

Summary
The pathogenic Yersinia species share a conserved type III secretion system, which delivers cytotoxic effectors known as Yops into target mammalian cells. In all three species, YopK (also called YopQ) plays an important role in regulating this process. In cell culture infections, yopK mutants inject higher levels of Yops, leading to increase cytotoxicity; however, in vivo the same mutants are highly attenuated. In this work, we investigate the mechanism behind this paradox. Using a β-lactamase reporter assay to directly measure the effect of YopK on translocation, we demonstrated that YopK controls the rate of Yop injection. Furthermore, we find that YopK cannot regulate effector Yop translocation from within the bacterial cytosol. YopE is also injected into host cells and was previously shown to contribute to regulation of the injectisome. In this work we show that YopK and YopE work at different steps to regulate Yop injection, with YopK functioning independently of YopE. Finally, by expressing YopK within tissue culture cells, we confirm that YopK regulates translocation from inside the host cell, and we show that cells pre-loaded with YopK are resistant to Yop injection. These results suggest a novel role for YopK in controlling the Yersinia type III secretion system.
Introduction
Yersinia pestis, the causative agent of plague, has been responsible for an estimated 175 million deaths (Dennis et al., 1999). A plasmid-encoded type III secretion system (TTSS) is essential for virulence and is used by the bacterium to translocate cytotoxic effector proteins, known as Yops (Yersinia outer proteins), into host cells during infection. The TTSS is essentially a molecular syringe, or injectisome, made up of over 20 structural proteins all of which are essential for Yop translocation (Cornelis, 2002a). All of the TTSS components as well as the Yops and regulatory factors are encoded on an ∼70 kb plasmid pCD1 (Ben-Gurion and Shafferman, 1981). Effector Yops disrupt signalling within the host cell to prevent phagocytosis, induce apoptosis and evade the immune response (Cornelis, 2002b; Viboud and Bliska, 2005). Y. pestis has two closely related enteropathogenic species, Yersinia enterocolitica and Yersinia pseudotuberculosis, that share the conserved virulence plasmid and effector Yops.
The suite of effector Yops translocated into host cells includes six proteins: Yops E, H, M, J, T and YpkA. YopE is a GTPase-activating protein (GAP) that causes rapid disruption of the actin cytoskeleton (Black and Bliska, 2000) and has also been implicated in regulation of TTSS injection (Aili et al., 2008; Mejia et al., 2008). YopH is a powerful protein tyrosine phosphatase that targets several host signalling pathways and is injected at high levels similar to YopE (Guan and Dixon, 1990; Persson et al., 1995). YopM is an essential virulence factor (Leung et al., 1990) that has several leucine-rich repeats and a nuclear localization signal (Boland et al., 1998; Skrzypek et al., 1998); however, its primary mechanism during infection is unknown. It is translocated at moderate levels and has been shown to bind and activate host kinases RSK1 and PRK2 (McDonald et al., 2003) as well as reduce natural killer populations after intravenous mouse infection (Kerschen et al., 2004). YopJ and YopT are cysteine proteases that inhibit immune signalling factors MAPK and NF-κB (Orth, 2002) and cleave RhoA family GTPases (Zumbihl et al., 1999) respectively.
YopK, also called YopQ in Y. enterocolitica, is a TTSS substrate and important virulence factor (Holmstrom et al., 1995a,b). It is conserved across the pathogenic Yersinia species; however, it exhibits no sequence homology to other known proteins and has no evident functional domains. YopK has been shown to play a role in downregulating effector Yop translocation as a yopK mutant strain injects effector Yops at greatly increased levels during cell culture infections (Holmstrom et al., 1995a,b). Likewise, overexpression of YopK results in decreased injection of effectors into mammalian culture cells. Despite increased injection of the cytotoxic Yops, Yersinia yopK mutants are severely attenuated in mouse models of infection (Straley and Bowmer, 1986; Mulder et al., 1989; Straley and Cibull, 1989; Holmstrom et al., 1995a). These phenotypes suggest that tight regulation of effector Yop injection during infection is critical to pathogenesis.
Data from haemolytic assays suggest that YopK might work in conjunction with the translocation pore formed by YopB, YopD and LcrV (Cornelis, 2002a) to regulate the pore size (Hakansson et al., 1996; Holmstrom et al., 1997). The pore proteins are essential for translocation to occur (Hakansson et al., 1996; Neyt and Cornelis, 1999; Pettersson et al., 1999; Lee et al., 2000), and ΔyopK mutants appear to generate larger pores than those from strains expressing YopK (Holmstrom et al., 1997). Additional work has demonstrated a role in regulating pore formation for another effector, YopE. Y. pseudotuberculosis strains lacking YopE appear to create pores in host cells that lead to haemolysis and lactate dehydrogenase (LDH) release (Holmstrom et al., 1997; Viboud and Bliska, 2001; Marenne et al., 2003; Aili et al., 2008). Although both YopE and YopK appear to have roles in regulating pores formed during Yersinia infection, how these functions are co-ordinated is unknown.
Although it appears that YopK acts at the level of the translocation pore, attempts to determine the location of YopK during infection have failed until recently. YopK is expressed at lower levels than the other effector Yops making it difficult to determine localization using traditional microscopy approaches (Holmstrom et al., 1995a; 1997). Recently, Garcia et al. developed a new method to monitor delivery of Yops into host cells during infection (Garcia et al., 2006). After fusing a portion of glycogen synthase kinase (GSK) to the C-terminus of YopK, they demonstrated that YopK–GSK was phosphorylated during tissue culture infection. This provided the first evidence that YopK was injected into host cells. However, many questions regarding the specific localization and function of YopK during infection remain unresolved. In this study, we used a β-lactamase (Bla) reporter system to gain additional insight into the function of YopK by quantifying Yop injection on a cell-by-cell basis (Marketon et al., 2005). Using this approach, we show that YopK is responsible for regulating the rate of Yop translocation into tissue culture cells. Importantly, this regulatory activity of YopK occurs from within the targeted host cells and is independent of the regulatory activity of YopE. These results demonstrate an important function for YopK, which is crucial for Yersinia pathogenesis.
Results
The fluorescent Bla reporter assay as a tool to study YopK function
YopK regulates Yop injection by the TTSS; however, little is understood about the mechanism. Techniques that have been used to study YopK, such as haemolysis or LDH release (Holmstrom et al., 1997; Aili et al., 2008; Brodsky et al., 2010), indicate host cell permeability associated with cytotoxicity and therefore are an indirect measure of Yop injection. Digitonin fractionation of infected cells can indicate the amount of each Yop delivered to host cells, but only at the population level (Holmstrom et al., 1997; Aili et al., 2008; Brodsky et al., 2010). Immunofluorescence gives similar information on an individual cell basis (Holmstrom et al., 1997), but this is not a high-throughput assay and only reliably detects Yops, such as YopE and YopH, which are translocated at high levels. In contrast, the β-lactamase (Bla) reporter assay used to monitor Yop injection (Marketon et al., 2005) is more sensitive compared with immunofluorescence due to the enzymatic activity of the reporter. In this assay, Bla is introduced through TTSS-mediated translocation of a Yop-Bla reporter during cell culture infection (Marketon et al., 2005). Mammalian cells are then dyed with CCF2-AM, a fluorescent compound composed of a fluorescein moiety connected to a coumarin group by a β-lactam ring, which gives rise to green fluorescence (518 nm) under violet laser excitation (409 nm). As Bla cleaves the β-lactam linkage, fluorescence resonance energy transfer (FRET) is disrupted and blue light (447 nm) is emitted. Using this approach, one can couple the sensitivity of the fluorescence assay with the power of flow cytometry to enable (i) direct measurement of Yop translocation, (ii) analysis of large populations of infected cells, (iii) unbiased quantification of the fluorescent signal and (iv) the ability to analyse the injection levels of each cell in the population.
Figure 1A shows a schematic of the YopM-Bla reporter assay used as a quantitative tool to measure Yop injection. CHO cells serve as the negative control population, as these cells lack Bla and only fluoresce green after incubation with the CCF2-AM Bla substrate. CMV-bla CHO cells were used as a positive control as these constitutively express Bla and therefore fluoresce blue after incubation with CCF2-AM. These two populations were used to set the single colour flow cytometry gates that distinguish blue (injected) from green (uninjected) cells, as shown in Fig. 1B. As a negative control for TTSS-mediated delivery of Bla, we used Bla fused to the C-terminus of glutathione-S-transferase (Gst), a cytosolic protein that is not secreted by the TTSS (Marketon et al., 2005). Infections with Y. pestis carrying Gst-Bla are performed in conjunction with every experiment to determine the background level of blue fluorescence from infected cells (Fig. 1, column 4). When cells were infected with Y. pestis carrying the YopM-Bla reporter (Fig. 1, column 5), translocation of YopM-Bla caused the cells to fluoresce blue as the CCF2-AM substrate was cleaved. Initially, the cells that received YopM-Bla contain a mixture of cleaved and intact CCF2-AM molecules, leading to cells that were double-positive for green and blue fluorescence (aqua). As more CCF2-AM is cleaved, the cells in the population continue shifting from aqua (double-positive) to blue (single-positive) fluorescence until all the dye is cleaved. Both aqua and blue cells represent injected cells, and the relative levels of each within the total population of stained cells can be displayed in stacked bar graph format, as shown in Fig. 1C. When the infections are synchronized, it is possible to distinguish cells that have been injected with different amounts of the YopM-Bla reporter.
Fig. 1.
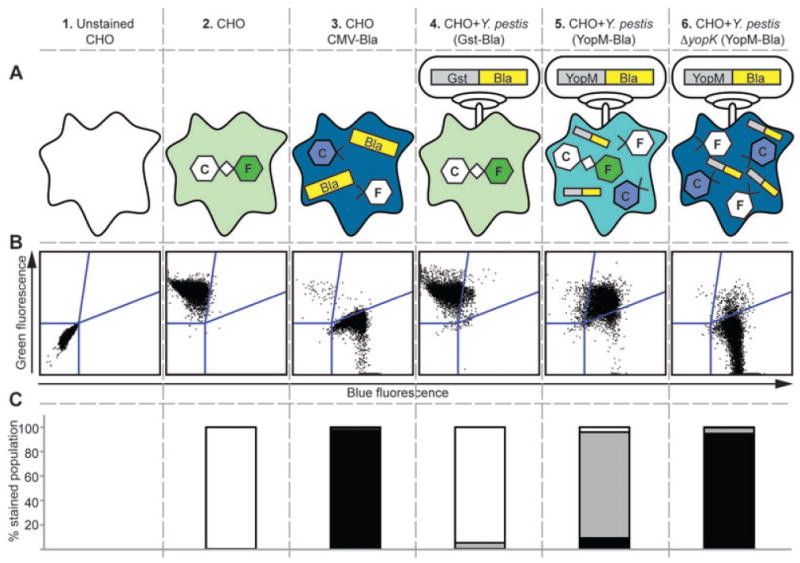
Schematic of quantitative Bla reporter assay. (A) shows the nature of the cell populations being analysed. (B) displays raw flow cytometry data. In (C), the flow cytometry data are quantified in stacked bar graph format. Columns 1–3 show uninfected samples used to establish both compensation settings and gates for flow cytometry. Unstained CHO cells determine the population negative for fluorescence. The rest of the samples have been incubated with CCF2-AM, a compound fluorophore composed of Coumarin (C) and Fluorescein (F) groups connected via a β-lactam ring. A CHO cell line that constitutively expresses Bla (CHO CMV-Bla) is used to create a gate for cells with only blue fluorescence (column 3). The gate between green and blue (upper right quadrant) is for cells with low-level injection that have incomplete cleavage of CCF2-AM and exhibit both blue and green fluorescence, referred to as aqua. Columns 4–6 show CHO cells infected with WT or ΔyopK Y. pestis expressing the Gst-Bla control or the YopM-Bla reporter. The flow cytometry data are shown in stacked bar graph format as % stained population, referring to the percentage of cells within the population of stained cells that exhibit either green (white bars), aqua (grey bars) or blue (black bars) fluorescence.
