

Abstract
Previous studies have demonstrated that Notch signaling regulates endochondral and intramembranous bone formation by controlling cell proliferation and differentiation. Notch signaling has also been shown to regulate healing in a variety of tissues. The objective of this study was to characterize and compare activation of the Notch signaling pathway during endochondral and intramembranous bone fracture healing using tibial fracture and calvarial defect injury models, respectively. Bilateral tibial fractures or bilateral 1.5 mm diameter calvarial defects were created in mice, and tissues were harvested at 0, 5, 10 and 20 days post-fracture. Gene expression of Notch signaling components was upregulated during both tibial fracture and calvarial defect healing, with expression generally higher during tibial fracture healing. The most highly expressed ligand and receptor during healing, Jag1 and Notch2 (specifically the activated receptor, known as NICD2), were similarly localized in mesenchymal cells during both modes of healing, with expression decreasing during chondrogenesis, but remaining present in osteoblasts at all stages of maturity. Results suggest that in addition to embryological bone development, Notch signaling regulates both endochondral and intramembranous bone healing.
Keywords: Notch signaling, bone fracture healing
Introduction
Bone regeneration occurs through a series of spatiotemporal events that recapitulate many aspects of embryological development [1, 2]. Long bones such as the tibia develop and heal primarily through endochondral ossification (indirect bone formation on a cartilage intermediate), whereas bones such as the calvarium develop and heal through intramembranous ossification (direct bone formation) [3]. A number of growth factor pathways, including bone morphogenetic protein (BMP) and Wnt signaling, have been widely demonstrated to be required for fracture healing and have also been shown to promote regeneration [4–9]. However, despite the importance of these pathways, the significance of other growth factor pathways that regulate bone healing is not as well described.
Notch signaling is a developmentally conserved pathway that mediates the development of stem and progenitor cell populations in many tissues. Activation of the canonical Notch signaling pathway occurs through direct cell-to-cell contact. When one of four Notch ligands, Jagged (Jag) 1,2 and Delta-like (Dll) 1,4, interacts with one of four Notch receptors, Notch1-4, a two-stage proteolytic event liberates the Notch intracellular domain (NICD) which then translocates to the nucleus and binds with co-activators to initiate transcription of Notch target gene families Hes and Hey.
Notch gain of function mutations in the murine mesenchymal lineage result in enhanced cell proliferation while inhibiting differentiation, which prevents mature endochondral and intramembranous bone development [10, 11]. Alternatively, loss of Notch signaling in the mesenchymal lineage results in enhanced osteoprogenitor differentiation and early endochondral bone formation, which is rapidly lost during aging due to depletion of the progenitor pool [12, 13]. Notch signaling in osteoblasts has also been shown to negatively regulate osteoclast behavior [10, 13–15]. Collectively, these studies demonstrate that the Notch signaling pathway regulates endochondral and intramembranous bone formation.
Although Notch signaling has been shown to regulate tissue repair in a variety of tissues [16–21], an extensive characterization of Notch signaling during bone fracture healing has not been reported. Therefore, the objective of this study was to rigorously characterize and compare activation of the Notch signaling pathway during endochondral and intramembranous bone regeneration, using tibial fracture healing (TF) as a model of endochondral bone repair and calvarial defect healing (CD) as a model of intramembranous bone repair.
Methods
Experimental Design
All in vivo protocols were approved by the IACUC. Bilateral tibial fractures or bilateral calvarial defects were created in 8–11 week old male C57Bl/6 mice to evaluate Notch signaling during endochondral and intramembranous bone healing, respectively. Specimens were harvested at 0, 5, 10 and 20 days post-fracture (dpf). Quantitative real-time polymerase chain reaction (qRT-PCR) was used to quantify gene expression of Notch pathway components including ligands (Jag1,2, Dll1,4), receptors (Notch1–4), and target genes (Hes1, Hey1,2,L) (n=4–5). Immunohistochemistry was used to identify cell types that express the Jag1 ligand and the activated form of the Notch2 receptor, called the Notch2 intracellular domain (NICD2).
