

Abstract
Background
Hemorrhagic shock activates cellular stress signals and can lead to systemic inflammatory response, organ injury, and death. We have previously shown that treatment with histone deacetylase inhibitors (HDACIs) significantly improves survival in lethal models (60% blood loss) of hemorrhage. The aim of the current study was to examine whether these protective effects were due to attenuation of mitogen activated protein kinase (MAPK) signaling pathways, which are known to promote inflammation and apoptosis.
Study Design
Wistar-Kyoto rats (250–300g) were subjected to 40% blood loss and randomized to treatment with: 1) HDACI valproic acid (VPA 300mg/kg IV; volume = 0.75 ml/kg), or 2) vehicle control (0.75 ml/kg of 0.9% saline). Animals were sacrificed at 1, 4 and 20 hours (n=3–4/group/timepoint), and lung samples were analyzed by Western blotting for expression of active (phosphorylated) and inactive forms of c-Jun N terminal Kinase (JNK) and p38 MAPK. Myeloperoxidase (MPO) activity was measured in lung tissue 20 hours after hemorrhage as a marker of neutrophil infiltration. Normal animals (n=3) served as shams.
Results
Hemorrhaged animals demonstrated significant increases in phosphorylated p38 at 1 hour, phosphorylated JNK at 4 hours, and increased MPO activity at 20 hours (p<0.05 compared to sham). VPA treatment significantly (p <0.05) attenuated all of these changes.
Conclusions
Hemorrhagic shock activates pro-inflammatory MAPK signaling pathways and promotes pulmonary neutrophil infiltration, affects that are significantly attenuated by VPA treatment. This may represent a key mechanism through which HDACIs decrease organ damage and promote survival in hemorrhagic shock.
Keywords: acute lung injury, inflammation, hemorrhage, valproic acid, histone deacetylase inhibitors, stress signals
Introduction
Hemorrhage is the leading cause of morbidity and mortality in trauma patients and a major cause of secondary complications including acute lung injury (ALI), systemic inflammatory response syndrome, and multi organ dysfunction syndrome (MODS). Hemorrhagic shock triggers a unique cellular phenotype through alternations at genomic and proteomic levels (1). Acutely, hemorrhage activates several inflammatory and immunologic signaling pathways. Systemically, these pathways promote recruitment of neutrophils and release of inflammatory cytokines (2, 3). Within hypoperfused tissues extracellular, danger associated molecular patterns (DAMPs) trigger intracellular changes in the phosphorylation status of proteins and modulate the epigenetic accessibility of DNA (4–6). Downstream, these signals alter the gene expression profile of the injured cell, creating a competition between pro-survival and pro-death signals that ultimately determines the fate of the cell. One key pathway in hemorrhage-induced cellular injury is the mitogen activated protein (MAP) kinase pathway. These proteins: extracellular signal related protein kinase 1/2 (ERK1/2), c JUN N-terminal kinase (JNK), and p38 protein kinase, are globally expressed and known to be key regulators of stress-mediated cell fate decisions (7- 9).
Focusing on the cellular pathophysiology of hemorrhagic shock, our team has explored the efficacy of pharmacologic resuscitation with histone deacetylase inhibitors (HDACIs) as a treatment strategy. HDACIs alter the acetylation status of cytoplasmic and nuclear proteins and therefore have the potential to modulate the genomic and proteomic changes induced by hemorrhage. We have previously shown that HDACIs can dramatically improve survival in lethal models of hemorrhagic shock in rat (10, 11) and swine (12), and in murine models of septic shock (13, 14). At the level of cell signaling, HDACI treatment can increase acetylation of β-catenin and transcription of the pro-survival protein bcl-2 in neurons subjected to hypoxic insult (15). It can also attenuate inflammatory cytokine mRNA expression, and reduce the activation of pro-inflammatory protein kinases (including hepatic MAP kinase proteins ERK and p38) in mice subjected to severe sepsis (16). In rodent models of hemorrhage, HDACIs have also been shown to attenuate ERK 1/2 activation (11).
Yet the molecular events underlying the ability of HDACIs to shift the balance from pro-death to pro-survival during shock remains incompletely characterized. Since we had used lethal models of septic and hemorrhagic shock in previous studies, delayed tissue analyses were influenced by a clear survival bias (differential survival rates in the treated and control animals). Therefore, the objective of the present study was to extend our previous work (11) and employ a sub-lethal model of hemorrhagic shock to better understand the influence of HDACIs on MAP kinase pathway activation and neutrophil infiltration in the lung.
We specifically chose HDACI valproic acid (VPA) for several reasons: VPA has well-established HDACI activity (17), it is an FDA-approved drug with a known safety and side effect profiles, it is readily available, and has been successfully used in our previous survival studies of hemorrhagic shock in rodents and swine. We elected to study the lung because pulmonary inflammation and acute lung injury are early and deleterious consequence of hemorrhage (18, 19). Moreover, lung is often the target for a ‘first hit’ priming event in traumatic injury, which may predispose patients to secondary insults including ALI, sepsis, and MODS (20–3). Our hypothesis was that treatment with valproic acid, an HDACI, would attenuate post-hemorrhagic shock pulmonary inflammation.