The ΔyopK phenotype is readily seen using this approach. When the KIM5 parent (WT) carrying the YopM-Bla reporter was used to infect CHO cells, the majority of cells displayed aqua fluorescence, which represents low levels of YopM-Bla injection in each cell. Infection with the ΔyopK mutant resulted in a similar number of CHO cells injected; however, there was a greatly increased proportion of blue cells (high levels of YopM-Bla injection) to aqua cells (Fig. 1, columns 5 and 6). This demonstrates that the ΔyopK mutant injects more YopM-Bla into each CHO cell, indicating a role for YopK in regulating Yop delivery into host cells. Furthermore, the data illustrate the utility of the Bla reporter assay as a quick, sensitive and quantitative way to analyse Yop translocation.
To confirm that the phenotype seen for the yopK mutant was not specific to the YopM-Bla reporter, we generated several additional reporters by fusing Bla to YopN, YopJ and YopH. These constructs were introduced into both KIM5 (WT) and MEL27 (ΔyopK) and the resulting strains were used to infect CHO cells for 3 h, followed by CCF2-AM staining and flow cytometry to assess Yop translocation. All Bla reporters were translocated into host cells (Fig. S1), although the levels of injection varied between the reporters with YopN-Bla being injected at the lowest levels and YopH-Bla injected at the highest levels. This trend was consistent between WT and ΔyopK infections. Importantly, cells infected with the yopK mutant displayed higher levels of Yop injection per cell compared with WT, regardless of the reporter. In contrast, the yscU mutant, which cannot secrete or inject Yops (Marketon et al., 2005), could not deliver any of the Bla reporters into CHO cells, so all cells within the population remained green. These data demonstrate that the yopK mutant delivers more Yops into each cell compared with WT, and they also show that the YopM-Bla reporter can be used as a representative Yop for the purposes of quantifying translocation in this assay.
YopK controls the rate of Yop translocation
The above data demonstrate that cells infected with a yopK mutant receive more Yops per cell. Since the infections are synchronized, this implies that YopK controls the rate at which Yops are injected into host cells. To confirm this hypothesis, a time-course of infection was performed to compare the ability of WT and the yopK mutant to deliver YopM-Bla over time. As shown in Fig. 2, low levels of injection for about 10% of the cells could be seen within 1 h post infection for both WT and ΔyopK, and after 2 h nearly 100% of the cells had been injected. Thus, the kinetics of initiating the injection process appear to be equivalent for both strains. When comparing the relative levels of YopM-Bla injected into host cells, it is clear that there is an increase in the amount of Yops delivered into each cell over time as judged by the increased proportion of blue to aqua cells as time progresses, and this is true for both WT and ΔyopK infections. However, cells infected with the yopK mutant show a greater proportion of blue cells much earlier in the time-course (within 1.5 h), and this trend continues over time until all of the CCF2-AM substrate has been cleaved, at which point ∼100% of the ΔyopK-infected host cells show high levels of YopM-Bla injection compared with ∼40% of WT-infected cells. The striking difference in the proportion of blue to aqua cells over time indicates that YopK functions in regulating the rate of Yop translocation.
Fig. 2.
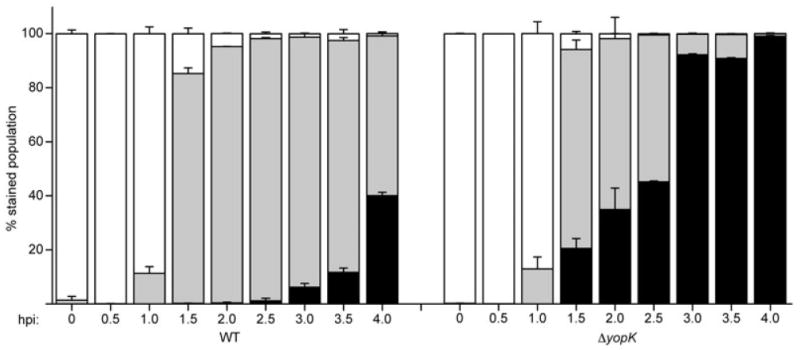
YopK controls the rate of translocation. WT and ΔyopK Y. pestis carrying YopM-Bla were used to infect CHO cells at an moi = 10. At the indicated times post infection, cells were stained with CCF2-AM and analysed by flow cytometry. Each infection was carried out in triplicate and samples were averaged with standard deviation error bars. White bars: green cells (uninjected), grey bars: aqua cells (low-level injection), black bars: blue cells (high-level injection).
To confirm that the increase in the high levels of injection (blue cells) is not simply an artefact of the enzymatic amplification of the signal due to continual cleavage of CCF2-AM, we repeated the time-course experiment in order to determine whether the protein levels of the Bla reporter correlate with the fluorescent signal of cleaved CCF2-AM. CHO cells were infected with WT Y. pestis carrying either the YopM-Bla or the YopH-Bla reporter, and at 1.5 or 4 h post infection an aliquot of each infection was stained and analysed by flow cytometry. The remainder of each infection was subjected to digitonin fractionation, which selectively permeabilizes host cells while leaving bacteria intact. With this method, soluble cytoplasmic proteins within host cells are released into the digitonin-soluble fraction, while the digitonin pellet contains host membranes, organelles and attached bacteria (Tabrizi and Robins-Browne, 1993; Lee et al., 1998; Ruckdeschel et al., 2006; Aili et al., 2008). Western blots of fractionated cells show that the cytoplasmic eukaryotic protein, p130cas, was found in digitonin supernatants, whereas the cytoplasmic bacterial protein RpoA (RNA polymerase alpha subunit) remained within the pellet fractions (Fig. 3A). These fractionation controls demonstrate complete lysis of host cells but not bacterial cells. We then assessed the levels of Yop injection using antibodies to Bla, YopM and YopH to probe for the levels of injection of the YopM-Bla and YopH-Bla reporters as well as the native YopM and YopH proteins. As expected, the control cells that constitutively express Bla from a CMV promoter showed a strong Bla signal in the digitonin supernatant fraction. For cells infected with WT carrying Bla reporters, there is a strong Bla signal in the digitonin pellets, indicating expression of the reporter within adherent bacteria. The Yop-Bla signals in the supernatant fractions from the 1.5 h time point are very faint; however by 4 h there is substantially more Bla present in the supernatants. These results indicate that more of each reporter was injected into host cells over time. Similar results showing accumulation of Yop proteins within host cells were obtained for the native YopH and YopM proteins. When analysed by flow cytometry (Fig. 3B), the same 1.5 h time points show low levels of injection (aqua) in 62% and 73% of the cell population for YopM-Bla and YopH-Bla respectively. By 4 h post infection, there is a dramatic increase the levels of both reporters in host cells. Over 99% of the cells were injected at that time, and the majority of those injected cells showed high levels of injection (blue cells = 68% for YopM-Bla and 91% for YopH-Bla). These data correspond to the immunoblotting results, and together the data suggest that the increase in blue fluorescence measured by flow cytometry in this assay indeed represents increased levels of Yops translocated into host cells.
Fig. 3.
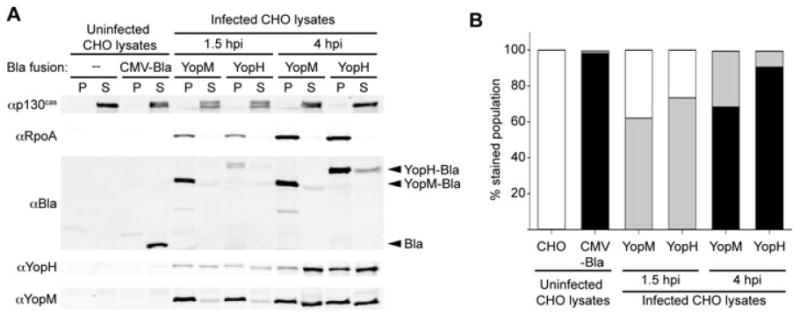
Accumulation of Yops within host cells. WT Y. pestis carrying either YopM-Bla or YopH-Bla were used to infect CHO cells at an moi = 10. At 1.5 and 4 h post infection (hpi), a portion of the cells were stained with CCF2-AM and analysed by flow cytometry (B). The remaining cells were permeabilized with digitonin to separate soluble cytoplasmic proteins including injected Yops (supernatant fraction, S) from membranes, organelles and adherent bacteria (pellet fraction, P). The proteins in each fraction were visualized by immunoblotting (A). p130cas (cytoplasmic eukaryotic protein) and RpoA (cytoplasmic bacterial protein) serve as fractionation controls. Because the data in (B) are from the same individual infections shown in (A), error bars are not available.
Non-secretable YopK cannot regulate the TTSS
With the knowledge that YopK is injected into host cells and that it is required for regulation of Yop injection, we wanted to determine whether injection of YopK was necessary for the ability of YopK to function as a regulator. To address this question, we created two plasmids: one that expressed native YopK and the other expressing a non-secretable form of YopK (Gst–YopK under the native yopK promoter). Placing Gst at the N-terminus of YopK effectively blocks the secretion signal so that YopK remains within the bacterium (Cambronne et al., 2004; Sorg et al., 2006). The plasmids were transformed into both WT Y. pestis and a ΔyopK mutant carrying either the YopM-Bla reporter or the Gst-Bla control plasmid. All strains were tested for correct protein expression and localization by secretion assay and Western blot. As shown in Fig. S2, Gst–YopK was expressed but not secreted, while native YopK was secreted into the media as expected. Expression and secretion of the YopM-Bla reporter and other TTSS proteins (YopM, YopN, YopE, YscD) was not affected by the presence of the plasmids overexpressing YopK or Gst–YopK.
These strains were then used in an injection assay to determine whether gst–yopK can complement the ΔyopK mutant (Fig. 4). CHO cells were infected with each strain at an moi of 10, stained with CCF2-AM and then analysed by flow cytometry. Initially apparent was the typical difference in injection phenotypes between the empty WT and ΔyopK strains. When YopK was absent there was a dramatic increase in injection of the YopM-Bla reporter, as demonstrated by the increased proportion of blue to aqua cells. Only ectopic expression of native YopK could reduce the amount of YopM-Bla injection into CHO cells in both the WT and ΔyopK backgrounds (P < 0.001). Strains expressing Gst–YopK resembled their respective empty parents; thus a non-secretable form of YopK could not complement the yopK mutant. This suggests that YopK must be injected into the host cell, not just present within the bacterium, to regulate translocation.