Tibial Fracture (TF) Procedure
Closed, transverse, mid-diaphyseal bilateral tibial fractures were created similar to previously published methods [22]. Briefly, under isoflurane anesthesia, a small incision was made medial to the tuberosity. A canal was punctured through the cortex using a 26-gauge needle, and a 0.009-inch diameter rod was inserted through the length of the intramedullary canal. The incision was closed with surgical glue. Fractures were created using a custom made three-point bending apparatus. Radiographs were generated to verify correct pin placement and fracture location (Faxitron X-Ray) (Supplemental Figure 1A). 0.05 mg/kg of buprenorphine was administered subcutaneously once after surgery. Mice recovered on heating pads and were fed ad libitum.
Calvarial Defect (CD) Procedure
Bilateral 1.5 mm diameter calvarial defects were created similar to previously published methods [23]. Under isoflurane anesthesia, the mouse was placed into stereotaxic equipment (Stoelting) and a sterile tegaderm drape (3M Health Care) was applied to the cranium after hair removal (Nair, Church & Dwight). A midline incision exposed the parietal bones, and a 1.5 mm diameter biopsy punch (Premier) was used to create a defect in the central portion of each parietal bone, leaving the surrounding periosteum intact (Supplemental Figure 1B). PBS was used to hydrate the tissue. The incision was closed with 5-0 prolene non-absorbable sutures (Ethicon). 0.05 mg/kg of buprenorphine was administered subcutaneously once after surgery. Mice recovered on heating pads and were fed ad libitum.
Quantitative Gene Expression
Fractured tibial calluses were dissected from the surrounding soft tissue at 5, 10 and 20 dpf. Uninjured diaphyseal bone, flushed of marrow, served as 0 dpf controls. Calvarial defects were dissected at 5, 10 and 20 dpf using a 3 mm diameter punch to excise the defect and surrounding bone tissue. Uninjured calvarial bone was similarly dissected for 0 dpf controls. Tissue was placed in Qiazol lysis reagent (Qiagen) and homogenized using the Tissue Tearor (BioSpec Products). mRNA was extracted using the Qiagen miRNeasy Mini Kit with DNase digestion to remove DNA contamination. RNA yield was determined spectrophotometrically. 1 μg of mRNA was reverse transcribed into cDNA using the Applied Biosystems High Capacity RNA-to-cDNA Kit. Gene expression was quantified from 0.5 μl of cDNA in 10 μl of Power SYBR Green PCR Master Mix (Applied Biosystems) using a 7500 Fast Real-Time PCR system (Applied Biosystems). For each gene of interest, samples were run in duplicate with several controls per primer set to verify that the measured signal was not due to DNA contamination or primer dimer binding. Proper amplicon formulation was confirmed by melt curve analysis.
Fracture healing involves a temporally changing profile of cells derived from different lineages. Although there is no ideal housekeeping gene for normalization across different cell types, a series of genes were identified that show minimal variation in expression [24]. We included three of those genes, run in duplicate and averaged together, as our housekeeping control:β-actin, which regulates cell motility; ornithine decarboxylase antizyme (OAZ1), which regulates polyamine synthesis; and 40S ribosomal protein 29 (RPS29), a component of the 40S ribosomal subunit that regulates protein synthesis. qRT-PCR data is presented as relative gene expression to housekeeping control, calculated using the formula 2−ΔC(t), where ΔC(t) is the difference in C(t) values between the gene of interest and the average of all three housekeeping genes.
Immunohistochemistry (IHC) and Histology
Tissue was fixed in 4% paraformaldehyde at 4ºC for 2–3 days, decalcified in a 4% hydrochloric acid 4% formic acid solution, paraffin embedded, and sectioned into 5 μm longitudinal slices. For Jag1 and NICD2 IHC, sections were deparaffinized and gradually hydrated. Sections were treated with blocking serum (5% donkey, 4% BSA, 0.1% Triton-X 100, 0.05% Tween 20) for 60 minutes at room temperature. Primary antibodies goat Jag1 (Santa Cruz sc-6011, 1:100) and rabbit cleaved NICD2 (Millipore 07–1234, 1:100) were incubated in a dilution buffer (2% BSA, 0.25% Triton-X 100) overnight at 4ºC in a humidified chamber. Control sections were treated with goat IgG (Santa Cruz sc-2028, 1:200) or rabbit IgG (Santa Cruz sc-2027, 1:200) to match the concentration of the appropriate antibody. Sections were then treated with 3% H2O2 for 30 minutes at room temperature, followed by biotinylated secondary antibody donkey anti-goat (Santa Cruz sc-2043, 1:200) or donkey anti-rabbit (Santa Cruz sc-2089, 1:200) for 30 minutes at room temperature, and finally streptavidin-HRP (Abcam ab7403, 1:500) for 30 minutes at room temperature. Sections were developed with DAB (Vector SK-4100) and counterstained with Hematoxylin. Additional sections were stained with Hematoxylin and Eosin (H&E) for 15 and 2.5 minutes, respectively, or 0.1% Safranin O and 0.03% Fast Green (SafO) for 5 minutes each to visualize tissue structure and cell morphology. Slides were imaged in brightfield with an Olympus BX51. Color images were acquired with a Spot RT3 2 megapixel camera.