Materials and Methods
Animals
All experimental procedures in this study were approved by the Institutional Animal Care and Use Committee and complied with the policies detailed in The Guide for the Care and Use of Laboratory Animals (7th ed., National Academies Press, 1996). Male Wistar Kyoto rats (249–305 grams) were purchased from Harlan (Indianapolis, IN), and given food and water ad libitum.
Anesthesia and Instrumentation
On each day of experimentation, Valproic acid (VPA; Calbiochem San Diego, CA) solution was freshly prepared: solid VPA was dissolved in sterile filtered water to create a 400 mg/mL solution. Anesthesia was induced using 4% isoflurane (Abbott Laboratories, North Chicago, IL) mixed with air in an induction chamber and delivered via a nose cone scavenging system. Animals were fitted with this nose cone and allowed to breathe spontaneously. Anesthesia was maintained by delivering 0.7–1.2% isoflurane through the nose cone using a veterinary multi-channel anesthesia delivery system and vaporizer (Kent Scientific Corporation, Torrington, CT). Body temperature was maintained with a heating lamp. Bupivicaine (AstraZeneca, Wilmington, DE) was injected (0.2mL of 0.75%) for local anesthesia.
An incision was made over the left femoral vessels, the femoral artery and vein were dissected, and both vessels were cannulated with PE50 polyethelyne catheters (Clay Adams, Sparks, MD). The venous cannula was used for hemorrhage and drug treatment while the arterial catheter was connected to the Ponemah Physiology Platform (Grould Instrument Systems, Valley View, OH) for continuous hemodynamic monitoring.
Sub-lethal hemorrhagic shock protocol
The volume of hemorrhage was calculated in proportion to each animal’s estimated total blood volume as follows: estimated total blood volume (mL) = weight (g) x 0.06 (mL/g) + 0.77 (23). Venous blood was withdrawn from the animal using Kent Scientific infusion and withdrawal syringe pumps (Kent Scientific Corporation, Torrington, CT). In order to study cellular signaling, a moderate (sub-lethal) hemorrhagic shock model was used. This sub-lethal hemorrhage model allowed all rats in treated and untreated groups to survive to the specified (1, 4 and 20 hour) time point and allow reliable inter-group comparisons without a survival bias.
After obtaining baseline (BL) arterial blood samples, 40% of total blood volume was withdrawn over 10 minutes, followed by un-resuscitated shock for 30 minutes. Post-shock (PS) arterial blood samples were obtained. Animals were randomly assigned to receive treatment with either: 1) valproic acid (VPA 300mg/kg IV; volume = 750 microliters/kg; given over 5 minutes), or 2) vehicle control (750 microliter/kg of 0.9% saline IV; given over 5 minutes). Treatment was followed by 200 microL of 0.9% saline flush given over 15 minutes.
Since this is a sublethal model, all of the animals survived the observation period and were quite active during this time. Thus, after treatment and saline flush, the arterial catheter was removed and femoral artery was ligated, the venous catheter was clamped and left in place so that it could be accessed to draw blood at the time of sacrifice, the skin was closed with silk suture, and animals were allowed to recover from anesthesia under close observation in their cages for the duration of the experiment. While this prevented us from long term hemodynamic monitoring, it enabled us to observe the animals’ recovery without the complicating effects of anesthesia and indwelling catheters.
Rats were sacrificed at 1h, 4h, and 20h (n=3–4/group/time point) after completion of treatment. At the time of sacrifice, venous blood samples were collected and organs were harvested. Lung tissue was rinsed with cold saline, frozen in liquid nitrogen, and stored at −80°C. All blood samples were analyzed using the Stat Profile 2 Blood Gas and electrolyte Analyzer (Nova Biomedical, Waltham, MA).
Valproic acid dosing
As in our previous studies, we chose to a high dose of VPA, 300mg/kg, approximately 10 times higher than is currently used clinically, so that we could guarantee a sufficient biologic response (histone acetylation). At this dose there is some risk VPA toxicity including CNS depression and hepatotoxicity (24). We have not, however, observed any deleterious effects of high dose VPA treatment in any of our studies. Moreover, we contend that the benefits of VPA treatment to soldiers and civilians suffering from severe hemorrhage likely outweigh its risks.
Western blotting
Lung tissue (50mg wet weight per sample) was homogenized manually using a glass hand-held homogenizer, and whole cell extracts were prepared using the Whole Cell Extraction Kit (Millipore, Temecula, CA) according to the manufacturer’s instructions. Whole cell extracts were used for the detection of ERK1/2, phospho-ERK1/2, JNK, phospho-JNK, p38 and phospho-p38. Nuclear extracts of the lung (50mg wet weight per sample) were prepared using the proteoExtract subcellular proteome extraction kit (Calbiochem, La Jolla, CA) according to the manufacturer’s instructions. Nuclear extracts were used for detection of ac-H3K9. Total protein concentration for each sample was determined by the Bradford method (Bio-Rad laboratories, Hercules, CA).
