course of effects on Input (A), Early Intracortical (B), and Late Intracortical (C) current sinks are shown for males (P36–P40; left column) and females (P26–P30; middle column). ACSF (□) or MLA (■, 10 nm at fluid port) was injected intracortically over 10 min (shaded area). Nicotine was injected systemically at the vertical lines (0 min). Right column, Mean current sink magnitude (average of 5 time points) before (□) and after (■) nicotine in the presence of ACSF (males, n = 6; females, n = 8) or MLA (males, n = 7; females, n = 6) for males (left side) and females (right side). Error bars: ±SEM. *p < 0.05, before versus after nicotine, paired t test.
These antagonist data suggest that α4β2* nAChRs located in A1 mediate enhancement of CF-evoked intracortical processing, while both α4β2* and α7 nAChRs mediate enhancement of non-CF-evoked intracortical processing. Administration of antagonists to A1 did not block enhancement of the CF-evoked Input current sink, likely due to dependence on subcortical nAChRs.
Discussion
Our results demonstrate how nicotinic regulation of tone-evoked CSD profiles in mouse A1 changes across adolescent development. The effects of nicotine are informative in two general ways. First, differential regulation of response components helps distinguish underlying neural circuits, thereby identifying independent substrates of acoustic processing. At least four distinct thalamocortical and intracortical circuits are implicated in the processing of CF and spectrally distant non-CF stimuli (Fig. 8). Second, the results identify developmental functions of nAChRs: over the course of adolescence, nicotinic regulation of tone-evoked responses develops rapidly and peaks, before declining thereafter. Remarkably, nicotinic regulation in females develops more quickly, peaks earlier, and declines more abruptly and fully than in males. These results elucidate potential mechanisms of (1) acoustic processing, (2) cognitive enhancement by nicotine or endogenous ACh, and (3) enhanced susceptibility of adolescents to nicotine abuse.
Figure 8.

Model of information processing in A1 and its regulation by nicotine. A, CF stimuli activate auditory thalamic (MGv) neurons, which provide input to layers 3/4 in A1 (1). The thalamocortical input evokes intracolumn intracortical activity (2; column indicated by shaded area), followed by possibly intercolumn activity (4). Non-CF stimuli similarly activate thalamocortical input and subsequent intracolumn activity, which then rapidly propagates to the CF site via long-distance, “horizontal” projections (3). Horizontal projections also, though more slowly, recruit intercolumn activity (4), which may overlap partially, though not fully (see Discussion), with similar CF-elicited activity. B, In early-adolescent females, late-adolescent males, and adult males, nicotine enhances (1, 2, and 4), but suppresses or does not affect (3). Receptors containing α4β2, but not α7, nAChR subunits mediate enhancement of 1 (Kawai et al., 2007) and 2 (moreover, sensitivity to DHβE was qualitatively greater in females than in males) and CF-evoked activity (4). Both α4β2* and α7 nAChRs mediate enhancement of non-CF-evoked activity (4). The receptor mechanism underlying nicotinic suppression of 3 is not clear. C, In early-adolescent males and late-adolescent and adult females, nicotine enhances 1 and suppresses 3, but has little effect on 2 and 4.
Neural circuits that mediate acoustic processing in auditory cortex
We used five temporal windows to characterize CF- and non-CF-evoked responses: CF-evoked Input, Early Intracortical, and Late Intracortical phases, and non-CF-evoked Early and Late phases. To estimate thalamocortical input, we measured the first 3 ms of the layer 3/4 sink (Input), thus capturing the initial rise of the sink but stopping well short of its peak. Recent work indicates that the peak reflects mostly local activity within cortex (Kaur et al., 2005; Happel et al., 2010) and therefore is more properly included in our Early Intracortical phase (3–20 ms after sink onset). Our results, showing differential effects of nicotine and DHβE on Input and Early Intracortical phases, further support the use of these temporal windows to identify distinct response components. Similarly, nicotine had differential effects on the CF-evoked Early Intracortical phase (enhancement) and the non-CF-evoked Early phase (suppression or no effect), despite their similar ∼20 ms time windows, indicating that the two responses reflect different synaptic circuits. This observation is consistent with the idea that the CF tones recruit intracolumn microcircuits, whereas non-CF-evoked activity propagates rapidly across cortex (horizontally) to the CF site from a distant site where the non-CF stimulus is CF (Kaur et al., 2005; Liu et al., 2007; Happel et al., 2010). Finally, nicotine enhanced both CF- and non-CF-induced late intracortical responses (30–80 ms postonset), but via different receptor mechanisms (see Cellular mechanisms of nicotinic regulation, below). Thus, the long-latency intracortical activity evoked by CF vs non-CF stimuli involves at least partly separate neural populations.