Statistical Analysis
Significance was assessed by one-way ANOVAs comparing the effect of time on gene expression during TF and CD separately, followed by Tukey’s post-hoc test. Pairwise t-tests were made to evaluate the level of gene expression during TF vs. CD at each time point.
Results
Validation of TF and CD as models for EO and IO, respectively
Stabilized tibial fractures have been shown to heal primarily through endochondral ossification, whereas calvarial defects have been shown to heal via intramembranous ossification. We further set out to verify these injuries as appropriate models to study endochondral and intramembranous bone repair by quantifying gene expression of Col2, a marker of cartilage formation, and Ocn, a marker of bone formation, and by analyzing SafO histology for cartilage formation.
During tibial fracture healing (TF), Col2 was transiently upregulated, whereas Ocn was initially downregulated and then upregulated later (Supplemental Figure 2). Histology confirmed extensive cartilage in the callus at 10 dpf which was replaced with bone through endochondral ossification by 20 dpf (results not shown). During calvarial defect healing (CD), Col2 expression did not change, whereas Ocn was upregulated. The absence of cartilage formation confirmed by histology verifies healing through intramembranous ossification.
Comparison of Notch gene expression over time during TF and CD
Tissue was collected at 0, 5, 10 and 20 dpf for quantitation of Notch ligand, receptor and target gene expression. All Notch genes examined were upregulated over time during TF (Figure 1). Generally, the most highly expressed ligand, receptor and target gene during TF (relative to each other) were Jag1, Notch2 and Hes1, whereas the least expressed were Dll4, Notch4 and Hey2. The ligand, receptor and target gene that showed the greatest change (upregulation) during TF (relative to 0 dpf) were Jag2 (71-fold, 10 dpf), Notch4 (19-fold, 10 dpf) and Hes1 (172-fold, 10 dpf).
Figure 1.

Gene expression of Notch ligands (left), receptors (middle) and target genes (right) during TF (white bars) and CD (grey bars). # indicates a significant difference between TF vs. CD at a given time point (p<0.05). A common letter above any two bars indicates a significant difference between those time points during TF (a,b,c) or CD (x,y,z) (p<0.05). Data is presented as relative gene expression to the housekeeping genes, calculated using the formula 2− ΔC(t) (arbitrary units).
Only Jag1, Notch2 and Notch4 were upregulated over time during CD. However, consistent with TF, the most highly expressed ligand, receptor and target gene during CD (relative to each other) were Jag1, Notch2 and Hes1, whereas the least expressed were Dll1, Notch4 and Hey2. The ligand, receptor and target gene that showed the greatest change (upregulation) during CD (relative to 0 dpf) were Jag1 (4.2-fold, 10 dpf), Notch4 (11-fold, 20 dpf) and HeyL (2.4-fold, 10 dpf).
Comparison of Notch gene expression during TF vs. CD at each time point
We next compared the level of expression for each gene (relative to housekeeping gene expression) during TF vs. CD at each time point (0, 5, 10 and 20 dpf). Basal expression levels (0 dpf) of Jag1, Dll4, Notch1, Notch2, Notch3, Hes1, Hey1, Hey2, and HeyL were higher in uninjured calvaria. No genes were expressed higher in uninjured tibiae (Figure 1).
After injury (5, 10, 20 dpf), a greater number of genes were more highly expressed (relative to housekeeping gene expression) during TF compared to CD. Jag2, Dll1, Notch1, Notch3 and Notch4 were greater during TF, whereas Notch2 and Hey2 were greater during CD. Hes1 was the only gene to show variable expression during both CD and TF at different time points.
Identification of cells that express Jag1 and NICD2 during TF
Consistent with previous studies investigating mesenchymal tissues [12, 13, 25], Jag1 and Notch2 were the predominantly expressed ligand and receptor during both TF and CD at all time points. Therefore, using IHC, we identified cells that express the Jag1 ligand and the activated form of the Notch2 receptor, called the Notch2 intracellular domain (NICD2), which is indicative of activated Notch signaling.