Equal amounts of proteins (about 100 μg per lane) were separated by sodium dodecyl sulphate-polyacrylamide gel electrophoresis (SDS-PAGE) on 12% polyacrylamide gels and transferred onto nitrocellulose membranes (Bio-Rad Laboratories). The membranes were blocked in 0.05% phosphate buffered saline-Tween 20 (PBS-T) containing 5% milk (Bio-Rad Laboratories) and then incubated with the primary antibody diluted in PBS-T containing 5% bovine serum albumin (Sigma-Aldrich) at 4°C overnight. Primary antibodies were purchased as follows: ERK1/2, phosho-ERK1/2, JNK, phospho-JNK, p38 and phospho-p38 from Cell Signaling Technology (Danvers, MA), histone 3 acetylated at lysine 9 (AcH3K9) from Millipore (Temecula, CA), and β-actin from Sigma-Aldrich (St. Louis, MO). The primary antibody was detected by incubating membranes with horseradish peroxidase-coupled second antibody (1:3000 in PBST with 5% milk) at room temperature for 2 hours. Chemiluminescence detection was performed by using Western Lighting Chemiluminescence Reagant Plus (Perkin Elmer LAS, Boston, Mass). Films were developed using a standard photographic procedure and detected bands were quantitatively analyzed by densitometer scanning using the VersaDoc Imaging System (Bio-Rad Laboratories).
Myeloperoxidase Activity
Lung myeloperoxidase (MPO) levels were quantified using the rat MPO enzyme-linked immunosorbent assay (ELISA) kit (Cell Sciences, Canton, MA). Rat lung tissue (50 mg per sample) was homogenized using a glass hand-held homoginizer in 1 mL of a lysis buffer (200 mM NaCl, 5 mM EDTA, 10 mM Tris, 10% glycine, 1 mM phenylmethylsulfonyl fluoride, 1 μg/mL leupeptide, 28 μg/mL aprotinin). The samples were centrifuged twice at 1,500 x g at 4 ºC for 15 min, and supernatants were analyzed for MPO levels by following the manufacturer’s instructions.
Sample Size and Statistical Analysis
We selected our sample based upon our maximum western blotting capacity of 10 samples in a single gel. Given this limitation, we elected to study 3 animals in our sham group, 3 in our vehicle control group, and 4 animals in our VPA group in order to avoid the imprecision introduced by pooling data from separate western blots.
All continuous variables are expressed as means ± standard error of the mean (SEM). Data were analyzed using SPSS statistical software (SPSS, Chicago, IL). Differences between 3 or more groups were assessed using one way analysis of variance (ANOVA) followed by Bonferroni post hoc testing for multiple comparisons. The independent samples t-test was used for comparisons between 2 groups. A paired-sample t-test was used to compare two variables within a group. For all analyses, statistical significance was defined as p < 0.05.
Results
Sub-lethal Hemorrhage
Animal survival in our model of sublethal (40% blood loss) hemorrhage was 100% in all groups. Sublethal hemorrhage resulted in hypotension (MAP post shock = 29 mmHg) as well as evidence of global hypoperfusion (post shock serum lactate levels : 4.7 ±0.5 and 4.6 ±0.2 mmol/L in vehicle and VPA treated animals respectively). Lactic acidosis corrected spontaneously by the end of the experiment (serum lactate levels at 4 hours: 0.8 ±0.2 and 1.3 ±0.3 mmol/L in vehicle and VPA treated animals respectively). There were no significant differences in the heart rate, blood pressure measurements, or lactic acidosis between the two groups at BL; small but statistically significant differences in baseline Hgb and pH between animals were noted but did not persist throughout the experiment (Table 1).
Table 1.
Selected variables from the sub-lethal (40%) hemorrhagic shock protocol are shown.
| Variable | Group | Baseline (BL) (arterial) | Post-Shock (PS) (arterial) | Post Tx (PT) (arterial) | Sacrifice (SA) (venous) |
|---|---|---|---|---|---|