Based on these data, we propose a model of the circuitry underlying auditory information processing in mouse A1 (Fig. 8). CF tones induce thalamocortical synaptic inputs in layers 3/4 (Input phase; Fig. 8A, labeled 1) and subsequent intracolumn activities within local circuits in the input layers (Early Intracortical; Fig. 8A, labeled 2), followed by population activity that may involve recurrent projections and propagate across cortex (Late Intracortical; Fig. 8A, labeled 4). Non-CF inputs to cortex recruit fast intracortical, intercolumn activity (Early phase; Fig. 8A, labeled 3) and slower intercolumn activity that may share features and circuitry with CF-evoked Late Intracortical activity (Late; Fig. 8A, labeled 4). Slow (Late) intracortical activity elicited by CF vs non-CF stimuli may be similar, but are not identical given the differential effects of nAChR antagonists. Some elements in this model are similar to that recently proposed by Happel et al. (2010). Moreover, previous work using the in vitro thalamocortical slice has demonstrated that subcortical stimulation elicits short latency “on-focus” and “off-focus” responses in A1, with each followed by slowly propagating, cell assembly-like activity (Metherate and Cruikshank, 1999). The in vitro responses may relate to the present CF- and non-CF-evoked early responses followed by slower intracortical activity, thereby providing a useful model for understanding mechanisms of processing.
Age and sex differences in nicotinic regulation of auditory processing
The two major findings of this study are that nicotinic regulation of auditory processing peaks during adolescence, yet differs, in terms of developmental time course, between sexes. Nicotinic enhancement of thalamocortical inputs peaked earlier in females than in males, yet continued into adulthood for both sexes. Nicotinic enhancement of early intracolumn and late intercolumn activity also peaked earlier in females than in males, yet this effect persisted into adulthood only in males. Thus, nicotinic regulation of intracortical activity varied across adolescent development and between sexes, even though basic functional connectivity matured early in adolescence and did not differ between sexes (Fig. 2). These developmental changes in auditory processing, therefore, result from changes in nicotinic regulation.
Another major finding is that, in contrast to the enhancement of most response phases, nicotine had little effect on, or even suppressed, non-CF-induced early intercolumn horizontal projections. Since short latency responses determine frequency-receptive fields in A1 (Kaur et al., 2004; Liu et al., 2007), our findings suggest that nicotine increases receptive field selectivity (Liang et al., 2006; Metherate, 2011). The extent and mechanism of enhanced selectivity differed with age and sex, depending on the degree to which thalamocortical and intracortical activity was enhanced and early intracortical activity was suppressed. It appears that nicotine-enhanced “filtering” would be more prominent in late-adolescent and adult males and early-adolescent females, and weaker in early-adolescent males and late-adolescent and adult females (Fig. 8B,C).