Jag1 and NICD2 were expressed in identical cell populations that participate in endochondral bone repair during TF (Figure 2). Interestingly, it appears that more cells stain positive for NICD2 than Jag1 (non-statistical comparison). At 5 dpf, undifferentiated mesenchymal cells undergo rapid proliferation to produce a fibrovascular callus. These cells are largely Jag1 and NICD2 positive (Figure 2A, black arrows), though isolated cells appear negative (white arrows). By 10 dpf, these progenitors gradually lose Jag1 and NICD2 expression as they differentiate into proliferative (Figure 2B), pre-hypertrophic (Figure 2C), and finally hypertrophic chondrocytes (Figure 2D) when they become largely Jag1 and NICD2 negative. During the cartilage-to-bone transition at 10 dpf, mineralized cartilage is resorbed allowing for vascular invasion of the callus. Many vascular endothelial cells that penetrate the matrix are Jag1 and NICD2 positive (Figure 3). Surprisingly, terminal hypertrophic chondrocytes that populate the chondro-osseous junction and border the invading vasculature appear to re-express Jag1 and NICD2 (Figures 2E and 3). The vascular network mediates an influx of Jag1 and NICD2 positive osteoprogenitor cells that lay the initial osteoid matrix on top of the resorbing cartilage (Figure 2F). By 20 dpf, these cells differentiate into immature and mature osteoblasts to produce primary (Figure 2G) and remodeled bone (Figure 2H), and continue to overwhelmingly, but not completely, express Jag1 and NICD2. Osteocytes embedded in remodeled bone are both positive and negative for Jag1 and NICD2 (Figure 2H). IgG control slides show no positive staining (Supplemental Figure 3). Supplemental figure 4 provides further evidence of these observations during TF. Localization of Jag1 and NICD2 to terminal hypertrophic chondrocytes, areas of vascular invasion, and immature osteoblasts was also observed in growth plates of uninjured adult mice (Supplemental Figure 5). However, pre-hypertrophic chondrocytes appear to stain more negative in the growth plate than in the fracture callus.
Figure 2.

Jag1 and NICD2 are expressed in identical cell populations that participate in endochondral bone repair during TF. Undifferentiated mesenchymal cells (A) are largely positive (brown staining, black arrows), but expression gradually decreases as cells differentiate into proliferative (B), pre-hypertrophic (C), and hypertrophic chondrocytes (D), and then is re-expressed in terminal hypertrophic chondrocytes (E). Alternative to chondrogenesis, osteogenic cells at various stages of maturity, located in osteoid (F), primary (G) and remodeled bone formation (H) are mostly positive. Note that varying amounts of Jag1 and NICD2 negative cells are present in distinct cell population (white arrows). H&E and SafO images acquired at 200X magnification. Jag1 and NICD2 images acquired at 600X magnification.
Figure 3.
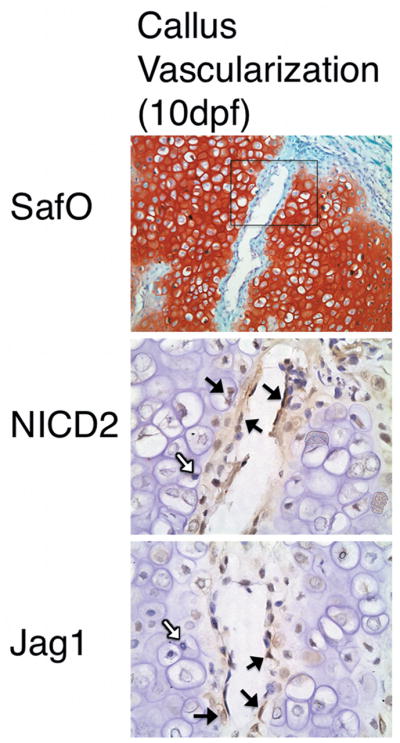
Jag1 and NICD2 are expressed in vascular endothelial cells invading the cartilage matrix, as well as terminal hypertrophic chondrocytes adjacent to the invading vasculature. Black arrows and brown staining indicate positive cells. White arrows indicate negative cells. SafO image acquired at 200X magnification. Jag1 and NICD2 images acquired at 600X magnification
Identification of cells that express Jag1 and NICD2 during CD
Jag1 and NICD2 were also expressed in identical cell populations that participate in intramembranous bone repair during CD (Figure 4). Following injury, periosteal-derived osteoprogenitors rapidly proliferate to re-establish a fibrous layer surrounding the defect (Figure 4A). At the same time, undifferentiated mesenchymal cells within the defect proliferate to produce fibrovascular tissue that initially fills the defect (Figure 4B). Cells that line the defect appear to have initiated early stages of osteogenesis. Consistent with TF, these cell populations are overwhelmingly, though not completely, Jag1 and NICD2 positive. Also consistent with TF, cells at various stages of osteogenic maturity continue to stain positive for Jag1 and NICD2 in areas of new (Figure 4C) and remodeled bone (Figure 4D). Furthermore, osteocytes embedded in remodeled bone are both positive and negative for Jag1 and NICD2 (Figure 4D). IgG control slides show no positive staining (Supplemental Figure 6). Supplemental figure 7 provides further evidence of these observations during CD. Localization of Jag1 and NICD2 was also observed in osteoblasts lining uninjured calvarial bone, and to a lesser extent periosteal-derived cells (Supplemental Figure 5). However, more osteocytes appear to stain negative in uninjured bone than in healing calvarium.