| HR (bpm) | Vehicle | 271± 25.6 | 240±19.9 | 265±14.3 | |
| VPA | 268± 21.8 | 235±23.1 | 261±15.6 | ||
| MAP (mmHg) | Vehicle | 95.435 ± 2.14 | 29.853 ± 0.75* | 66.28 ± 3.7 ** | |
| VPA | 94.753 ± 3.7 | 28.558 ± 3.9* | 73.084 ± 4.1 ** | ||
| Hgb (g/dL) | Sham | 12.5 ± 0.22 | -- | -- | |
| Vehicle 1h | 12.2 ± 0.35 | 9.3 ± 0.12* | 9.3 ± 0.32 | ||
| VPA 1h | 10.7 ± 0.35# | 9.2 ± 0.55 | 9.2 ± 0.58 | ||
| Vehicle 4h | 12.9 ± 0.06 | 10.47 ± 0.12* | 9.2 ± 0.39 | ||
| VPA 4h | 12.6 ± 0.30 | 10.1 ± 0.52* | 9.4 ± 0.23 | ||
| Vehicle 20h | 11.6 ± 0.37 | 9.6 ± 0.32 | 7.5 ± 0.72 | ||
| VPA 20h | 11.87 ± 0.38 | 8.7 ± 0.41 | 6.2 ± 0.88** | ||
| pH | Sham | 7.459 ± 0.005 | -- | -- | |
| Vehicle 1h | 7.463 ± 0.005 | 7.381 ± 0.004* | 7.309 ± 0.027 | ||
| VPA 1h | 7.47 ± 0.006 | 7.373 ± 0.011 | 7.309 ± 0.010 | ||
| Vehicle 4h | 7.461 ± 0.006 | 7.362 ± 0.001* | 7.384 ± 0.009** | ||
| VPA 4h | 7.467 ± 0.005 | 7.374 ± 0.007* | 7.418 ± 0.007** | ||
| Vehicle 20h | 7.45 ± 0.004## | 7.346 ± 0.010* | 7.472 ± 0.029** | ||
| VPA 20h | 7.476 ± 0.001 | 7.357 ± 0.008* | 7.506 ± 0.006** | ||
| Lactate (mmol/L) | Sham | 0.7 ± 0.15 | -- | -- | |
| Vehicle 1h | 0.9 ± 0.12 | 4.4 ± 0.40* | 4.6 ± 0.50 | ||
| VPA 1h | 0.7 ± 0.09 | 3.7 ± 1.30 | 5.5 ± 0.75 | ||
| Vehicle 4h | 0.3 ± 0.06 | 4.7 ± 0.46* | 0.8 ± 0.21** | ||
| VPA 4h | 0.4 ± 0.17 | 4.6 ± 0.21* | 1.3 ± 0.28** | ||
| Vehicle 20h | 0.7 ± 0.03 | 3.6 ± 0.25* | 1.4 ± 0.09** | ||
| VPA 20h | 0.7 ± 0.00 | 4.7 ± 0.52* | 0.8 ± 0.09** |
Mean arterial pressure (MAP) and heart rate (HR) values are shown at baseline (BL), following hemorrhage and un-resuscitated shock (post-shock; PS), and after treatment with either valproic acid (VPA) 300mg/kg IV or saline vehicle (post treatment; PT). Hemoglobin (Hgb), pH, and serum lactate are also shown at the time of sacrifice (SA). Animals were sacrificed at 1 h, 4h, or 20h. Sham animals served as normal controls. Data are shown as mean ± SEM. HR= heart rate; MAP = mean arterial pressure; Hgb = hemoglobin. At BL, differences in Hgb (#p<0.05 vs. Vehicle 1h) and pH (##p<0.05 vs. VPA 20h) were noted. There were no differences in MAP or lactate between groups at anytime point. Comparison of animals within a group at different time points yielded significant differences (*p<0.05 vs. BL and **p<0.05 vs. PS of the corresponding group).
Histone Protein Acetylation
At 1h after HS and treatment, there was a significant increase in acetylation of H3K9 among VPA treated animals compared to vehicle animals (Figure 1). This suggested that the dose of VPA was adequate to elicit a biological response (increased acetylation of a histone protein).
Figure 1.
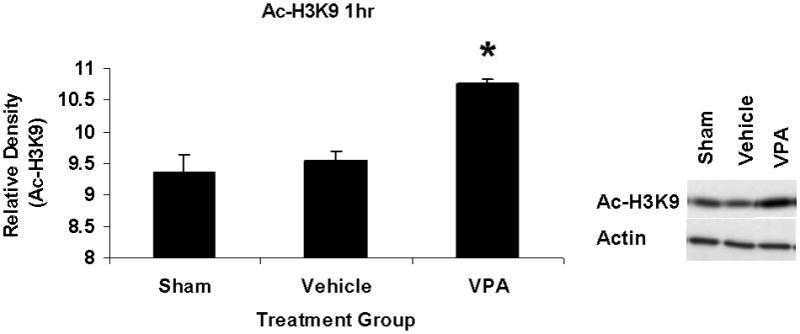
Effect of sublethal hemorrhage and VPA treatment on acetylation of histone 3 at Lysine 9 (Ac-H3K9) in the lung 1h after treatment with VPA 300mg/kg IV or normal saline vehicle. The nuclear fraction of lung tissue was assessed for Ac-H3K9 by western blotting. Data are shown as mean densitometry units +/− SEM. VPA treatment increased acetylation of H3K9 (*p<0.05 vs. vehicle). Actin was used as a protein loading control. N=3 per group.
MAP Kinase Protein Phosphorylation
Hemorrhagic shock resulted in increased ERK1/2 phosphorylation at 1 and 4 hours compared to sham (p<0.05), but no significant difference at 20 hours (Figure 2). JNK phosphorylation remained at sham levels 1 hour following hemorrhage in all groups but increased significantly at 4 hours compared to sham (p<0.05) then returned to sham levels at 20 hours in all groups (Figure 3). Hemorrhage increased p38 phosphorylation at 1 hour in vehicle control animals compared to sham animals (p<0.05); phospho-p38 levels in all groups were similar to sham animals at 4 hours (Figure 4). Total ERK1/2, JNK, or p38 protein levels did not differ between groups at any time point.
Figure 2.