Nicotinic enhancement of thalamocortical transmission
Nicotinic enhancement of thalamocortical input is widely hypothesized to depend on presynaptic nAChRs (Gil et al., 1997; Clarke, 2004). This hypothesis is supported by a dense band of nicotine binding sites, presumably α4β2*nAChRs, in layers 3/4 of primary sensory cortex in some species (for rat, see Clarke et al., 1984, 1985; London et al., 1985) (for cat, see Prusky et al., 1987; Parkinson et al., 1988), and by suppression of sensory responses in the thalamocortical input layers by an intracortical injection of a nicotinic antagonist, mecamylamine (for cat, see Parkinson et al., 1988; for rat, see Liang et al., 2006). In this study, however, intracortical injections of nicotinic antagonists had little effect on the Input phase in either sex, suggesting the absence of presynaptic nAChRs in mouse A1. Similarly, nicotine does not exert presynaptic modulation of monosynaptic thalamocortical synapses in adult mice in vitro (Kawai et al., 2007), nor do mice show a prominent band of nAChRs in layers 3/4 of A1 (binding of [3H]epibatidine and [125I]-α-bungarotoxin in 1-month-old mice, unpublished observations; binding of [18F]nifene in adult mice; J. Mukherjee and R. Kant, unpublished observations). Nicotinic enhancement of thalamocortical inputs could still occur, however, via axonal α4β2*nAChRs expressed along the thalamocortical pathway, as inferred from studies of mouse (Kawai et al., 2007; binding of [18F]nifene, J. Mukherjee and R. Kant, unpublished observations), rat (Pichika et al., 2006), and human (Ding et al., 2004). This is consistent with our interpretation that intracortical antagonist injection did not diffuse out of cortex to influence subcortical axonal nAChRs and, as a consequence, thalamocortical inputs in mouse. Thus, depending on species, axonal nAChRs and/or presynaptic nAChRs may underlie nicotinic enhancement of thalamocortical inputs.
If all thalamocortical axons possess axonal nAChRs and project to both layers 3/4 (main cortical input) and layers 5/6 (collateral input), one would expect to see nicotine enhancement of both current sinks. However, this was not the case; nicotinic effects on the Input phase typically were not accompanied by similar effects on the layer 5/6 collateral input. One explanation might be that axonal nAChRs are found only in a subset of thalamocortical afferents, which may not provide collateral inputs to layers 5/6, but future studies will be necessary to examine this.
Cellular mechanisms of nicotinic regulation
Our data indicate that α4β2*nAChRs mediate nicotinic regulation of thalamocortical and most intracortical processes evoked by CF and non-CF stimuli. However, non-CF-evoked late intracortical activity was regulated also by α7-nAChRs. Nicotine effects typically endured ∼30 min or longer (i.e., much longer than the ∼9 min half-life of nicotine in mice; DBA/2 and C57BL/6 strains, 1 mg/kg, s.c.; Siu and Tyndale, 2007). One possible mechanism of prolonged enhancement would be that a nicotine metabolite has functional effects similar to nicotine. However, the major metabolite of nicotine, cotinine, has an EC50 for α4β2*nAChR activation ∼550-fold higher than that of nicotine (based on the measurement of [3H]dopamine release from caudate synaptosomes) (O'Leary et al., 2008) and is therefore unlikely to be responsible for prolonged effects. Another possible mechanism is intracellular signaling downstream to activation of nAChRs. Recent studies implicate activation of ERK in mediating effects of nicotine in A1 (I. Intskirveli and R. Metherate, unpublished observations). Future studies are necessary to elucidate the precise mechanisms of nicotinic regulation.
Implications for cognitive function and adolescent smoking
Nicotinic regulation of sensory information processing may contribute to smoking dependence in adolescents. Nicotine and smoking improve attention in nonsmokers (see Introduction), an effect likely involving α4β2*nAChRs (Grottick and Higgins, 2000; Rezvani et al., 2011). Our finding of nicotinic filtering of sensory information may provide a neural substrate for the stimulus-filtering model of smoking-induced attentional improvement (Friedman et al., 1974; Kassel, 1997). As we demonstrate here, nicotinic filtering peaks in adolescence: early adolescence in females and late adolescence in males. Thus, during adolescence nicotine exposure may improve cognitive function maximally. These behavioral benefits may promote dependence in adolescents, earlier in females than in males (DiFranza et al., 2002).