Figure 4.

Jag1 and NICD2 are expressed in identical cell populations that participate in intramembranous bone repair during CD. Undifferentiated mesenchymal cells located in the periosteum (A) and adjacent to the defect site (B) are largely positive (brown staining, black arrows). As osteogenesis progresses, cells at various stages of maturity continue to stain positive in areas of new (C) and remodeled bone (D). Osteocytes (D) are both positive and negative. Note that Jag1 and NICD2 negative cells (white arrows) are present in each area. H&E images acquired at 400X magnification. Jag1 and NICD2 images acquired at 600X magnification.
Discussion
This is the first study to extensively characterize the Notch signaling pathway during endochondral and intramembranous bone fracture healing, which has previously been shown to be required for proper embryological bone development [10–13, 26]. Our results demonstrate that Notch signaling components are actively regulated during both endochondral and intramembranous fracture healing.
Consistent with previous studies, we identified Jag1 and Notch2 as the predominantly expressed ligand and receptor during TF and CD [12–14, 25]. This Notch ligand-receptor pair has been shown to primarily interact with one another in a variety of cell types [27]. We further identified Jag1 and activated Notch2 (NICD2) to be expressed in the same cell populations during endochondral and intramembranous repair. Jag1 and NICD2 expression is strong in undifferentiated mesenchymal cells, but gradually decreases during chondrogenesis. Previous studies have shown that transient activation of Notch components, including Jag1, is required in uncommitted mesenchymal progenitor cells both in vivo and in vitro, but must downregulate in order to initiate chondrogenesis [25, 28]. Furthermore, sustained activation of Notch signaling in committed chondrocytes (cells that express the Col2a1 promoter) inhibits both proliferation and differentiation [26]. Many studies have specifically shown that Notch negatively regulates the pre-hypertrophic to hypertrophic chondrocyte transition [13, 26, 28, 29]. NICD and its downstream target genes Hes1 and Hey1 are known to inhibit chondrogenic differentiation by binding to a Sox9 binding site on the Col2a1 promoter [26, 30]. Collectively, the data suggests decreased Notch signaling occurs during chondrogenic lineage commitment and hypertrophic maturation.
This is the first study to show that terminal hypertrophic chondrocytes have the ability to re-express Jag1 and NICD2 in areas that have been infiltrated by Jag1 and NICD2 positive vascular endothelial cells. This applies to the chondro-osseous junction in both the callus during endochondral fracture healing, and in the growth plate during endochondral bone formation. This is consistent with a previous study, which showed that although Notch signaling negatively regulates hypertrophic chondrocyte differentiation, it positively regulates the progression of hypertrophic chondrocytes to their terminal differentiation, identified by Mmp-13 expression, at the chondro-osseous junction in the growth plate [13]. The Notch signaling pathway is initiated through direct cell-to-cell contact. It is plausible that this re-activation is initiated by endothelial-mesenchymal cell interactions, whereas prior activation of Notch signaling was initiated by mesenchymal-mesenchymal cell interactions. However, more research is required to understand the mechanism of this re-activation as well as the functional significance of Notch signaling in terminal hypertrophic chondrocytes.