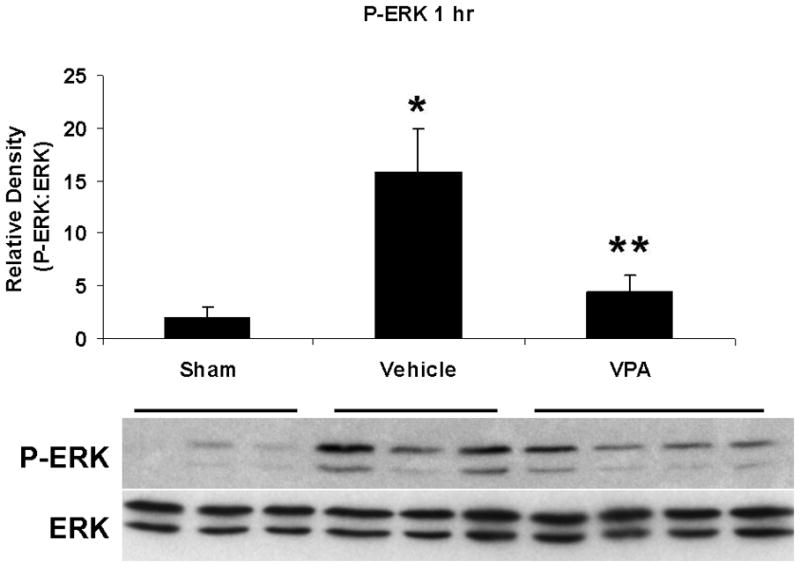
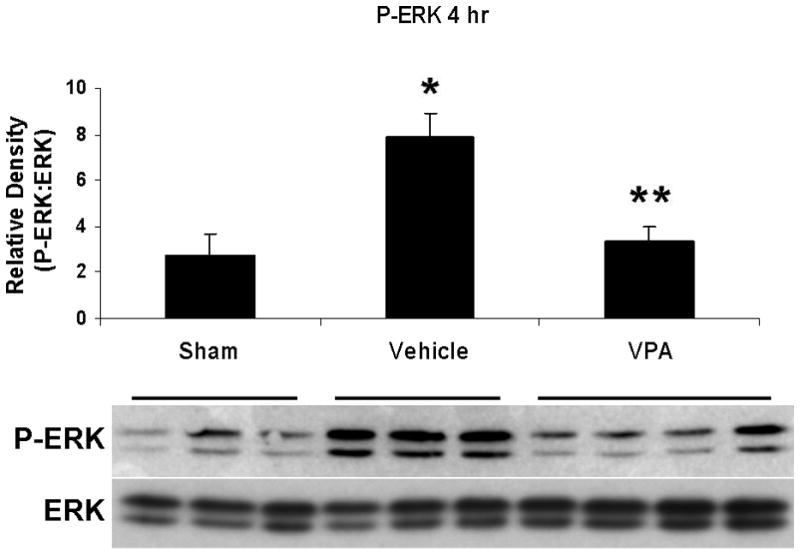
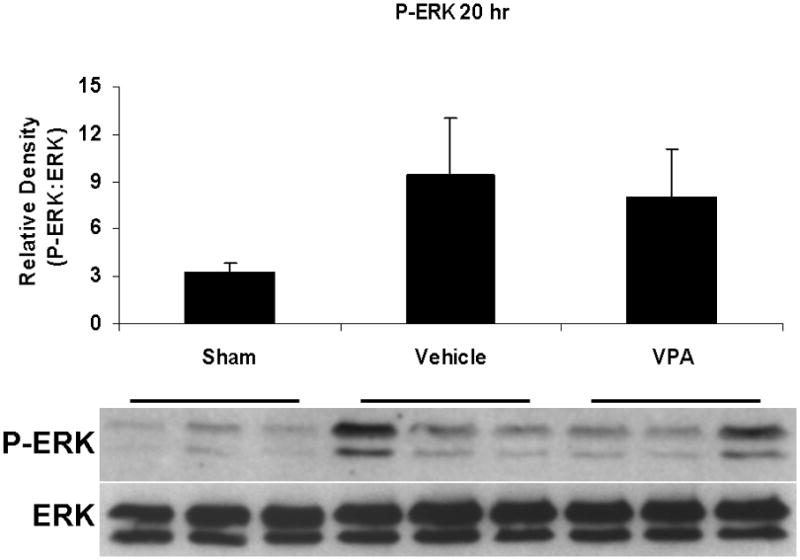
Effect of hemorrhage and VPA treatment on ERK activation (phosphorylation) in the lung at 1h, 4h, or 20h after treatment with VPA 300mg/kg IV or normal saline vehicle. Lung tissue whole cell lysate was analyzed for ERK1/2 phosphorylation (P-ERK). Data are shown as a mean ratio of P-ERK:ERK +/− SEM. A, B: Hemorrhage increased P-ERK expression at 1h and 4h (*p<0.05 vs. sham), whereas VPA treatment attenuated P-ERK at 1h and 4h (**p<0.05 vs. vehicle). C: No significant difference in P-ERK was observed at 20h. There were no differences in total ERK protein expression at each time point. N = 3 per sham and vehicle groups, n=4 for VPA treated groups.
Figure 3.