Footnotes
This work was supported by National Institutes of Health Grants R01 DA12929, R01 DC02967, and P30 DC08369 (to R.M.); and RO3 DC08204 (to H.K.). We thank Ronit Lazar and Marcos Cantu for technical assistance.
References
- Adriani W, Spijker S, Deroche-Gamonet V, Laviola G, Le Moal M, Smit AB, Piazza PV. Evidence for enhanced neurobehavioral vulnerability to nicotine during periadolescence in rats. J Neurosci. 2003;23:4712–4716. doi: 10.1523/JNEUROSCI.23-11-04712.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alkondon M, Albuquerque EX. Diversity of nicotinic acetylcholine receptors in rat hippocampal neurons. I. Pharmacological and functional evidence for distinct structural subtypes. J Pharmacol Exp Ther. 1993;265:1455–1473. [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Wonnacott S, Albuquerque EX. Blockade of nicotinic currents in hippocampal neurons defines methyllycaconitine as a potent and specific receptor antagonist. Mol Pharmacol. 1992;41:802–808. [PubMed] [Google Scholar]
- Anderson LA, Christianson GB, Linden JF. Mouse auditory cortex differs from visual and somatosensory cortices in the laminar distribution of cytochrome oxidase and acetylcholinesterase. Brain Res. 2009;1252:130–142. doi: 10.1016/j.brainres.2008.11.037. [DOI] [PubMed] [Google Scholar]
- Barth DS, Di S. Three-dimensional analysis of auditory-evoked potentials in rat neocortex. J Neurophysiol. 1990;64:1527–1536. doi: 10.1152/jn.1990.64.5.1527. [DOI] [PubMed] [Google Scholar]
- Breslau N, Peterson EL. Smoking cessation in young adults: age at initiation of cigarette smoking and other suspected influences. Am J Public Health. 1996;86:214–220. doi: 10.2105/ajph.86.2.214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broide RS, O'Connor LT, Smith MA, Smith JA, Leslie FM. Developmental expression of alpha 7 neuronal nicotinic receptor messenger RNA in rat sensory cortex and thalamus. Neuroscience. 1995;67:83–94. doi: 10.1016/0306-4522(94)00623-d. [DOI] [PubMed] [Google Scholar]
- Broide RS, Robertson RT, Leslie FM. Regulation of α7 nicotinic acetylcholine receptors in the developing rat somatosensory cortex by thalamocortical afferents. J Neurosci. 1996;16:2956–2971. doi: 10.1523/JNEUROSCI.16-09-02956.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buisson B, Gopalakrishnan M, Arneric SP, Sullivan JP, Bertrand D. Human α4β2 neuronal nicotinic acetylcholine receptor in HEK 293 cells: a patch-clamp study. J Neurosci. 1996;16:7880–7891. doi: 10.1523/JNEUROSCI.16-24-07880.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Millar WJ. Age of smoking initiation: implications for quitting. Health Rep. 1998;9:39–46. (Eng); 39–48(Fre) [PubMed] [Google Scholar]
- Clarke PB. Nicotinic modulation of thalamocortical neurotransmission. Prog Brain Res. 2004;145:253–260. doi: 10.1016/S0079-6123(03)45017-6. [DOI] [PubMed] [Google Scholar]
- Clarke PB, Pert CB, Pert A. Autoradiographic distribution of nicotine receptors in rat brain. Brain Res. 1984;323:390–395. doi: 10.1016/0006-8993(84)90320-2. [DOI] [PubMed] [Google Scholar]
- Clarke PB, Schwartz RD, Paul SM, Pert CB, Pert A. Nicotinic binding in rat brain: autoradiographic comparison of [3H]acetylcholine, [3H]nicotine, and [125I]-α-bungarotoxin. J Neurosci. 1985;5:1307–1315. doi: 10.1523/JNEUROSCI.05-05-01307.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curró Dossi R, Paré D, Steriade M. Short-lasting nicotinic and long-lasting muscarinic depolarizing responses of thalamocortical neurons to stimulation of mesopontine cholinergic nuclei. J Neurophysiol. 1991;65:393–406. doi: 10.1152/jn.1991.65.3.393. [DOI] [PubMed] [Google Scholar]