Alternative to chondrogenesis, Jag1 and NICD2 are expressed in osteogenic cells at all stages of differentiation. Although this is the first study to show this via histology in vivo, Notch signaling has previously been shown to perform pro-osteogenic functions in osteoblasts at all stages of differentiation. Activation of Notch signaling in uncommitted mesenchymal progenitors (cells that express the Prx1 promoter [31]) maintains cells in an undifferentiated state while stimulating proliferation [12, 13]. However, alternative to chondrogenesis, activation of Notch signaling in committed osteoprogenitors (Col3.6 promoter [32]) and immature osteoblasts (Col2.3 promoter [32]) continues to promote proliferation while inhibiting differentiation [10, 11, 14]. Notch pathway components have been shown to prevent early and late osteoblast differentiation by binding to Runx2 (NICD1, Hes1, Hey1) [10, 13, 33] and the Ocn promoter (Hes1) [34]. Interestingly, instead of directly regulating bone formation, activation of Notch signaling in mature osteoblasts (Ocn promoter) reduces bone resorption by inhibiting osteoclast differentiation [13–15]. Collectively, the data suggests that during endochondral and intramembranous fracture healing, elevated levels of Notch signaling in undifferentiated cells may serve to increase the number of progenitors available to differentiate and produce a mature tissue matrix, and that Notch signaling in mature osteoblasts maintains the tissue matrix through a negative feed-back of osteoclast-mediated bone resorption.
In addition to regulating osteoblast and chondrocyte behavior, Notch signaling also regulates angiogenesis, which is critical for fracture healing. Dll4 signaling through Notch1 has been shown to restrict angiogenesis [35], whereas Jag1 is pro-angiogenic [36]. Not surprisingly Jag1 was the only ligand upregulated during both TF and CD, whereas Dll4 was the least expressed ligand during TF. Notch4 has been shown to have a redundant angiogenic function to Notch1 [37]. Consistent with this, our data showed that Notch4 was the least expressed receptor during both TF and CD. However, Notch4 was one of only two receptors to be upregulated during both TF and CD, and also demonstrated the greatest fold change among all receptors relative to 0 dpf, suggesting that while redundant, it still may play an active role in the Notch-mediated angiogenic response during bone repair.
Previous studies have shown that bones derived from different embryological germ layers have distinct tissue matrix compositions [38]. The calvarium and tibia originate from the ectoderm and mesoderm, respectively [39], which may explain the difference in basal expression levels of Notch genes in those tissues. There are injury models that would allow for comparison of endochondral and intramembranous fracture healing using a single long bone, which would control for factors intrinsic to the tissue. It is possible that Notch signaling may not be equivalent during intramembranous ossification in all types of bone. However, in this study we show that expression of Notch components are equivalently localized in osteogenic cells regardless of germ layer origin, embryological development, or method of healing, which may suggest that similar results would be expected in all models of bone repair. Importantly, we chose our injury models in order to develop a broader understanding of Notch signaling with applications to both craniofacial and long bone skeletal regeneration.
In conclusion, this study demonstrates that Notch signaling is upregulated during endochondral and intramembranous bone repair, with expression generally greater during endochondral repair. Furthermore, Jag1 and NICD2 are expressed in identical cell populations during healing, with expression gradually decreasing during chondrogenesis, but remaining present at multiple stages of osteoblastogenesis. Targeting the Notch signaling pathway may ultimately provide a mechanism to enhance bone repair; however, much more research is required to understand the spatiotemporal effects of Notch signaling in mesenchymal, hematopoietic and vascular cells.
Supplementary Material
{kind=link}
Acknowledgments
This study was funded by the Osteosynthesis and Trauma Care (OTC) foundation, the peer-reviewed orthopaedic research program (PROP) of the congressionally directed medical research program (CDMRP), and the NIH/NIDCR NRSA F31DE020231. The authors would like to thank Jason Combs for his help with animal surgeries.