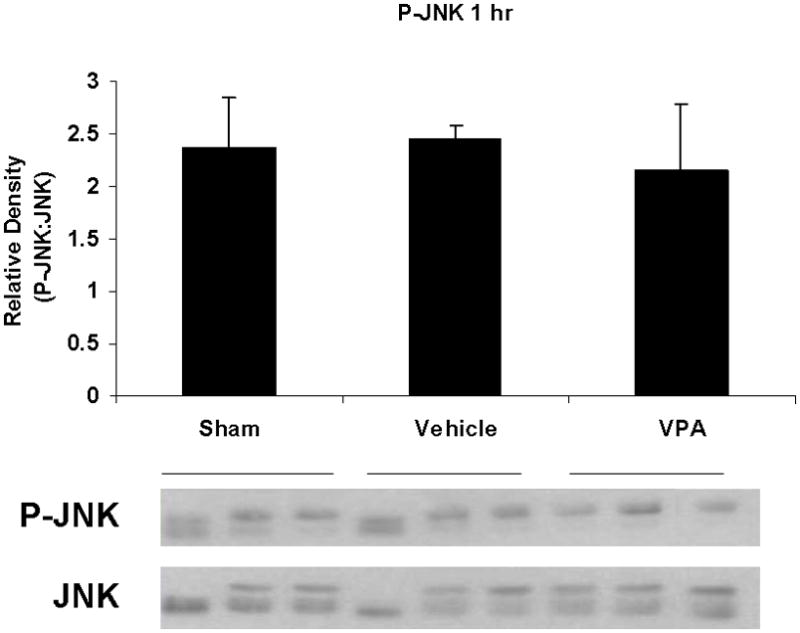
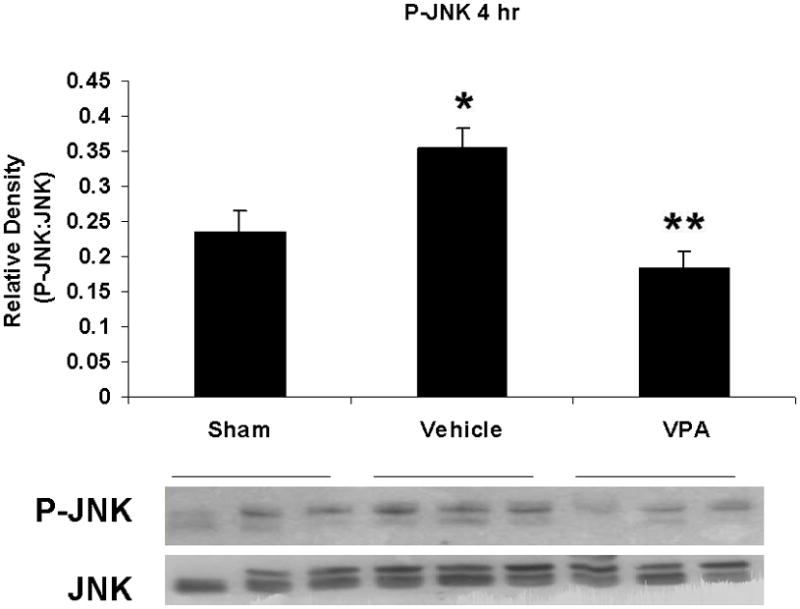
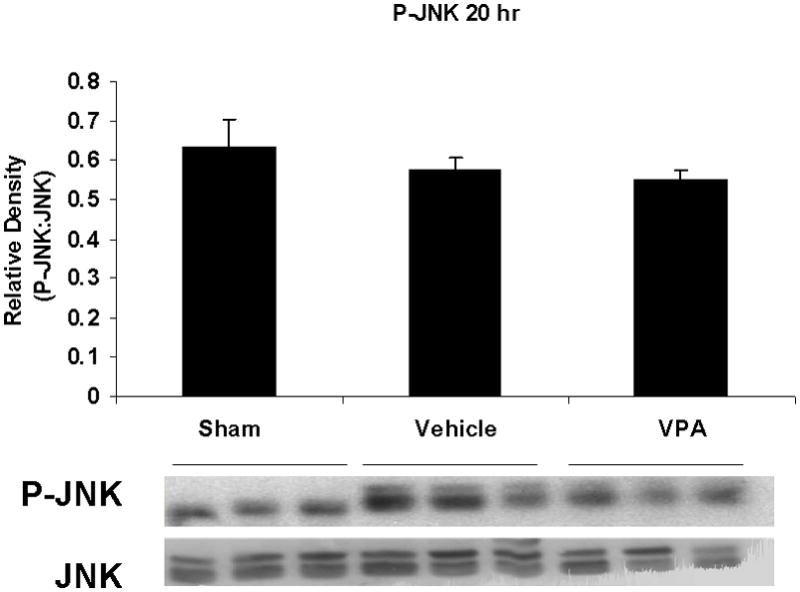
Effect of hemorrhage and VPA treatment on JNK activation (phosphorylation) in the lung at 1h, 4h, or 20h after treatment with VPA 300mg/kg IV or normal saline vehicle. Lung tissue whole cell lysate was analyzed for JNK phosphorylation (P-JNK). Data are shown as a mean ratio of P-JNK:JNK +/− SEM. B: Hemorrhage increased P-JNK expression at 4h (*p<0.05 vs. sham), whereas VPA treatment attenuated P-JNK at 4h (**p<0.05 vs. vehicle). A, C: No significant difference in P-JNK was observed at 1h and 20h. There were no differences in total JNK protein expression at each time point. N = 3 per sham and vehicle groups, n=4 for VPA treated groups.
Figure 4.