- DiFranza JR, Savageau JA, Rigotti NA, Fletcher K, Ockene JK, McNeill AD, Coleman M, Wood C. Development of symptoms of tobacco dependence in youths: 30 month follow up data from the DANDY study. Tob Control. 2002;11:228–235. doi: 10.1136/tc.11.3.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding YS, Fowler JS, Logan J, Wang GJ, Telang F, Garza V, Biegon A, Pareto D, Rooney W, Shea C, Alexoff D, Volkow ND, Vocci F. 6-[18F]Fluoro-A-85380, a new PET tracer for the nicotinic acetylcholine receptor: studies in the human brain and in vivo demonstration of specific binding in white matter. Synapse. 2004;53:184–189. doi: 10.1002/syn.20051. [DOI] [PubMed] [Google Scholar]
- Drasdo A, Caulfield M, Bertrand D, Bertrand S, Wonnacott S. Methyllycaconitine: a novel nicotinic antagonist. Mol Cell Neurosci. 1992;3:237–243. doi: 10.1016/1044-7431(92)90043-2. [DOI] [PubMed] [Google Scholar]
- Foulds J, Stapleton J, Swettenham J, Bell N, McSorley K, Russell MA. Cognitive performance effects of subcutaneous nicotine in smokers and never-smokers. Psychopharmacology (Berl) 1996;127:31–38. doi: 10.1007/BF02805972. [DOI] [PubMed] [Google Scholar]
- Friedman J, Horvath T, Meares R. Tobacco smoking and a stimulus barrier. Nature. 1974;248:455–456. doi: 10.1038/248455a0. [DOI] [PubMed] [Google Scholar]
- Fuchs JL. [125I]alpha-bungarotoxin binding marks primary sensory area developing rat neocortex. Brain Res. 1989;501:223–234. doi: 10.1016/0006-8993(89)90640-9. [DOI] [PubMed] [Google Scholar]
- Gil Z, Connors BW, Amitai Y. Differential regulation of neocortical synapses by neuromodulators and activity. Neuron. 1997;19:679–686. doi: 10.1016/s0896-6273(00)80380-3. [DOI] [PubMed] [Google Scholar]
- Grottick AJ, Higgins GA. Effect of subtype selective nicotinic compounds on attention as assessed by the five-choice serial reaction time task. Behav Brain Res. 2000;117:197–208. doi: 10.1016/s0166-4328(00)00305-3. [DOI] [PubMed] [Google Scholar]
- Happel MF, Jeschke M, Ohl FW. Spectral integration in primary auditory cortex attributable to temporally precise convergence of thalamocortical and intracortical input. J Neurosci. 2010;30:11114–11127. doi: 10.1523/JNEUROSCI.0689-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harkrider AW, Champlin CA. Acute effect of nicotine on non-smokers: II. MLRs and 40-Hz responses. Hear Res. 2001a;160:89–98. doi: 10.1016/s0378-5955(01)00346-x. [DOI] [PubMed] [Google Scholar]
- Harkrider AW, Champlin CA. Acute effect of nicotine on non-smokers: III. LLRs and EEGs. Hear Res. 2001b;160:99–110. doi: 10.1016/s0378-5955(01)00347-1. [DOI] [PubMed] [Google Scholar]
- Harkrider AW, Champlin CA, McFadden D. Acute effect of nicotine on non-smokers: I. OAEs and ABRs. Hear Res. 2001;160:73–88. doi: 10.1016/s0378-5955(01)00345-8. [DOI] [PubMed] [Google Scholar]