Fig. 4.
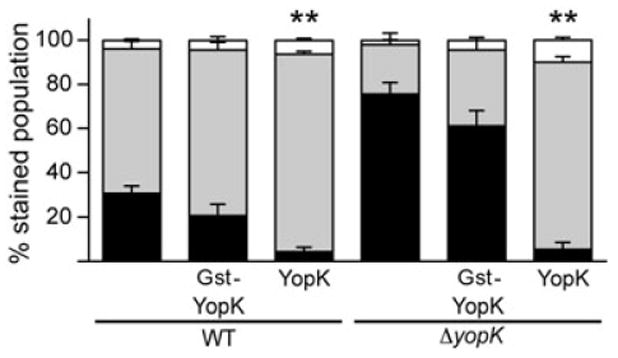
Gst–YopK does not complement ΔyopK. Plasmids expressing Gst–YopK or YopK were transformed into WT and ΔyopK Y. pestis carrying YopM-Bla. These strains were used to infect CHO cells at an moi = 10 for ∼3 h, followed by CCF2-AM staining and flow cytometry. Each infection is carried out in triplicate and samples are averaged with standard deviation error bars. Student's t-test was performed to demonstrate significant differences in high-level injection (blue cells) relative to empty parent strains (**P < 0.001).
Expression of YopK within eukaryotic cells
To confirm that YopK functions from within host cells to regulate the TTSS and to further elucidate the role of YopK, we attempted to express YopK directly within tissue culture cells. A eukaryotic YopK expression vector was created using phmKeima-Red-MClinker, which constitutively expresses the red fluorescent protein Keima. Keima has a large stokes shift with excitation at 440 nm and emission at 620 nm (Kogure et al., 2008). This allows Keima excitation and detection concurrent with the CCF2-AM fluorophore. yopK was cloned into phmKeima-Red-MCLinker as a C-terminal fusion to keima. After transient transfection into CHO cells, expression and fluorescence of the Keima–YopK fusion protein was verified by Western blot and flow cytometry respectively (Fig. 5). Western blots of transfected CHO lysates consistently showed a band at ∼48 kDa with both Keima and YopK antibodies corresponding to full-length Keima–YopK along with several smaller bands that are likely degradation products (Fig. 5A). The measured red fluorescence of the fusion protein indicated a transfection efficiency of approximately 15% with red fluorescence well above background levels (Fig. 5B).
Fig. 5.
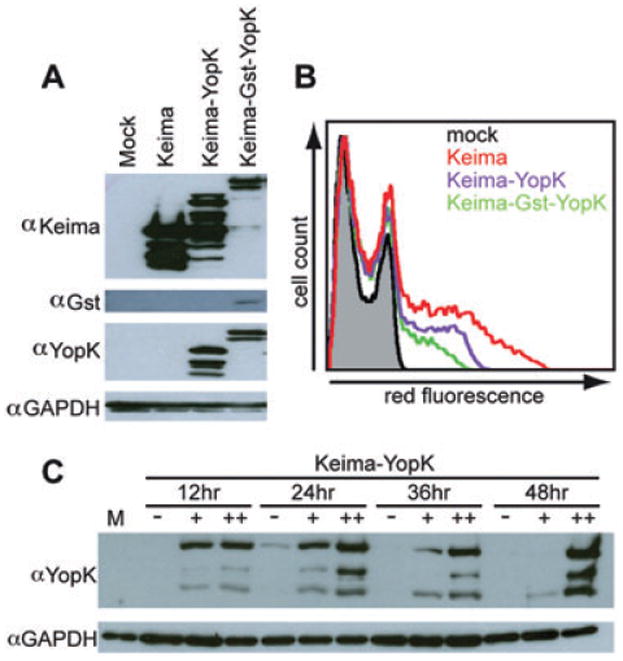
Characterization of eukaryotic YopK expression vectors. A. CHO cells transfected for 24 h were lysed, subjected to SDS-PAGE and Western blotted with antibody raised against YopK, Keima, Gst and p130cas as a loading control. B. Twenty-four-hour transfected CHO cells were suspended and analysed via flow cytometry for red fluorescence. The histogram shows the traces of a negative mock-transfected population (black), cells carrying the empty vector expressing Keima (red), cells expressing Keima–YopK (purple) and cells expressing Keima–Gst–YopK (green). C. CHO cells transfected for times ranging from 12 to 48 h were divided into populations with no (−), low (+) or high (++) red fluorescence using FACS. Each sample was lysed, subjected to SDS-PAGE and Western blotted to visualize YopK and loading control GAPDH.
To ensure that red fluorescence corresponded directly to YopK expression, CHO cells were transfected with the plasmid expressing Keima–YopK for 12–48 h before analysis. Each sample was then sorted based on no, low or high levels of red fluorescence with a fluorescence-activated cell sorter (FACS). Collected samples were analysed by SDS-PAGE and Western blot. Robust YopK expression could be detected as early as 12 h and continued throughout the time-course. However, at 48 h, YopK expression was only evident in the high red fluorescence population and the majority of the YopK signal was present as degradation products (Fig. 5C). YopK expression was absent in non-fluorescent cells, as expected. At 24–36 h post transfection there was abundant YopK expression with very little degradation products making it the optimal time for expression of Keima–YopK.
YopK expressed within host cells can regulate the TTSS
After confirming that YopK can be expressed within CHO cells, we wanted to determine whether the protein was functional and would complement a ΔyopK mutant during infection. To this end, transfected CHO cells expressing either Keima or Keima–YopK for 36 h were infected with either WT or ΔyopK Y. pestis carrying the YopM-Bla reporter (Fig. 6A). After 3 h of infection, the cells were stained with CCF2-AM and the red fluorescence of transfection was measured in conjunction with the blue and green fluorescence of CCF2-AM by flow cytometry. Cells expressing Keima (the empty parent vector) are the control population, but it is also important to note that due to a transfection rate of ∼15%, the rest of the population (∼85%) acts as an internal untransfected control. CHO cells were first analysed with a size exclusion gate determined by propidium iodide staining of a duplicate sample. This separates by size live cells from dead/necrotic cells. The percentage of live cells shows little variance between Keima and Keima–YopK transfected cells, suggesting that YopK expression does not significantly disrupt CHO cell viability (data not shown). The red fluorescence of Keima or Keima–YopK was then analysed to separate transfected and untransfected cells. Each population was then separately analysed for fluorescence of CCF2-AM. CHO cells expressing Keima alone had a YopM-Bla injection phenotype indistinguishable from untransfected populations showing that transfection alone does not alter the injection level of the reporter (data not shown). Importantly, cells expressing Keima–YopK showed a significant decrease (P < 0.001) in levels of YopM-Bla injection when compared with the Keima control (Fig. 6B). Note that in this experiment the total number of injected cells (aqua + blue) was much lower for CHO cells expressing YopK, unlike the experiment in Fig. 4 where the total number of injected cells was similar between WT and ΔyopK infections. This difference is likely due to ‘preloading’ the CHO cells with functional YopK proteins that can immediately send a negative feedback signal to the infecting Y. pestis bacteria. These data demonstrate that Keima–YopK can complement the yopK mutant during infection. Furthermore, YopK expression in the host cell is sufficient for regulation of the TTSS of infecting Y. pestis.
Fig. 6.
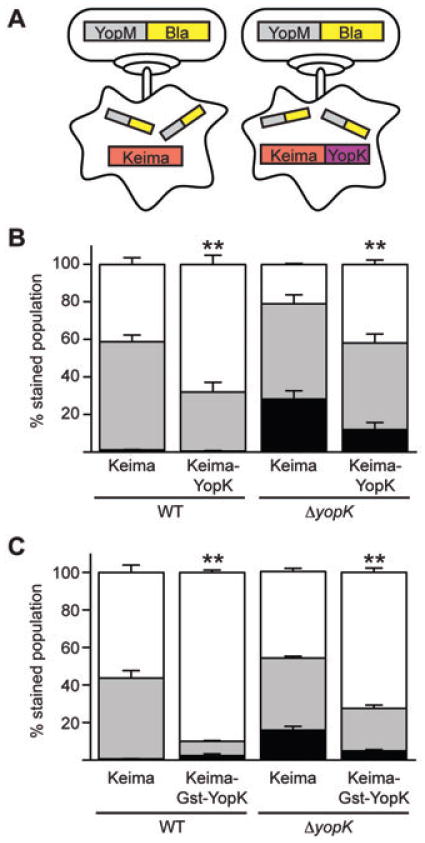
Keima–YopK negatively regulates effector Yop translocation. A. Schematic of transfection of CHO cells with plasmids expressing Keima or Keima–YopK followed by infection with Y. pestis and injection of the YopM-Bla reporter. B and C. CHO cells expressing either Keima, Keima–YopK or Keima–Gst–YopK were infected with WT or ΔyopK Y. pestis carrying the YopM-Bla reporter. The CHO cells were then stained with CCF2-AM and analysed by flow cytometry for red, green and blue fluorescence. Infections were performed in triplicate and are shown as averages with standard deviations. Student's t-test was performed to demonstrate significant differences in total injection levels (aqua + blue cells) for strains expressing Keima–YopK or Keima–Gst–YopK relative to the Keima-only controls (**P < 0.001). The data shown are representative of one data set, and the experiment was performed at least twice.
Keima–Gst–YopK expressed within host cells is functional
To ensure that fusion of Gst to the N-terminus of YopK (Fig. 4) did not disrupt its regulatory function, an additional eukaryotic expression vector was created in which gst was cloned between the keima and yopK ORFs. The complete Keima–Gst–YopK fusion could be detected on Western blots by antibodies to Keima, Gst and YopK (Fig. 5A). Fewer degradation products were seen for the Keima–Gst–YopK hybrid compared with the Keima–YopK fusion, suggesting increased stability imparted by the Gst fusion (Fig. 5A). Red fluorescence was measured by flow cytometry and was slightly less intense than Keima–YopK, probably due to lower efficiency of transfection and the increased size of the protein product (Fig. 5B).
We next tested whether Keima–Gst–YopK was functional. CHO cells were transiently transfected and allowed to express the YopK fusions for 36 h, followed by infection with WT and ΔyopK Y. pestis carrying the YopM-Bla reporter. Infected cells were stained with CCF2-AM and analysed by flow cytometry as before.As shown in Fig. 6C, cells expressing Keima–Gst–YopK showed a significant reduction (P < 0.001) in total injection compared with cells expressing Keima alone. These results are similar to those seen when testing the Keima–YopK fusion (Fig. 6B), and perhaps show more regulatory activity than Keima–YopK alone, possibly due to the increased stability of the Keima– Gst–YopK fusion. These data suggest that the lack of complementation observed with Gst–YopK (Fig. 4) was due to its inability to be translocated into host cells and not due to a dominant-negative effect exerted by the Gst fusion. Furthermore, the data confirm the role of YopK in regulating TTSS injection from inside the host cell.