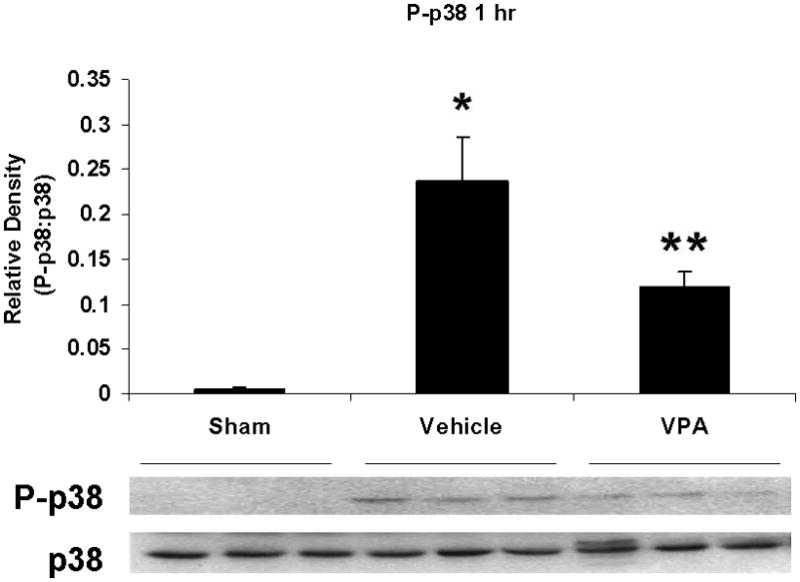
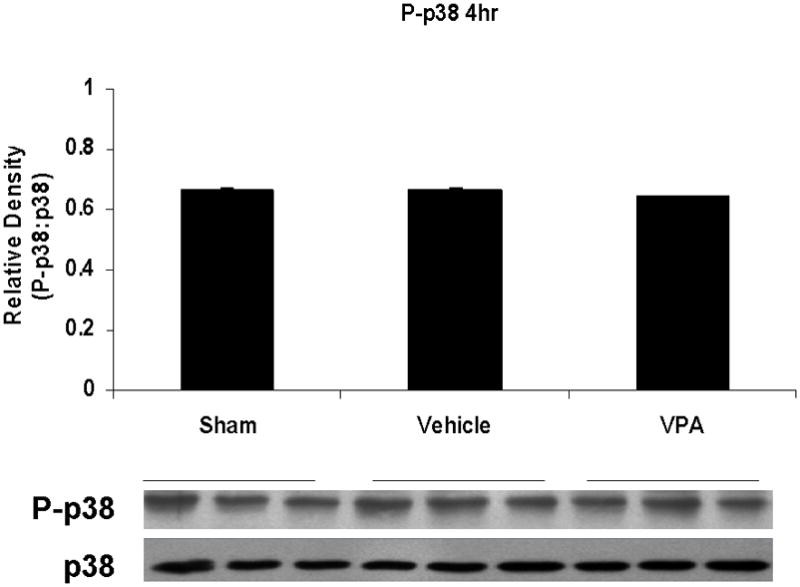
Effect of hemorrhage and VPA treatment on p38 activation (phosphorylation) in the lung at 1h, 4h, or 20h after treatment with VPA 300mg/kg IV or normal saline vehicle. Lung tissue whole cell lysate was analyzed for p38 phosphorylation (P-p38). Data are shown as a mean ratio of P-p38:p38 +/− SEM. A: Hemorrhage increased P-p38 expression at 1h (*p<0.05 vs. sham), whereas VPA treatment attenuated P-p38 at 1h (**p<0.05 vs. vehicle). B: No significant difference in P-p38 protein levels was observed at 4h. N = 3 per sham and vehicle groups, n= 4 for VPA treated groups.
Post-shock administration of VPA significantly (P<0.05) attenuated HS-induced ERK1/2 phosphorylation at 1 and 4 hours (Figure 2A, 2B), JNK phosphorylation at 4 hours (Figure 3B), and p38 phosphorylation at 1 hour (Figure 4A). VPA treated animals had similar ERK1/2, JNK, and p38 phosphorylation levels as sham animals. In figure 5, levels of phosphorylated to total protein relative to sham are graphically represented for vehicle control and VPA treated animals in order to highlight the differences between phosphorylation of ERK1/2, JNK, and p38 in these two groups over the 20 hour time course.
Figure 5.
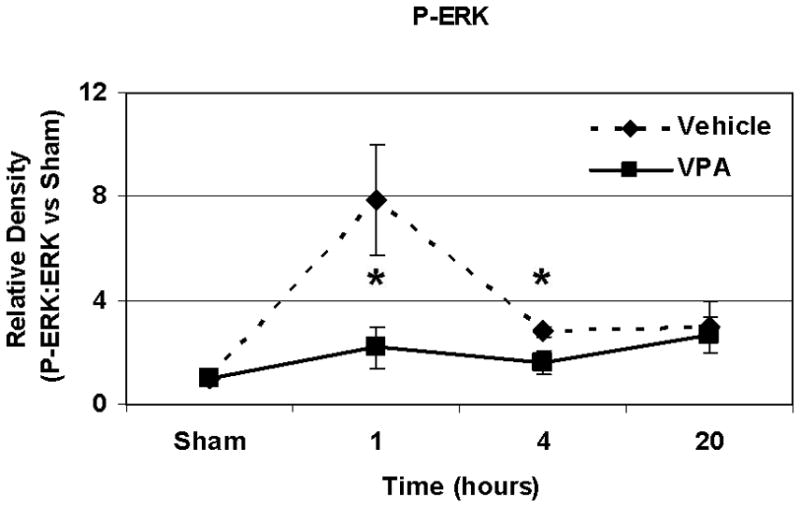
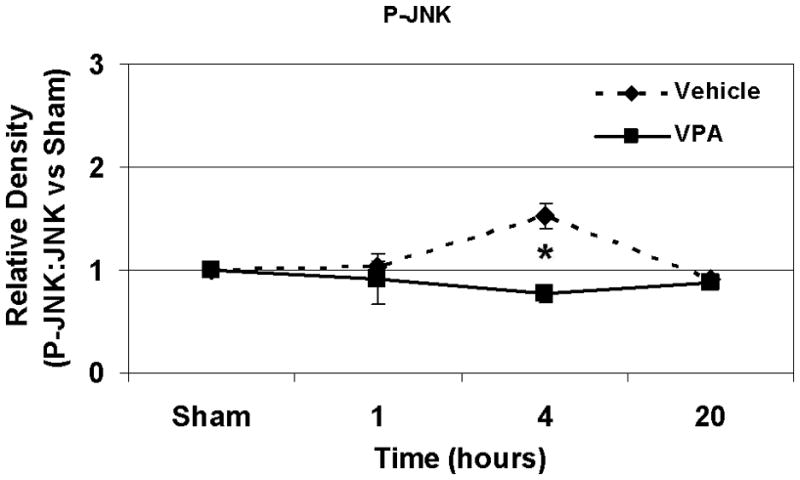
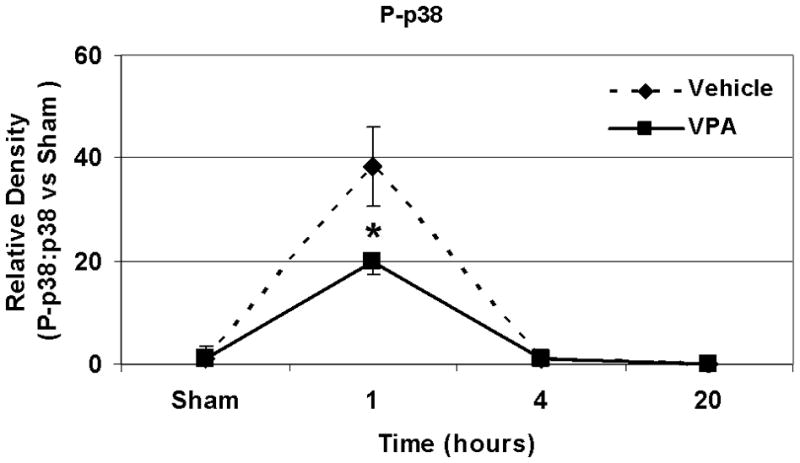
A, B, C: Effect of hemorrhage and VPA treatment on P-ERK, P-JNK and p38 expression respectively over a 20 hour time course. Data for animals treated with VPA and normal saline vehicle control are shown as phosphorylated MAP kinase protein: total MAP kinase protein normalized to sham phosphorylated MAP kinase protein: total MAP kinase protein levels at 1h, 4h, and 20 h for P-ERK, P-JNK, and P-p38 (*p<0.05 vs. vehicle). N = 3 per sham and vehicle groups, n= 4 for VPA treated groups.