References
- 1.Vortkamp A, et al. Recapitulation of signals regulating embryonic bone formation during postnatal growth and in fracture repair. Mech Dev. 1998;71(1–2):65–76. doi: 10.1016/s0925-4773(97)00203-7. [DOI] [PubMed] [Google Scholar]
- 2.Ferguson C, et al. Does adult fracture repair recapitulate embryonic skeletal formation? Mech Dev. 1999;87(1–2):57–66. doi: 10.1016/s0925-4773(99)00142-2. [DOI] [PubMed] [Google Scholar]
- 3.Einhorn TA. The cell and molecular biology of fracture healing. Clin Orthop Relat Res. 1998;(355 Suppl):S7–21. doi: 10.1097/00003086-199810001-00003. [DOI] [PubMed] [Google Scholar]
- 4.Tsuji K, et al. BMP2 activity, although dispensable for bone formation, is required for the initiation of fracture healing. Nat Genet. 2006;38(12):1424–9. doi: 10.1038/ng1916. [DOI] [PubMed] [Google Scholar]
- 5.Yu YY, et al. Bone morphogenetic protein 2 stimulates endochondral ossification by regulating periosteal cell fate during bone repair. Bone. 2010;47(1):65–73. doi: 10.1016/j.bone.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Friedlaender GE, et al. Osteogenic protein-1 (bone morphogenetic protein-7) in the treatment of tibial nonunions. J Bone Joint Surg Am. 2001;83-A(Suppl 1 Pt 2):S151–8. [PMC free article] [PubMed] [Google Scholar]
- 7.Govender S, et al. Recombinant human bone morphogenetic protein-2 for treatment of open tibial fractures: a prospective, controlled, randomized study of four hundred and fifty patients. J Bone Joint Surg Am. 2002;84-A(12):2123–34. doi: 10.2106/00004623-200212000-00001. [DOI] [PubMed] [Google Scholar]
- 8.Minear S, et al. Wnt proteins promote bone regeneration. Sci Transl Med. 2010;2(29):29ra30. doi: 10.1126/scitranslmed.3000231. [DOI] [PubMed] [Google Scholar]
- 9.Komatsu DE, et al. Modulation of Wnt signaling influences fracture repair. J Orthop Res. 2010;28(7):928–36. doi: 10.1002/jor.21078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Engin F, et al. Dimorphic effects of Notch signaling in bone homeostasis. Nat Med. 2008;14(3):299–305. doi: 10.1038/nm1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tao J, et al. Osteosclerosis owing to Notch gain of function is solely Rbpj-dependent. J Bone Miner Res. 2010;25(10):2175–83. doi: 10.1002/jbmr.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dong Y, et al. RBPjkappa-dependent Notch signaling regulates mesenchymal progenitor cell proliferation and differentiation during skeletal development. Development. 2010;137(9):1461–71. doi: 10.1242/dev.042911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hilton MJ, et al. Notch signaling maintains bone marrow mesenchymal progenitors by suppressing osteoblast differentiation. Nat Med. 2008;14(3):306–14. doi: 10.1038/nm1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zanotti S, et al. Notch inhibits osteoblast differentiation and causes osteopenia. Endocrinology. 2008;149(8):3890–9. doi: 10.1210/en.2008-0140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bai S, et al. NOTCH1 regulates osteoclastogenesis directly in osteoclast precursors and indirectly via osteoblast lineage cells. J Biol Chem. 2008;283(10):6509–18. doi: 10.1074/jbc.M707000200. [DOI] [PubMed] [Google Scholar]
- 16.Gude NA, et al. Activation of Notch-mediated protective signaling in the myocardium. Circ Res. 2008;102(9):1025–35. doi: 10.1161/CIRCRESAHA.107.164749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oya S, et al. Attenuation of Notch signaling promotes the differentiation of neural progenitors into neurons in the hippocampal CA1 region after ischemic injury. Neuroscience. 2009;158(2):683–92. doi: 10.1016/j.neuroscience.2008.10.043. [DOI] [PubMed] [Google Scholar]
- 18.Okamoto R, et al. Requirement of Notch activation during regeneration of the intestinal epithelia. Am J Physiol Gastrointest Liver Physiol. 2009;296(1):G23–35. doi: 10.1152/ajpgi.90225.2008. [DOI] [PubMed] [Google Scholar]
- 19.Hayes S, et al. Notch signaling regulates regeneration in the avian retina. Dev Biol. 2007;312(1):300–11. doi: 10.1016/j.ydbio.2007.09.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chigurupati S, et al. Involvement of notch signaling in wound healing. PLoS One. 2007;2(11):e1167. doi: 10.1371/journal.pone.0001167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Conboy IM, et al. Notch-mediated restoration of regenerative potential to aged muscle. Science. 2003;302(5650):1575–7. doi: 10.1126/science.1087573. [DOI] [PubMed] [Google Scholar]