YopE regulation of the TTSS
YopE is an effector Yop that also has been implicated in regulating type III secretion, possibly through regulating the opening or closing of the translocation pore (Aili et al., 2008; Mejia et al., 2008).YopE contains a GAP domain that is essential for virulence in the closely related Y. pseudotuberculosis (Black and Bliska, 2000; Aili et al., 2002). The GAP domain stimulates Rho GTPases to hydrolyse GTP thus keeping G proteins in a continuously inactive state, resulting in depolymerization of actin and subsequent cell rounding (Black and Bliska, 2000; von Pawel-Rammingen et al., 2000). yopE mutants have been assigned a ‘hypertranslocation phenotype’, similar to the phenotype of a ΔyopK mutant (Aili et al., 2008). Using digitonin fractionation, it was shown that both YopE and YopH were translocated into host cells at higher levels when cells were infected with ΔyopE or Y. pseudotuberculosis expressing YopE-GAP- (Black and Bliska, 2000; Aili et al., 2008). This suggests that the GAP activity of YopE is required for regulation of Yop injection. The digitonin fractionation approach differs from the Bla reporter assay in that it cannot analyse injection on a cell-by-cell basis, but rather analyses the entire population as a whole.
Because YopK and YopE have been implicated in similar functions during Yersinia infection, we wanted to further assess the role of each protein using the Bla reporter system. A single ΔyopE mutant and a double ΔyopEK mutant were created towards this end. A ΔyopH mutant was also created as a negative control to ensure that deleting any single effector Yop would not alter Bla reporter injection. Secretion assays were performed for each strain to determine whether the mutations affected Yop expression or secretion (Fig. S3). As expected, the mutants expressed and secreted native Yops and the YopM-Bla reporter at levels similar to the KIM5 parent. Next, injection assays were performed to assess the injection phenotype of each mutant strain compared with WT Y. pestis. CHO cells were infected with each Y. pestis mutant strain, stained with CCF2-AM and analysed by flow cytometry for blue and green fluorescence. The experiment was repeated on three separate days to ensure accurate measurement of injection phenotypes. The total number of injected cells (aqua + blue) was similar for infections with each strain (Fig. 7A). However, the ΔyopE mutant displayed a higher proportion of blue cells compared with WT, indicating an increased rate of YopM-Bla injection (P < 0.001). Likewise, the ΔyopK mutant resulted in more blue cells, as expected, but the phenotype of ΔyopE was consistently less severe compared with ΔyopK (P < 0.001). Statistical comparison of the samples using one-way ANOVA with Tukey post hoc test shows that WT, ΔyopK and ΔyopE have distinctly different injection phenotypes. The ΔyopEK double mutant is statistically indistinguishable from ΔyopK suggesting that regulation by YopK is taking place either upstream or independent of YopE activity. The ΔyopH mutant is not statistically different from WT showing that merely knocking out one of the many effector Yops does not alter the injection of the Bla reporter.
Fig. 7.
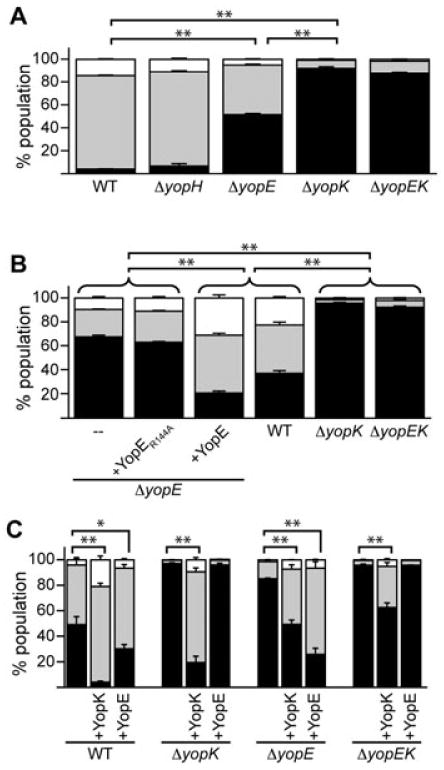
Regulation of translocation by YopK and YopE. A. CHO cells were infected with WT or mutant Y. pestis strains carrying the YopM-Bla reporter at moi = 10 for ∼3 h. Cells were stained with CCF2-AM and injection was quantified by flow cytometry. Infections were performed in triplicate and the entire experiment was repeated at least twice. The data shown are a representative data set from one experiment showing average injection levels and standard deviation error bars. High-level injection (blue cells) was compared using one-way ANOVA with Tukey post hoc test. B. ΔyopE Y. pestis strains were complemented with plasmids expressing either YopE or a GAP-deficient YopE (YopER144A), used to infect CHO cells, and injection of the YopM-Bla reporter was measured. Assays and analysis were performed as in (A). C. Y. pestis strains were transformed with plasmids expressing YopK or YopE in addition to the YopM-Bla reporter and used to infect CHO cells as in (A). Student's t-test was performed to demonstrate significant differences in high-level injection relative to empty parent strains. For all panels, *P < 0.01, **P < 0.001.
It has been demonstrated that the GAP domain of YopE is essential for pore formation, which is linked to a hypertranslocation phenotype in Y. pseudotuberculosis (Aili et al., 2008). To investigate the role of YopE's GAP activity on translocation directly using our Bla reporter system, we created two yopE bacterial complementation plasmids. The first contains yopE and its chaperone sycE (Wattiau and Cornells, 1993) under their native promoter. The second introduced a point mutation in YopE that changed the essential arginine residue in the arginine finger motif at residue 144 to alanine. This mutation has been shown to completely disrupt the GAP activity of YopE in Y. pseudotuberculosis (von Pawel-Rammingen et al., 2000). ΔyopE Y. pestis was complemented with plasmids expressing either native YopE or the YopER144A mutant, and secretion assays were performed (Fig. S4). Although there is a slight reduction in expression of some Yops when YopE or the YopE-GAP mutant was overexpressed, secretion of Yops was unaffected. We therefore proceeded to evaluate the effect of the YopE constructs on injection of the YopM-Bla reporter. Complementing the ΔyopE mutant with native YopE resulted in an injection phenotype similar to WT (Fig. 7B). Complementing with the YopER144A mutant, however, resulted in an injection phenotype that mirrored the parent ΔyopE strain, which injected YopM-Bla at higher levels than WT (P < 0.001). This shows that the GAP domain of YopE is essential for regulation of Yop injection in Y. pestis.
Relationship of YopK and YopE in regulating Yop translocation
While both YopK and YopE have a role in TTSS regulation as shown by their Yop injection phenotypes (Fig. 7A and B), how these proteins interact to regulate injection is unknown. To begin to tease apart this question, each mutant strain was transformed with plasmids expressing either YopK or YopE along with the YopM-Bla reporter plasmid. Again, overexpression of YopE slightly reduced expression levels of some Yops, but secretion was not affected (Fig. S5). As expected, overexpression of YopK did not affect either expression or secretion of Yops. The strains were then used in an injection assay to assess translocation levels. When WT was transformed with the YopK plasmid, effectively an overexpression of YopK, there was a dramatic decrease in the levels of blue cells (high-level injection of YopM-Bla) compared with the empty parent (P < 0.001), while overexpression of YopE yielded an intermediate phenotype (P < 0.01) (Fig. 7C). Complementation of ΔyopK with the YopK plasmid dramatically decreased the proportion of highly injected blue cells within the population, while complementation with the YopE plasmid had no effect. Surprisingly, introduction of the YopK plasmid into ΔyopE partially complemented the mutant, while the YopE plasmid provided full complementation (P < 0.001). Lastly, the YopK plasmid partially complemented the double ΔyopEK mutant (P < 0.001), while the YopE plasmid had no effect on the injection phenotype. These data support the hypothesis either that YopK functions upstream of YopE in the regulation of Yop translocation, or that YopK works independently of YopE.
To further investigate the relationship between YopK and YopE, we once again utilized the Keima–YopK eukaryotic expression vector. CHO cells were transfected with plasmids expressing Keima or Keima–YopK and then infected with Y. pestis strains carrying the YopM-Bla reporter to evaluate Yop translocation. Total injection levels (aqua + blue cells) were low for the WT infection, intermediate for the ΔyopE infection and higher for the ΔyopK and ΔyopEK infections, as expected (Fig. 8). When Keima–YopK was present in CHO cells, total injection was significantly decreased compared with Keima transfection alone, and this was true for all infections. This shows that YopK expressed in host cells does indeed reduce Yop translocation during infection with each of the Y. pestis strains tested. More importantly, the results demonstrate that YopK works independently of YopE as the presence of YopE in the host cell is not necessary for YopK to exert its negative control on the TTSS.
Fig. 8.
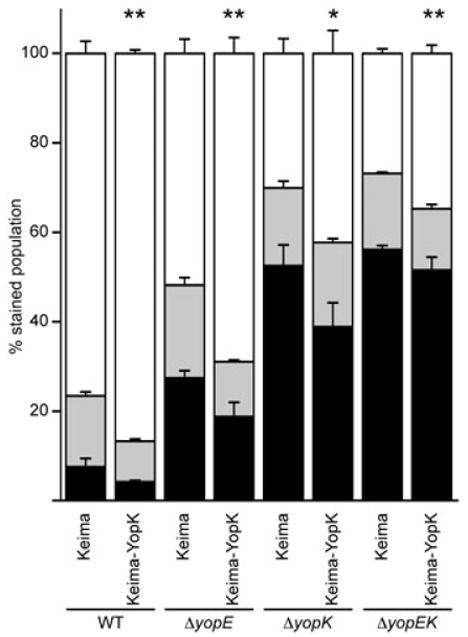
YopK regulation of TTSS is independent of YopE. CHO cells were transfected with plasmids expressing Keima or Keima–YopK and then infected with Y. pestis strains carrying the YopM-Bla reporter as done in Fig. 6. Infections were performed in triplicate and are shown as average with standard deviation. Student's t-test was performed to demonstrate significant differences in total injection levels (aqua + blue cells) for strains expressing Keima–YopK relative to the Keima-only controls (**P < 0.005, *P < 0.05). The data shown are representative of one data set and the experiment was performed at least twice.
Discussion
Previous studies have used indirect methods or population-based methods to determine whether YopK regulates effector injection, sometimes generating conflicting results (Holmstrom et al., 1997; Aili et al., 2008; Brodsky et al., 2010). Here we use the YopM-Bla reporter to expand upon previously published data that suggested YopK negatively regulates effector translocation into host cells (Holmstrom et al., 1997; Aili et al., 2008). The Bla reporter system allows us to directly measure translocation of an effector Yop into individual host cells during infection and analyse large infected populations in a high-throughput manner. We chose to focus on translocation of YopM, which is injected at sufficient levels to be easily detected and plays no role in modulating pore size or the injection process. This allows unique insight into YopK regulation of effector translocation. Consistent with previous reports, our assay shows increased levels of translocation in yopK mutants; however, our data clarify the phenotype by showing that ΔyopK infection results in the same number of injected cells as WT, but a higher amount of YopM-Bla is injected into each cell (Figs 1–3). The ΔyopK phenotype extended to injection of other Yops as well (Fig. S1 and Fig. 3), suggesting this phenomenon applies to all Yops, not just the YopM-Bla reporter. These data also indicate that YopK influences the rate of Yop translocation. Importantly, we have demonstrated for the first time that this regulatory activity of YopK is performed within host cells. By blocking the injection signal of YopK with an N-terminal Gst fusion, we have shown that a bacterially located YopK cannot regulate injection of YopM-Bla (Fig. 4). This is the first evidence that YopK plays an active role in regulating translocation from within host cells.