- Heath CJ, Picciotto MR. Nicotine-induced plasticity during development: modulation of the cholinergic system and long-term consequences for circuits involved in attention and sensory processing. Neuropharmacology. 2009;56(Suppl 1):254–262. doi: 10.1016/j.neuropharm.2008.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen LK, Krystal JH, Mencl WE, Westerveld M, Frost SJ, Pugh KR. Effects of smoking and smoking abstinence on cognition in adolescent tobacco smokers. Biol Psychiatry. 2005;57:56–66. doi: 10.1016/j.biopsych.2004.10.022. [DOI] [PubMed] [Google Scholar]
- Jacobsen LK, Picciotto MR, Heath CJ, Frost SJ, Tsou KA, Dwan RA, Jackowski MP, Constable RT, Mencl WE. Prenatal and adolescent exposure to tobacco smoke modulates the development of white matter microstructure. J Neurosci. 2007a;27:13491–13498. doi: 10.1523/JNEUROSCI.2402-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobsen LK, Slotkin TA, Mencl WE, Frost SJ, Pugh KR. Gender-specific effects of prenatal and adolescent exposure to tobacco smoke on auditory and visual attention. Neuropsychopharmacology. 2007b;32:2453–2464. doi: 10.1038/sj.npp.1301398. [DOI] [PubMed] [Google Scholar]
- Johnson JL, Ratner PA, Tucker RS, Bottorff JL, Zumbo B, Prkachin KM, Shoveller J. Development of a multidimensional measure of tobacco dependence in adolescence. Addict Behav. 2005;30:501–515. doi: 10.1016/j.addbeh.2004.07.006. [DOI] [PubMed] [Google Scholar]
- Kassel JD. Smoking and attention: a review and reformulation of the stimulus-filter hypothesis. Clin Psychol Rev. 1997;17:451–478. doi: 10.1016/s0272-7358(97)00032-9. [DOI] [PubMed] [Google Scholar]
- Kaur S, Lazar R, Metherate R. Intracortical pathways determine breadth of subthreshold frequency receptive fields in primary auditory cortex. J Neurophysiol. 2004;91:2551–2567. doi: 10.1152/jn.01121.2003. [DOI] [PubMed] [Google Scholar]
- Kaur S, Rose HJ, Lazar R, Liang K, Metherate R. Spectral integration in primary auditory cortex: laminar processing of afferent input, in vivo and in vitro. Neuroscience. 2005;134:1033–1045. doi: 10.1016/j.neuroscience.2005.04.052. [DOI] [PubMed] [Google Scholar]
- Kawai H, Lazar R, Metherate R. Nicotinic control of axon excitability regulates thalamocortical transmission. Nat Neurosci. 2007;10:1168–1175. doi: 10.1038/nn1956. [DOI] [PubMed] [Google Scholar]
- Kumar A, Schliebs R. Postnatal laminar development of cholinergic receptors, protein kinase C and dihydropyridine-sensitive calcium antagonist binding in rat visual cortex. Effect of visual deprivation. Int J Dev Neurosci. 1992;10:491–504. doi: 10.1016/0736-5748(92)90050-a. [DOI] [PubMed] [Google Scholar]
- Le Houezec J, Halliday R, Benowitz NL, Callaway E, Naylor H, Herzig K. A low dose of subcutaneous nicotine improves information processing in non-smokers. Psychopharmacology (Berl) 1994;114:628–634. doi: 10.1007/BF02244994. [DOI] [PubMed] [Google Scholar]
- Levin ED. Nicotinic receptor subtypes and cognitive function. J Neurobiol. 2002;53:633–640. doi: 10.1002/neu.10151. [DOI] [PubMed] [Google Scholar]
- Levin ED, Conners CK, Silva D, Hinton SC, Meck WH, March J, Rose JE. Transdermal nicotine effects on attention. Psychopharmacology (Berl) 1998;140:135–141. doi: 10.1007/s002130050750. [DOI] [PubMed] [Google Scholar]
- Liang K, Poytress BS, Chen Y, Leslie FM, Weinberger NM, Metherate R. Neonatal nicotine exposure impairs nicotinic enhancement of central auditory processing and auditory learning in adult rats. Eur J Neurosci. 2006;24:857–866. doi: 10.1111/j.1460-9568.2006.04945.x. [DOI] [PubMed] [Google Scholar]