- 22.Taylor DK, et al. Thrombospondin-2 influences the proportion of cartilage and bone during fracture healing. J Bone Miner Res. 2009;24(6):1043–54. doi: 10.1359/jbmr.090101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Aalami OO, et al. Applications of a mouse model of calvarial healing: differences in regenerative abilities of juveniles and adults. Plast Reconstr Surg. 2004;114(3):713–20. doi: 10.1097/01.prs.0000131016.12754.30. [DOI] [PubMed] [Google Scholar]
- 24.de Jonge HJ, et al. Evidence based selection of housekeeping genes. PLoS One. 2007;2(9):e898. doi: 10.1371/journal.pone.0000898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Oldershaw RA, et al. Notch signaling through Jagged-1 is necessary to initiate chondrogenesis in human bone marrow stromal cells but must be switched off to complete chondrogenesis. Stem Cells. 2008;26(3):666–74. doi: 10.1634/stemcells.2007-0806. [DOI] [PubMed] [Google Scholar]
- 26.Mead TJ, Yutzey KE. Notch pathway regulation of chondrocyte differentiation and proliferation during appendicular and axial skeleton development. Proc Natl Acad Sci U S A. 2009;106(34):14420–5. doi: 10.1073/pnas.0902306106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shimizu K, et al. Mouse jagged1 physically interacts with notch2 and other notch receptors. Assessment by quantitative methods. J Biol Chem. 1999;274(46):32961–9. doi: 10.1074/jbc.274.46.32961. [DOI] [PubMed] [Google Scholar]
- 28.Watanabe N, et al. Suppression of differentiation and proliferation of early chondrogenic cells by Notch. J Bone Miner Metab. 2003;21(6):344–52. doi: 10.1007/s00774-003-0428-4. [DOI] [PubMed] [Google Scholar]
- 29.Crowe R, Zikherman J, Niswander L. Delta-1 negatively regulates the transition from prehypertrophic to hypertrophic chondrocytes during cartilage formation. Development. 1999;126(5):987–98. doi: 10.1242/dev.126.5.987. [DOI] [PubMed] [Google Scholar]
- 30.Grogan SP, et al. Repression of chondrogenesis through binding of notch signaling proteins HES-1 and HEY-1 to N-box domains in the COL2A1 enhancer site. Arthritis Rheum. 2008;58(9):2754–63. doi: 10.1002/art.23730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Logan M, et al. Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis. 2002;33(2):77–80. doi: 10.1002/gene.10092. [DOI] [PubMed] [Google Scholar]
- 32.Kalajzic I, et al. Use of type I collagen green fluorescent protein transgenes to identify subpopulations of cells at different stages of the osteoblast lineage. J Bone Miner Res. 2002;17(1):15–25. doi: 10.1359/jbmr.2002.17.1.15. [DOI] [PubMed] [Google Scholar]
- 33.Zamurovic N, et al. Coordinated activation of notch, Wnt, and transforming growth factor-beta signaling pathways in bone morphogenic protein 2-induced osteogenesis. Notch target gene Hey1 inhibits mineralization and Runx2 transcriptional activity. J Biol Chem. 2004;279(36):37704–15. doi: 10.1074/jbc.M403813200. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Y, et al. The Notch-responsive transcription factor Hes-1 attenuates osteocalcin promoter activity in osteoblastic cells. J Cell Biochem. 2009;108(3):651–9. doi: 10.1002/jcb.22299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hellstrom M, et al. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature. 2007;445(7129):776–80. doi: 10.1038/nature05571. [DOI] [PubMed] [Google Scholar]
- 36.Benedito R, et al. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell. 2009;137(6):1124–35. doi: 10.1016/j.cell.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 37.Krebs LT, et al. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev. 2000;14(11):1343–52. [PMC free article] [PubMed] [Google Scholar]
- 38.van den Bos T, et al. Differences in matrix composition between calvaria and long bone in mice suggest differences in biomechanical properties and resorption: Special emphasis on collagen. Bone. 2008;43(3):459–68. doi: 10.1016/j.bone.2008.05.009. [DOI] [PubMed] [Google Scholar]
- 39.Chung UI, et al. Distinct osteogenic mechanisms of bones of distinct origins. J Orthop Sci. 2004;9(4):410–4. doi: 10.1007/s00776-004-0786-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.