With the creation of a eukaryotic YopK expression vector, the function of YopK expressed within the host cell could be analysed for the first time. We have shown that when YopK is expressed exclusively in the host cell bacterial injection of the YopM-Bla reporter is significantly reduced (Fig. 6). In addition, the total number of host cells injected with the YopM-Bla reporter is reduced when the host cells are ‘pre-loaded’ with YopK. This is the first evidence that YopK can mediate a negative feedback signal from within the host cell to adherent extracellular bacteria. Recent work by Brodsky et al. has shown a role for YopK in preventing caspase-1 activation and subsequent inflammasome activation, although it was not demonstrated that this activity resulted specifically from a YopK function within host cells (Brodsky et al., 2010). Knowing that YopK regulates from inside the host cells opens a myriad of possibilities previously unforeseen for interactions between YopK and various eukaryotic proteins as well as other bacterial proteins.
YopK may regulate injection through interaction with the translocon pore complex. Several reports have implicated YopK in regulation of pore formation. A Y. pseudotuberculosis yopK mutant was evaluated for haemoglobin release from sheep erythrocytes and LDH release from HeLa cells to assess lytic pore formation (Holmstrom et al., 1997; Aili et al., 2008). These assays generally measure osmotic cell lysis due to pore formation, but the relationship of these lytic pores that are being measured to the translocon pores that are connected to the TTSS is not well defined. Since these assays do not measure translocation directly, the conclusions gained from them regarding regulation of Yop injection must be carefully considered. Measuring haemoglobin release, Holmstrom et al. (1997) reported that a yopK mutant showed increased lytic activity, similar to that of a yopHyopMyopE triple mutant, whereas a single yopE mutant was similar to the parent strain that displayed little activity. In contrast, using the LDH release assay, the yopE single mutant showed very high levels of lytic activity, whereas the yopK mutant was only slightly higher than the basal level displayed by wild type (Aili et al., 2008). Despite this discrepancy, the lytic phenotypes of yopE mutants and multi-yop mutants as measured by the LDH release assay seem to be consistent throughout the literature (Viboud and Bliska, 2001; Marenne et al., 2003; Viboud et al., 2006; Aili et al., 2008). Various models have been proposed that suggest the pores created by multi-yop mutants may be due to empty translocon channels, that YopE GAP activity modulates pore size by influencing actin rearrangement surrounding the pores, and that YopK contributes to pore regulation in some manner. However, a recent report showed phenotypes for parent and multi-yop mutants that were opposite of previous studies (Brodsky et al., 2010). Altogether, these differences highlight the importance of exercising caution when interpreting results from various lytic assays and making the distinction between lytic pores and translocation channels when extrapolating to formulate a model.
The contrasting YopK phenotypes, as measured by lytic assays, leave questions about the function of YopK; however, additional experiments suggest a role for YopK in translocation regulation. Digitonin fractionation experiments support a role for YopK interaction with the translocon pore complex, since YopK was found in the digitonin pellet fraction rather than in the soluble host cytosolic fraction (Lee et al., 1998). The pellet fraction contains host cell membranes, organelles and adherent bacteria. Recent work by Brodsky et al. has suggested an interaction between YopB and YopK using immunoprecipitation (Brodsky et al., 2010). These data support a model whereby YopK is injected into host cells, interacts with the pore complex within the host membrane, and either slows translocation of effector Yops or sends a negative feedback signal to the adherent bacterium. Our data further support this model by showing that YopK regulates from within the host cell, a yopK mutant has an increased rate of injection compared with WT, and that YopK expression in host cells reduces the number of host cells injected.
Despite reports indicating that YopK and YopE may have overlapping functions in regulating pore formation, we present evidence that the mechanisms of the two proteins are distinct from one another. First, the ΔyopK and ΔyopE mutants have distinctly different injection phenotypes as shown in Fig. 7. Second, the fact that YopK expressed in host cells reduces injection from a ΔyopE mutant clearly shows that YopK regulation of the TTSS is independent of YopE (Fig. 8). Furthermore, YopE has been shown to act on Rho G-proteins to mediate actin rearrangement leading to the formation of actin halos surrounding sites of bacterial contact (Viboud and Bliska, 2001; Mejia et al., 2008). Here we provide direct evidence that the GAP activity of YopE is integral to its ability to regulate translocation, indicating that actin rearrangement may contribute to translocation efficiency (Fig. 7). Thus, it seems that YopE's regulation of the injection process may be indirect, while YopK may directly influence the translocation pore.
Clearly, tight regulation of effector translocation is essential to pathogenesis. Yersinia spp. have been shown to target immune cells for Yop injection in vivo (Marketon et al., 2005; Köberle et al., 2009). The preferred targeting of innate immune cells would cripple the host's ability to mount a response to the infection. Furthermore, YopK is correlated with reduced inflammasome activation (Brodsky et al., 2010), which could be due to its ability to downregulate injection of other effector Yops. In addition, secreted LcrV is known to suppress an inflammatory response (Nakajima et al., 1995; Nedialkov et al., 1997; Brubaker, 2003). It is likely that the collective functions of YopK and YopE co-ordinate the effector translocation process to prevent over-injection of cytotoxic Yops in order to induce ultimate cell death, while also minimizing excessive inflammatory signalling. That, along with the actions of LcrV, would allow the bacteria to create a nutrient-rich necrotic lesion in which to replicate and achieve high bacterial loads before the host is alerted to the infection. Indeed, for Y. pestis this represents a very successful virulence strategy.
Experimental procedures
Bacterial strains and media
Stains used in this study are shown in Table 1. Y. pestis KIM5, an attenuated variant of the Y. pestis mediaevalis strain KIM lacking the 102 kb pgm locus (Brubaker, 1969), was propagated on Heart Infusion Agar (HIA) plates at 26°C for 2 days. Overnight cultures were grown in Heart Infusion Broth (HIB) at 26°C. Antibiotics were added as appropriate to a final concentration of 20 μg ml−1 chloramphenicol (Cm), 50 μg ml−1 kanamycin (Km) or 50 μg ml−1 ampicillin (Ap). Escherichia coli strains were propagated in Luria–Bertani broth or agar, supplemented with 30 μg ml−1 Cm, 100 μg ml−1 Ap or 50 μg ml−1 Km, at 37°C.
Table 1. Strains and plasmids used in this study.
| Strain or plasmid | Relevant phenotype | Citation |
|---|---|---|
| Y. pestis | ||
| KIM5 | Wild-type parent strain, attenuated Y. pestis mediaevalis strain lacking the pgm locus | Brubaker (1969) |
| MEL27 | ΔyopK | This work |
| MEL19 | ΔyopE | This work |
| DEW1 | ΔyopE, ΔyopK | This work |
| DEW2 | ΔyopH | This work |
| Plasmid | ||
| pHSG576 | Low-copy-number bacterial expression vector | Takeshita et al. (1987) |
| pCR2.1 | TA cloning vector | Invitrogen |
| pMM25 | For creation of yopE knockouts | This work |
| pMM83 | YopM with C-terminal Bla fusion expressed from yopM promoter on pHSG576 backbone | Marketon et al. (2005) |
| pMM91 | GST with C-terminal Bla fusion expressed from yopM promoter on pHSG576 backbone | Marketon et al. (2005) |
| pMM111 | YopH with C-terminal Bla fusion expressed from yopN promoter on pHSG576 backbone | This work |
| pMM112 | YopJ with C-terminal Bla fusion expressed from yopN promoter on pHSG576 backbone | This work |
| PMM118 | YopN with C-terminal Bla fusion expressed from yopN promoter on pHSG576 backbone | This work |
| pUC19 | Bacterial cloning vector, high copy number | Fermentas Life Sciences |
| pMM206 | yopK under native promoter on pUC19 backbone | This work |
| pMM208 | yopE under native promoter on pUC19 backbone | This work |
| pMM209 | pMM206 with N-terminal GST fusion to YopK | This work |
| phmKeima-Red-MCLinker | humanized monomeric Keima-Red fluorescent protein expression vector | MBL International Corp. |
| pRD1 | yopK ORF inserted as a C-terminal fusion to keima | This work |
| pRD3 | gst cloned into pRD1 at the N-terminus of yopK | This work |
| pRD7 | pMM208 carrying yopER144A | This work |
| pKD13 | Source of KmR cassette | Datsenko and Wanner (2000) |
| pWL204 | Carries λ red recombination genes | Lathem et al. (2007) |
| pLB001 | Source of Flp recombinase, created from pFLPs, ApR | This work |
| pBR322 | Source of β-lactamase ORF | Promega |
| pFLP3 | Source of Flp recombinase, TcR, ApR | Choi et al. (2005) |
| pHis-YopQ | yopK ORF cloned into pQE30 to express His-YopK | This work |
| pQE30 | Bacterial expression vector with N-terminal 6× His tag | Qiagen |
| pGEX-2TK | Source of Gst ORF | Amersham |
| pLC28 | Suicide vector | Cheng et al. (1997) |
| pKD3 | Source of CmR cassette | Datsenko and Wanner (2000) |
| pKD46 | Carries λ red recombination genes | Datsenko and Wanner (2000) |
| pCP20 | Source of Flp recombinase | Cherepanov and Wackernagel (1995) |
| pMM1 | Used for curing pCP20 | Marketon et al. (2005) |
| pGH3 | rpoA ORF cloned into pDEST17 to express His-RpoA | This work |
| pGH5 | yopE ORF cloned into pDEST17 to express His-YopE | This work |
| pMM230 | yopM ORF cloned into pDEST17 to express His-YopM | This work |
| pDest17 | Bacterial expression vector with N-terminal 6× His tag | Invitrogen |
| pENTR/D-TOPO | Cloning vector | Invitrogen |
Strain and plasmid construction
Plasmids and primers used in this study are shown in Tables 1 and 2. Fusion of Yops (YopH, YopJ, YopN) to the mature portion of TEM-1 β-lactamase was created by amplifying Yop ORFs from pCD1 as fragments containing engineered NdeI and KpnI sites at the 5′ and 3′ ends, respectively, using the primers YopHORF-NdeI and YopHORF-KpnI, YopJORF-NdeI and YopJORF-KpnI, YopNORF-NdeI and YopNORF-KpnI. All ORFs were cloned behind the yopN promoter, which was amplified from pCD1 as a fragment containing EcoRI and NdeI sites at the 5′ and 3′ ends, respectively, using the primers YopNpro-EcoRI and YopNpro-NdeI. The Bla fragment was amplified from pBR322 (Promega) with engineered KpnI and BamHI sites at its 5′ and 3′ ends as described previously (Marketon et al., 2005). PCR fragments encoding the yopN promoter, Yop ORFs and Bla were cloned between EcoRI and BamHI sites of the low-copy vector pHSG576 (Takeshita et al., 1987), resulting in pMM111 (YopH-Bla), pMM112 (YopJ-Bla), pMM118 (YopN-Bla).