- Liu BH, Wu GK, Arbuckle R, Tao HW, Zhang LI. Defining cortical frequency tuning with recurrent excitatory circuitry. Nat Neurosci. 2007;10:1594–1600. doi: 10.1038/nn2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- London ED, Waller SB, Wamsley JK. Autoradiographic localization of [3H]nicotine binding sites in the rat brain. Neurosci Lett. 1985;53:179–184. doi: 10.1016/0304-3940(85)90182-x. [DOI] [PubMed] [Google Scholar]
- Mackesy-Amiti ME, Fendrich M, Goldstein PJ. Sequence of drug use among serious drug users: typical vs atypical progression. Drug Alcohol Depend. 1997;45:185–196. doi: 10.1016/s0376-8716(97)00032-x. [DOI] [PubMed] [Google Scholar]
- McCormick DA, Prince DA. Actions of acetylcholine in the guinea-pig and cat medial and lateral geniculate nuclei, in vitro. J Physiol. 1987;392:147–165. doi: 10.1113/jphysiol.1987.sp016774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metherate R. Nicotinic acetylcholine receptors in sensory cortex. Learn Mem. 2004;11:50–59. doi: 10.1101/lm.69904. [DOI] [PubMed] [Google Scholar]
- Metherate R. Functional connectivity and cholinergic modulation in auditory cortex. Neuorsci Biobehav Rev. 2011 doi: 10.1016/j.neubiorev.2010.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metherate R, Cruikshank SJ. Thalamocortical inputs trigger a propagating envelope of gamma-band activity in auditory cortex in vitro. Exp Brain Res. 1999;126:160–174. doi: 10.1007/s002210050726. [DOI] [PubMed] [Google Scholar]
- Mulle C, Changeux JP. A novel type of nicotinic receptor in the rat CNS characterized by patch-clamp techniques. J Neurosci. 1990;10:169–175. doi: 10.1523/JNEUROSCI.10-01-00169.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Müller-Preuss P, Mitzdorf U. Functional anatomy of the inferior colliculus and the auditory cortex: current source density analyses of click-evoked potentials. Hear Res. 1984;16:133–142. doi: 10.1016/0378-5955(84)90003-0. [DOI] [PubMed] [Google Scholar]
- O'Leary K, Parameswaran N, McIntosh JM, Quik M. Cotinine selectively activates a subpopulation of alpha3/alpha6beta2 nicotinic receptors in monkey striatum. J Pharmacol Exp Ther. 2008;325:646–654. doi: 10.1124/jpet.108.136838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson D, Kratz KE, Daw NW. Evidence for a nicotinic component to the actions of acetylcholine in cat visual cortex. Exp Brain Res. 1988;73:553–568. doi: 10.1007/BF00406614. [DOI] [PubMed] [Google Scholar]
- Picciotto MR. Nicotine as a modulator of behavior: beyond the inverted U. Trends Pharmacol Sci. 2003;24:493–499. doi: 10.1016/S0165-6147(03)00230-X. [DOI] [PubMed] [Google Scholar]
- Pichika R, Easwaramoorthy B, Collins D, Christian BT, Shi B, Narayanan TK, Potkin SG, Mukherjee J. Nicotinic alpha4beta2 receptor imaging agents: part II. Synthesis and biological evaluation of 2-[18F]fluoro-3-[2-((S)-3-pyrrolinyl)methoxy]pyridine (18F-nifene) in rodents and imaging by PET in nonhuman primate. Nucl Med Biol. 2006;33:295–304. doi: 10.1016/j.nucmedbio.2005.12.017. [DOI] [PubMed] [Google Scholar]
- Provost SC, Woodward R. Effects of nicotine gum on repeated administration of the Stroop test. Psychopharmacology (Berl) 1991;104:536–540. doi: 10.1007/BF02245662. [DOI] [PubMed] [Google Scholar]
- Prusky GT, Shaw C, Cynader MS. Nicotine receptors are located on lateral geniculate nucleus terminals in cat visual cortex. Brain Res. 1987;412:131–138. doi: 10.1016/0006-8993(87)91447-8. [DOI] [PubMed] [Google Scholar]