Table 2. Primers used in this study.
| Primer name | Sequence |
|---|---|
| YopHKO_P1 | 5′-GAAGCAGCTCCAGCCTACACCATGCTTCCCTCCTTAATTAAATACACGCC |
| YopHKO_P4 | 5′-GGTCGACGGATCCCCCGGAATTGTAAATATTTATTCCTATGAGTAAATAAA |
| YopH upstream | 5′-GCATCCGTCCGGTGGCCGTAGGCCG |
| YopH downstream | 5′-ACTACATACACACTGGGAAAATCTC |
| GST-BamHI-For | 5′-AAGGATCCTCCCCTATACTAGGTTAT |
| GST-BamHI-Rev | 5′-AAGGATCCGTCACGATGAATTCCCGG |
| YopK-KpnI-For | 5′-AAGGTACCTTTATTAAAGATACTTATAACATGCGT |
| YopQ-Hind-Rev | 5′-AAAAGCTTTCATCCCATAATACATTCTTGATC |
| PKD13Kan.P1 | 5′-GTGTAGGCTGGAGCTGCTTC |
| PKD13Kan.P4 | 5′-ATTCCGGGGATCCGTCGACC |
| YopE_R144A For | 5′-GAGTGATACTGCCGGCAAGAGGGCCGCTGC |
| YopE_R144A Rev | 5′-GCAGCGGCCCTCTTGCCGGCAGTATCACTC |
| yopNpro-EcoRI | 5′-AAGAATTCACCCACCCCAACCTGAT |
| yopNpro-Ndel | 5′-AACATATGAACTACTCCCTGAGATG |
| yopNORF-NdeI | 5′-AACATATGACGACGCTTCATAACC |
| YopNORF-KpnI | 5′-AAGGTACCGAAAGGTCGATCGCCATT |
| YopHORF-NdeI | 5′-AACATATGAACTTATCATTAAGCGAT |
| YopHORF-KpnI | 5′-AAGGTACCGCTATTTAATAATGGTCG |
| YopJORF-NdeI | 5′-AACATATGATCGGACCAATATCACAA |
| YopJORF-KpnI | 5′-AAGGTACCTACTTTGAGAAGTGTTTT |
| yopK-upstream | 5′-AAGCTTTGGCCTTGTGCAT |
| yopK-downstream | 5′-GCATGCCTCTGGTGTCAGTGCAGGAT |
| GST-YopK-L | 5′-GTTTATAAAGTAAATTTGGGAGTAGTAACTATGTCCCCTATACTAGGTTATTG |
| GST-YopK-R | 5′-AGCACGCATGTTATAAGTATCTTTAATAAAATCCGATTTTGGAGGATGGTCGC |
| yopE-upstream | 5′-AGATCTGACGCTTTCGCGATGAATGC |
| yopE-downstream | 5′-AAGCTTTTTGCCGTTATAAATG |
| yopEKO-SalI | 5′-AAGTCGACGGTAATTCTGATAGTG |
| yopEKO-Hind1 | 5′-AAAAGCTTGCAGGGGCAGTGATG |
| yopEKO-Hind2 | 5′-AAAAGCTTGAGATCAAAGGGCTGG |
| yopEKO-XbaI | 5′-AATCTAGAGATGAAGCCTCAAAGG |
| yopQKO-P1 | 5′-GTTTATAAAGTAAATTTGGGAGTAGTAACTATGAGTGTAGGCTGGAGCTGCTTC |
| yopQKO-P2 | 5′-TCAACTACCATATCCCAAACTCTTTAATATAGCTTCACATATGAATATCCTCCTTAG |
| pFLP3_tet_forward | 5′-TTCTTGCGGAGAACTGTGAATGCG |
| pFPL3_tet_reverse | 5′-GGTGCCTGACTGCGTTAGCAATTT |
| pFLP3_tet_forward A | 5′-CTGTTGCATGGGCATAAAGTTGCC |
| pFLP3_tet_reverse A | 5′-ACACGCACTATGCCGTTCTTCTCA |
| YopQ-Bam-For | 5′-AAGGATCCTTTATTAAAGATACTTATAACATGCGT |
| RpoA_TopoFor | 5′-CACCCAGGGTTCTGTGACAGAG |
| RpoA_TopoRev | 5′-TTACTCGTCAGCAATGCT |
| YopM_TopoFor | 5′-CACCTTCATAAATCCAAGAAAT |
| YopM_TopoRev | 5′-TACTCAAATACATCATCT |
| YopE_TopoFor | 5′-CACCAAAATATCATCATTTATT |
| YopE_TopoRev | 5′-TCACATCAATGACAGTAA |
To construct pMM206, a 3 kb fragment containing the yopK ORF and native promoter was amplified from pCD1 using the primers yopK-upstream and yopK-downstream, and the blunt PCR product was cloned into the SmaI site of pUC19. To construct pMM209 (Gst–YopK), the Gst ORF from pGEX-2TK (Amersham) was amplified using the primers GST-YopK-L and GST-YopK-R, which generated a product containing gst flanked by homology to the 5′ end of yopK. The PCR product was then used as the primers for a second PCR using pMM206 as the template, which resulted in the insertion of gst at the 5′ end of the yopK ORF thereby creating a Gst–YopK fusion.
To construct pMM208, a 2.5 kb fragment containing the yopE and sycE ORFs and native promoter was amplified from pCD1 using the primers yopE-upstream and yopE-downstream, and the blunt PCR product was cloned into the SmaI site of pUC19. The yopE GAP mutant was created by quick change PCR using primers YopE_R144A For and YopE_R144A Rev to introduce a single codon mutation into the yopE ORF in pMM208, thus creating pRD7.
Fusion of the red fluorescent protein Keima to YopK was constructed by amplifying the yopK ORF using primers YopK-KpnI-For and YopQ-Hind-Rev. The PCR product containing KpnI and HindIII sites at the 5′ and 3′ ends, respectively, was cloned into the KpnI and HindIII sites of phmKeima-Red-MCLinker (MBL International Corporation), thereby creating pRD1 (Keima–YopK). pRD3 (Keima–Gst–YopK) was constructed by amplifying the gst ORF from pGEX-T2K with primers Gst-BamHI-For and Gst-BamHI-Rev. The PCR product containing BamHI sites at each end was cloned into the BamHI site within the linker region of pRD1.
MEL19 (ΔyopE) was created using allelic exchange as previously described (Cheng et al., 1997). Two fragments flanking yopE were amplified using the primers yopEKO-SalI and yopEKO-Hind1 as well as yopEKO-Hind2 and yopEKO-XbaI. The resulting fragments were cloned into pCR2.1 (Invitrogen) and then subcloned into pLC28 using a three-way ligation to yield pMM25. pMM25 was then electroporated into KIM5 and single-cross-over events were selected by resistance to Cm. CmR colonies were then grown in 5% sucrose to select for double-cross-over events to eliminate the inserted vector and drug cassette. Colonies were screened for loss of yopE by PCR using the yopEKO-SalI and yopEKO-XbaI primers. Mutants were identified by the presence of a smaller PCR product that could also be cut by HindIII.
MEL27 (ΔyopK) was created using λ Red-mediated recombination. The CmR cassette with flanking homology to yopK was amplified from pKD3 using the primers yopQKO-P1 and yopQKO-P2. The PCR product was electroporated into Y. pestis KIM5 containing pKD46 as previously described (Datsenko and Wanner, 2000). The resulting pCD1 plasmid carrying the CmR cassette was isolated and electroporated into KIM5 lacking pKD46 and absence of pKD46 was verified by sensitivity to Ap. pCP20 carrying the Flp recombinase (Datsenko and Wanner, 2000) was then introduced to eliminate the CmR cassette, creating an unmarked yopK deletion. pCP20 was cured by introducing pMM1 (Marketon et al., 2005), and then pMM1 was cured by plating on 5% sucrose. The resulting strain, MEL27, carries a yopK deletion and no antibiotic resistances.
DEW1 (ΔyopEΔyopK) was constructed by allelic exchange as described above for MEL19, creating a deletion of yopE in the MEL27 (ΔyopK) strain. The final strain, DEW1, carries unmarked deletions of yopE and yopK.
DEW2 (ΔyopH) was created by λ Red-mediated recombination, as previously described (Lathem et al., 2007), except that pLB001 was used as a source of FRT recombinase. Briefly, the KnR cassette flanked by FRT sites was amplified from pKD13 using primers PKD13Kan.P1 and PKD13Kan. P4. Next, primers YopH upstream and YopHKO_P1, and YopH downstream and YopHKO_P4 were used in PCR to amplify 500 bp of homologous regions on either side of YopH from genomic DNA. The PCR products were electroporated into KIM5 carrying pWL204 and expressing the λ red genes (Lathem et al., 2007). pWL204 was cured by growing colonies on 5% sucrose plates. The KnR cassette was resolved by electroporating pLB001, which contains the FRT recombinase, into the strain. pLB001 was created using deletion PCR to excise the tetracycline (Tc) resistance cassette from pFLP3 (Choi et al., 2005). Outward primers pFLP3_ tet_forward and pFLP3_tet_reverse were used to amplify plasmid pFLP3 and then the PCR product was ligated and transformed into DH5α. Resultant colonies were screened by Tc sensitivity, digestion with NdeI and EcoRI and PCR with primers outside the TcR cassette, pFLP3_tet_forward A and pFLP3_tet_reverseA. pLB001 was in turn cured by growth on 5% sucrose. The final strain, DEW2, carries an unmarked deletion of yopH.
Antibodies
The YopK antibody was created with expression of YopK from pHis-YopQ. The yopK ORF was amplified from pCD1 using primers YopQ-Bam-For and YopQ-Hind-Rev and cloned into expression vector pQE30 (Invitrogen) between the BamHI and HindIII restriction sites resulting in an N-terminal 6× His tag to YopK. The plasmid was transformed into XL1-Blue cells (Stratagene) and induced to express with 1 mM IPTG for 4 h. Cells were lysed with a French press in the presence of Complete protease inhibitor tabs (Roche) and the soluble fraction was run on a nickel column. Imidazole (200 mM) was used to elute His-tagged YopK from the column and the purified sample was sent to Cocalico Biologicals for antibody production in New Zealand white rabbits.
RpoA, YopE and YopM ORFs were expressed as C-terminal fusions to the His tag in pDEST 17. The ORFs were amplified from KIM5 genomic DNA using the primers RpoA_TopoFor and RpoA_TopoRev, YopE_TopoFor and YopE_TopoRev, YopM_TopoFor and YopM_TopoRev. The PCR products were cloned into pENTR/D-TOPO (Invitrogen) following manufacturer's instructions. The resulting plasmids were subjected to an LR Clonase reaction (Invitrogen) to recombine the ORFs into pDEST17 (Invitrogen), thereby generating pGH3, pGH5 and pMM230 to express the His-tagged proteins. pGH3 was given to the Cultivation and Bio-processing Facility (Indiana University, Bloomington) where it was transformed into BL21 <DE3> cells and expression of His-RpoA was induced with 0.2 mM IPTG overnight. Cells were lysed by sonication in buffer containing 20 mM Tris, pH 8.0, 300 mM NaCl, 10 mM imidazole, 5 mM β-mercaptoethanol, 0.1 mg ml−1 lysozyme, 4 mM MgCl2 and 100 μl of protease inhibitor cocktail, and the soluble fraction was purified by nickel column chromatography and eluted with an imidazole gradient. pGH5 and pMM230 were transformed into BL21 <DE3> (pLysS) cells and expression was induced with 1 mM IPTG for 3 h. Cells were lysed with Bug-Buster Reagent (Novagen) according to manufacturer's instructions. His-YopE was found primarily in inclusion bodies, so the insoluble fraction was collected and used for antibody production. His-YopM was purified from the soluble fraction by nickel chromatography and eluted with an imidazole gradient. All three proteins were sent to Covance Research Products for antibody production in New Zealand white rabbits.
Antibodies were purchased for GST (Affinity BioReagents), Keima (MBL international), Bla (Chemicon International), GAPDH (Novus Biologicals) and p130cas (BD Biosciences). The YscD, YopN and YopH antibodies were a gift from the laboratory of Olaf Schneewind.
Mammalian cell culture and transient transfection
CHO-K1 (ATCC) and CMV-Bla CHO-K1 (Invitrogen) were maintained in F12K (ATCC) supplemented with 10% heat-inactivated FBS (Cellgro). CMV-Bla CHO-K1 cells were supplemented with 1 mg ml−1 geneticin (Invitrogen). Transfections were performed using Lipofectamine 2000 (Invitrogen) as suggested by the manufacturer. CHO cells were seeded at 6.5 × 105 cells per well into six-well plates 1 day prior to transfection. Each transfection used 4 μg of plasmid DNA, 12.5 μl of Lipofectamine 2000 and 500 μl of OptiMEM (Invitrogen) per well. Cells were incubated for 12–48 h at 37°C, 5% CO2 to allow expression.
Yop secretion assay
Secretion assays were performed as previously described (Marketon et al., 2005). Bacteria are grown overnight at 26°C in HIB with antibiotics as appropriate, then subcultured 1:20 into modified M9. They were grown an additional 2 h at 26°C before transferring to 37°C for 3 h to induce secretion. Proteins in medium supernatant and bacterial pellet fractions were TCA precipitated and visualized by immunoblotting.
Injection assay
CHO cells were seeded at 6.5 × 105 per well of a six-well plate 1 day before infection. Y. pestis was grown to exponential phase at 26°C in HIB with antibiotics as appropriate and then transferred to 37°C for 1 h to induce expression of TTSS proteins. Bacteria were added at moi = 10 to CHO cells. Synchronization was achieved by quickly adding bacteria to cells within a multi-well plate and then briefly centrifuging the plate (500 g for 5 min) to facilitate contact between bacteria and host cells. Infected cells were incubated at 37°C, 5% CO2 for 30 min before the monolayer was washed with warmed F12K to remove non-adherent bacteria. Cells were further incubated at 37°C, 5% CO2 for 2.5–4 h until ∼90% cell rounding was observed. The cells were detached using trypsin/EDTA and then suspended in HBSSflow (1× HBSS, 0.5 mM EDTA, 25 mM HEPES, 2% BSA, pH 7.4) and strained using a 0.4 μm filter. CCF2-AM dye (6×; Invitrogen) was added to each sample to obtain a final concentration of 1× per manufacturer's instructions. HBSS (Gibco) was substituted for Solution C in the 6× dye to prevent autofluorescence interference during flow cytometry. Samples were incubated for 15 min at room temperature then transferred to ice until analysis. Each sample was then analysed on a BD FACSAria II cell sorter in the Indiana University Flow Cytometry Core Facility. Flow cytometry samples were first gated based on size exclusion as determined by propidium iodide (BD Biosciences) positive cells stained at a final concentration of 0.1 μg ml−1, which are either apoptotic or necrotic. Flow cytometry gates for green and blue fluorescence were then set using uninfected CHO cells (green gate, Fig. 1A, column 2) and a control CHO cell line (CMV-bla, Invitrogen) expressing β-lactamase constitutively (blue gate, Fig. 1A, column 3), both dyed with CCF2-AM. Finally, experimental samples were analysed for green and blue fluorescence. At least 10 000 live cells are measured for each sample. As time is a factor in the intensity of blue fluorescence, one sample from each triplicate infection is analysed in order. The order is repeated until all three samples have been analysed to ensure that the average injection of triplicate samples includes any inherent change in fluorescence that occurs over time. Since the kinetics of infection can vary from day to day, infected samples are prepared in triplicate and compared with controls run on the same day. Trends and statistical significance are calculated for the data collected each day and also compared with data collected from at least two independent trials to ensure the reproducibility of each injection phenotype.
Injection assays following transfection were similar with some modifications. CHO cells were seeded at 6.5 × 105 cells per well 1 day prior to transfection. CHO cells were transfected as described above and allowed to express recombinant proteins for 36 h before infection occurred. CHO cells were washed with warmed F12K, 10% FBS prior to infection. Additional control samples were included for setting gates and compensation on the Aria II. Transfected and mock-transfected CHO cells were used to set the gates for red fluorescence, while transfected CHO cells stained with CCF2-AM were used to adjust the compensation between Keima and fluorescein fluorescence. Cells were gated first for live population by size exclusion, and then analysed for red fluorescence followed by blue versus green analysis. At least 3000 transfection events were measured for each sample.
For digitonin fractionation experiments, CHO cells were infected and harvested for flow cytometry as described above. After an aliquot of cells was removed for CCF2-AM staining, the remainder of the cells were incubated with digitonin extraction buffer to release soluble cytoplasmic proteins from CHO cells, as previously described (Ramsby and Makowski, 2005). Proteins in the supernatant and pellet fractions were analysed by SDS-PAGE and immunoblotting. Antibodies to p130cas were used as a fractionation control for soluble cytoplasmic eukaryotic proteins, while RpoA was used a fractionation control for cytoplasmic bacterial proteins. Additional antibodies monitored the delivery of the YopM-Bla and YopH-Bla reporters, as well as native YopH and YopM.
Supplementary Material
Acknowledgments
We would like to acknowledge the Indiana University Bloomington Flow Cytometry Core Facility for use of the FACSAria II flow cytometer and Dr Pengyun Li in the Cultivation and Bioprocessing Facility for assistance in purifying proteins for antibody production. Special thanks to Lesley Bohman and Gwendolyn Humphreys for aid in creating strains and plasmids, and Coral Maldonado for cloning, expressing and purifying YopK. Thanks to other members of the Marketon laboratory for helpful discussions on the work and the manuscript. This work is supported by award number R21AI083660 from the NIAID to Melanie Marketon and NIH Training Grant T32 GMOO7757 support for Rebecca Dewoody. We acknowledge membership in and support provided by the Region V ‘Great Lakes’ RCE (NIH award 2-U54-AI-057153).
Footnotes
Supporting information: Additional supporting information may be found in the online version of this article.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- Aili M, Hallberg B, Wolf-Watz H, Rosqvist R. GAP activity of Yersinia YopE. Methods Enzymol. 2002;358:359–370. doi: 10.1016/s0076-6879(02)58102-7. [DOI] [PubMed] [Google Scholar]
- Aili M, Isaksson EL, Carlsson SE, Wolf-Watz H, Rosqvist R, Francis MS. Regulation of Yersinia Yop-effector delivery by translocated YopE. Int J Med Microbiol. 2008;298:183–192. doi: 10.1016/j.ijmm.2007.04.007. [DOI] [PubMed] [Google Scholar]
- Ben-Gurion R, Shafferman A. Essential virulence determinants of different Yersinia species are carried on a common plasmid. Plasmid. 1981;5:183–187. doi: 10.1016/0147-619x(81)90019-6. [DOI] [PubMed] [Google Scholar]
- Black DS, Bliska JB. The RhoGAP activity of the Yersinia pseudotuberculosis cytotoxin YopE is required for antiphagocytic function and virulence. Mol Microbiol. 2000;37:515–527. doi: 10.1046/j.1365-2958.2000.02021.x. [DOI] [PubMed] [Google Scholar]
- Boland A, Havaux S, Cornelis GR. Heterogeneity of the Yersinia YopM protein. Microb Pathog. 1998;25:343–348. doi: 10.1006/mpat.1998.0247. [DOI] [PubMed] [Google Scholar]
- Brodsky IE, Palm NW, Sadanand S, Ryndak MB, Sutterwala FS, Flavell RA, et al. A Yersinia effector protein promotes virulence by preventing inflammasome recognition of the type III secretion system. Cell Host Microbe. 2010;7:376–387. doi: 10.1016/j.chom.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker RR. Mutation rate to nonpigmentation in Pasteurella pestis. J Bacteriol. 1969;98:1404–1406. doi: 10.1128/jb.98.3.1404-1406.1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brubaker RR. Interleukin-10 and inhibition of innate immunity to Yersiniae: roles of Yops and LcrV (V antigen) Infect Immun. 2003;71:3673–3681. doi: 10.1128/IAI.71.7.3673-3681.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cambronne ED, Sorg JA, Schneewind O. Binding of SycH chaperone to YscM1 and YscM2 activates effector Yop expression in Yersinia enterocolitica. J Bacteriol. 2004;186:829–841. doi: 10.1128/JB.186.3.829-841.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng LW, Anderson DM, Schneewind O. Two independent type III secretion mechanisms for YopE in Yersinia enterocolitica. Mol Microbiol. 1997;24:757–765. doi: 10.1046/j.1365-2958.1997.3831750.x. [DOI] [PubMed] [Google Scholar]
- Cherepanov P, Wackernagel W. Gene disruption in Escherichia coli: TcR and KmR cassettes with the option of Flp-catalyzed excision of the antibiotic-resistance determinant. Gene. 1995;158:9–14. doi: 10.1016/0378-1119(95)00193-a. [DOI] [PubMed] [Google Scholar]