- Prusky GT, Shaw C, Cynader MS. The distribution and ontogenesis of [3H]nicotine binding sites in cat visual cortex. Brain Res. 1988;467:161–176. doi: 10.1016/0165-3806(88)90020-x. [DOI] [PubMed] [Google Scholar]
- Rezvani AH, Cauley M, Sexton H, Xiao Y, Brown ML, Paige MA, McDowell BE, Kellar KJ, Levin ED. Sazetidine-A, a selective α4β2 nicotinic acetylcholine receptor ligand: effects on dizocipline and scopolamine-induced attentional impairements in female Sprague-Dawley rats. Psychopharmacology (Berl) 2011;215:621–630. doi: 10.1007/s00213-010-2161-8. [DOI] [PubMed] [Google Scholar]
- Sacco KA, Bannon KL, George TP. Nicotinic receptor mechanisms and cognition in normal states and neuropsychiatric disorders. J Psychopharmacol. 2004;18:457–474. doi: 10.1177/0269881104047273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakata S, Harris KD. Laminar structure of spontaneous and sensory-evoked population activity in auditory cortex. Neuron. 2009;64:404–418. doi: 10.1016/j.neuron.2009.09.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siu EC, Tyndale RF. Characterization and comparison of nicotine and cotinine metabolism in vitro and in vivo in DBA/2 and C57BL/6 mice. Mol Pharmacol. 2007;71:826–834. doi: 10.1124/mol.106.032086. [DOI] [PubMed] [Google Scholar]
- Slotkin TA. Nicotine and the adolescent brain: insights from an animal model. Neurotoxicol Teratol. 2002;24:369–384. doi: 10.1016/s0892-0362(02)00199-x. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, MacKillop EA, Rudder CL, Ryde IT, Tate CA, Seidler FJ. Permanent, sex-selective effects of prenatal or adolescent nicotine exposure, separately or sequentially, in rat brain regions: indices of cholinergic and serotonergic synaptic function, cell signaling, and neural cell number and size at 6 months of age. Neuropsychopharmacology. 2007;32:1082–1097. doi: 10.1038/sj.npp.1301231. [DOI] [PubMed] [Google Scholar]
- Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev. 2000;24:417–463. doi: 10.1016/s0149-7634(00)00014-2. [DOI] [PubMed] [Google Scholar]
- Steinschneider M, Tenke CE, Schroeder CE, Javitt DC, Simpson GV, Arezzo JC, Vaughan HG., Jr Cellular generators of the cortical auditory evoked potential initial component. Electroencephalogr Clin Neurophysiol. 1992;84:196–200. doi: 10.1016/0168-5597(92)90026-8. [DOI] [PubMed] [Google Scholar]
- Vossel S, Thiel CM, Fink GR. Behavioral and neural effects of nicotine on visuospatial attentional reorienting in non-smoking subjects. Neuropsychopharmacology. 2008;33:731–738. doi: 10.1038/sj.npp.1301469. [DOI] [PubMed] [Google Scholar]
- Wesnes K, Warburton DM. Effects of smoking on rapid information processing performance. Neuropsychobiology. 1983;9:223–229. doi: 10.1159/000117969. [DOI] [PubMed] [Google Scholar]
- Whiting P, Schoepfer R, Lindstrom J, Priestley T. Structural and pharmacological characterization of the major brain nicotinic acetylcholine receptor subtype stably expressed in mouse fibroblasts. Mol Pharmacol. 1991;40:463–472. [PubMed] [Google Scholar]
- Wong ET, Holstad SG, Mennerick SJ, Hong SE, Zorumski CF, Isenberg KE. Pharmacological and physiological properties of a putative ganglionic nicotinic receptor, alpha 3 beta 4, expressed in transfected eucaryotic cells. Brain Res Mol Brain Res. 1995;28:101–109. doi: 10.1016/0169-328x(94)00189-l. [DOI] [PubMed] [Google Scholar]