- Choi K, Gaynor JB, White KG, Lopez C, Bosio CM, Karkhoff-Schweizer RR, Schweizer HP. A Tn7-based broad-range bacterial cloning and expression system. Nat Methods. 2005;2:443–448. doi: 10.1038/nmeth765. [DOI] [PubMed] [Google Scholar]
- Cornelis G. The Yersinia Ysc-Yop virulence apparatus. Int J Med Microbiol. 2002a;291:455–462. doi: 10.1078/1438-4221-00153. [DOI] [PubMed] [Google Scholar]
- Cornelis G. Yersinia type III secretion: send in the effectors. J Cell Biol. 2002b;158:401–408. doi: 10.1083/jcb.200205077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis DT, Gage K, Gratz N, Poand J, Tikhomirov E. Plague Manual: Epidemiology, Distribution, Surveillance and Control. Geneva: World Health Organization; 1999. [Google Scholar]
- Garcia JT, Ferracci F, Jackson MW, Joseph SS, Pattis I, Plano LRW, et al. Measurement of effector protein injection by type III and type IV secretion systems by using a 13-residue phosphorylatable glycogen synthase kinase tag. Infect Immun. 2006;74:5645–5657. doi: 10.1128/IAI.00690-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan KL, Dixon JE. Protein tyrosine phosphatase activity of an essential virulence determinant in Yersinia. Science. 1990;249:553–556. doi: 10.1126/science.2166336. [DOI] [PubMed] [Google Scholar]
- Hakansson S, Schesser K, Persson C, Galyov EE, Rosqvist R, Homble F, Wolf-Watz H. The YopB protein of Yersinia pseudotuberculosis is essential for the translocation of Yop effector proteins across the target cell plasma membrane and displays a contact-dependent membrane disrupting activity. EMBO J. 1996;15:5812–5823. [PMC free article] [PubMed] [Google Scholar]
- Holmstrom A, Rosqvist R, Wolf-Watz H, Forsberg A. Virulence plasmid-encoded YopK is essential for Yersinia pseudotuberculosis to cause systemic infection in mice. Infect Immun. 1995a;63:2269–2276. doi: 10.1128/iai.63.6.2269-2276.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmstrom A, Rosqvist R, Wolf-Watz H, Forsberg A. YopK, a novel virulence determinant of Yersinia pseudotuberculosis. Contrib Microbiol Immunol. 1995b;13:239–243. [PubMed] [Google Scholar]
- Holmstrom A, Pettersson J, Rosqvist R, Hakansson S, Tafazoli F, Fallman M, et al. YopK of Yersinia pseudotuberculosis controls translocation of Yop effectors across the eukaryotic cell membrane. Mol Microbiol. 1997;24:73–91. doi: 10.1046/j.1365-2958.1997.3211681.x. [DOI] [PubMed] [Google Scholar]
- Kerschen EJ, Cohen DA, Kaplan AM, Straley SC. The plague virulence protein YopM targets the innate immune response by causing a global depletion of NK cells. Infect Immun. 2004;72:4589–4602. doi: 10.1128/IAI.72.8.4589-4602.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köberle M, Klein-Günther A, Schütz M, Fritz M, Berchtold S, Tolosa E, et al. Yersinia enterocolitica targets cells of the innate and adaptive immune system by injection of Yops in a mouse infection model. PLoS Pathog. 2009;5:e1000551. doi: 10.1371/journal.ppat.1000551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kogure T, Kawano H, Abe Y, Miyawaki A. Fluorescence imaging using a fluorescent protein with a large Stokes shift. Methods. 2008;45:223–226. doi: 10.1016/j.ymeth.2008.06.009. [DOI] [PubMed] [Google Scholar]
- Lathem WW, Price PA, Miller VL, Goldman WE. A plasminogen-activating protease specifically controls the development of primary pneumonic plague. Science. 2007;315:509–513. doi: 10.1126/science.1137195. [DOI] [PubMed] [Google Scholar]
- Lee VT, Anderson DM, Schneewind O. Targeting of Yersinia Yop proteins into the cytosol of HeLa cells: one-step translocation of YopE across bacterial and eukaryotic membranes is dependent on SycE chaperone. Mol Microbiol. 1998;28:593–601. doi: 10.1046/j.1365-2958.1998.00822.x. [DOI] [PubMed] [Google Scholar]
- Lee VT, Tam C, Schneewind O. LcrV, a substrate for Yersinia enterocolitica type III secretion, is required for toxin targeting into the cytosol of HeLa cells. J Biol Chem. 2000;275:36869–36875. doi: 10.1074/jbc.M002467200. [DOI] [PubMed] [Google Scholar]
- Leung KA, Reisner BS, Straley SC. YopM inhibits platelet aggregation and is necessary for virulence of Yersinia pestis in mice. Infect Immun. 1990;58:3262–3271. doi: 10.1128/iai.58.10.3262-3271.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald C, Vacratsis PO, Bliska JB, Dixon JE. The Yersinia virulence factor YopM forms a novel protein complex with two cellular kinases. J Biol Chem. 2003;278:18514–18523. doi: 10.1074/jbc.M301226200. [DOI] [PubMed] [Google Scholar]
- Marenne MN, Journet L, Mota LJ, Cornelis GR. Genetic analysis of the formation of the Ysc-Yop translocation pore in macrophages by Yersinia enterocolitica: role of LcrV, YscF and YopN. Microb Pathog. 2003;35:243–258. doi: 10.1016/s0882-4010(03)00154-2. [DOI] [PubMed] [Google Scholar]
- Marketon MM, DePaolo RW, DeBord KL, Jabri B, Schneewind O. Plague bacteria target immune cells during infection. Science. 2005;309:1739–1741. doi: 10.1126/science.1114580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mejia E, Bliska JB, Viboud GI. Yersinia controls type III effector delivery into host cells by modulating Rho activity. PLoS Pathog. 2008;4:e3. doi: 10.1371/journal.ppat.0040003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulder B, Michiels T, Simonet M, Sory MP, Cornelis G. Identification of additional virulence determinants on the pYV plasmid of Yersinia enterocolitica W227. Infect Immun. 1989;57:2534–2541. doi: 10.1128/iai.57.8.2534-2541.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakajima R, Motin VL, Brubaker RR. Suppression of cytokines in mice by protein A–V antigen fusion peptide and restoration of synthesis by active immunization. Infect Immun. 1995;63:3021–3029. doi: 10.1128/iai.63.8.3021-3029.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedialkov YA, Motin VL, Brubaker RR. Resistance to lipopolysaccharide mediated by the Yersinia pestis V antigen–polyhistidine fusion peptide: amplification of interleukin-10. Infect Immun. 1997;65:1196–1203. doi: 10.1128/iai.65.4.1196-1203.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neyt C, Cornelis GR. Insertion of a Yop translocation pore into the macrophage plasma membrane by Yersinia enterocolitica: requirement for translocators YopB and YopD, but not LcrG. Mol Microbiol. 1999;33:971–981. doi: 10.1046/j.1365-2958.1999.01537.x. [DOI] [PubMed] [Google Scholar]
- Orth K. Function of the Yersinia effector YopJ. Curr Opin Microbiol. 2002;5:38–43. doi: 10.1016/s1369-5274(02)00283-7. [DOI] [PubMed] [Google Scholar]
- von Pawel-Rammingen U, Telepnev MV, Schmidt G, Aktories K, Wolf-Watz H, Rosqvist R. GAP activity of the Yersinia YopE cytotoxin specifically targets the Rho pathway: a mechanism for disruption of actin microfilament structure. Mol Microbiol. 2000;36:737–748. doi: 10.1046/j.1365-2958.2000.01898.x. [DOI] [PubMed] [Google Scholar]
- Persson C, Nordfelth R, Holmstrom A, Hakansson S, Rosqvist R, Wolf-Watz H. Cell-surface-bound Yersinia translocate the protein tyrosine phosphatase YopH by a polarized mechanism into the target cell. Mol Microbiol. 1995;18:135–150. doi: 10.1111/j.1365-2958.1995.mmi_18010135.x. [DOI] [PubMed] [Google Scholar]
- Pettersson J, Holmstrom A, Hill J, Leary S, Frithz-Lindsten E, von Euler-Matell A, et al. The V-antigen of Yersinia is surface exposed before target cell contact and involved in virulence protein translocation. Mol Microbiol. 1999;32:961–976. doi: 10.1046/j.1365-2958.1999.01408.x. [DOI] [PubMed] [Google Scholar]
- Ramsby M, Makowski G. Differential detergent fractionation of eukaryotic cells. In: Walker J, editor. The Proteomics Protocols Handbook. Totowa, NJ: Humana Press; 2005. pp. 37–48. [Google Scholar]
- Ruckdeschel K, Pfaffinger G, Trulzsch K, Zenner G, Richter K, Heesemann J, Aepfelbacher M. The proteasome pathway destabilizes Yersinia outer protein E and represses its antihost cell activities. J Immunol. 2006;176:6093–6102. doi: 10.4049/jimmunol.176.10.6093. [DOI] [PubMed] [Google Scholar]
- Skrzypek E, Cowan C, Straley SC. Targeting of the Yersinia pestis YopM protein into HeLa cells and intracellular trafficking to the nucleus. Mol Microbiol. 1998;30:1051–1065. doi: 10.1046/j.1365-2958.1998.01135.x. [DOI] [PubMed] [Google Scholar]
- Sorg JA, Blaylock B, Schneewind O. Secretion signal recognition by YscN, the Yersinia type III secretion ATPase. Proc Natl Acad Sci USA. 2006;103:16490–16495. doi: 10.1073/pnas.0605974103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straley SC, Bowmer WS. Virulence genes regulated at the transcriptional level by Ca2+ in Yersinia pestis include structural genes for outer membrane proteins. Infect Immun. 1986;51:445–454. doi: 10.1128/iai.51.2.445-454.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Straley SC, Cibull ML. Differential clearance and host–pathogen interactions of YopE- and YopK- YopL-Yersinia pestis in BALB/c mice. Infect Immun. 1989;57:1200–1210. doi: 10.1128/iai.57.4.1200-1210.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabrizi S, Robins-Browne R. Elimination of extracellular bacteria by antibiotics in quantitative assays of bacterial ingestion and killing by phagocytes. J Immunol Methods. 1993;158:201–206. doi: 10.1016/0022-1759(93)90215-s. [DOI] [PubMed] [Google Scholar]
- Takeshita S, Sato M, Toba M, Masahashi W, Hashimoto-Gotoh T. High-copy-number and low-copy-number plasmid vectors for lacZ alpha-complementation and chloramphenicol- or kanamycin-resistance selection. Gene. 1987;61:63–74. doi: 10.1016/0378-1119(87)90365-9. [DOI] [PubMed] [Google Scholar]
- Viboud GI, Bliska JB. A bacterial type III secretion system inhibits actin polymerization to prevent pore formation in host cell membranes. EMBO J. 2001;20:5373–5382. doi: 10.1093/emboj/20.19.5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viboud GI, Bliska JB. Yersinia outer proteins: role in modulation of host cell signaling responses and pathogenesis. Annu Rev Microbiol. 2005;59:69–89. doi: 10.1146/annurev.micro.59.030804.121320. [DOI] [PubMed] [Google Scholar]
- Viboud GI, Mejia E, Bliska JB. Comparison of YopE and YopT activities in counteracting host signalling responses to Yersinia pseudotuberculosis infection. Cell Microbiol. 2006;8:1504–1515. doi: 10.1111/j.1462-5822.2006.00729.x. [DOI] [PubMed] [Google Scholar]
- Wattiau P, Cornells GR. SycE, a chaperone-like protein of Yersinia enterocolitica involved in the secretion of YopE. Mol Microbiol. 1993;8:123–131. doi: 10.1111/j.1365-2958.1993.tb01209.x. [DOI] [PubMed] [Google Scholar]
- Zumbihl R, Aepfelbacher M, Andor A, Jacobi CA, Ruckdeschel K, Rouot B, Heesemann J. The cytotoxin YopT of Yersinia enterocolitica induces modification and cellular redistribution of the small GTP-binding protein RhoA. J Biol Chem. 1999;274:29289–29293. doi: 10.1074/jbc.274.41.29289. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.