Pulmonary Myeloperoxidase Levels
Myeloperoxidase levels were measured in lung tissue at 20 hours post-hemorrhage as a marker of neutrophil infiltration. Hemorrhage-induced lung MPO concentration was markedly increased compared with sham animals, whereas VPA treatment significantly attenuated this increase (p< 0.05; Figure 6).
Figure 6.
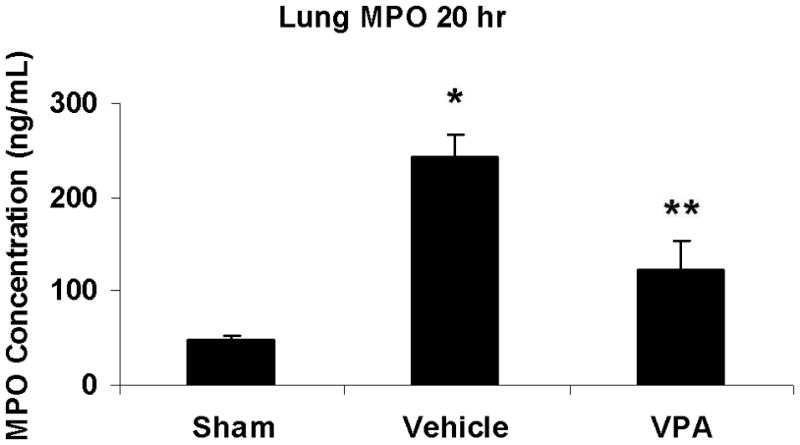
Effect of hemorrhage and VPA on myeloperoxidase levels in lung tissue at 20 hours post-hemorrhage. Data are presented as MPO concentration in ng/mL +/− SEM. Hemorrhage-induced lung MPO activity was markedly increased compared with sham animals (*p<0.05 vs. sham). VPA significantly attenuated this increase (**p<0.05 vs. vehicle). N = 3 per sham and 20 hr vehicle group, n= 4 for 20hr VPA group.
Discussion
We have previously shown that HDACIs can improve survival and attenuate inflammation in animal models of hemorrhagic shock, poly-trauma, and septic shock through modulation of cellular signaling pathways including the MAP kinase cascade. In sepsis, HDACI treatment attenuates activation (phosphorylation) of ERK and p38 MAP kinases in the liver and can reduce expression of inflammatory cytokines and myeloperoxidase (16). In a lethal model of hemorrhagic shock, HDACI treatment attenuates JNK activation and decreases levels of pro-apoptotic caspase 3 protein in the liver (15). Most recently, in a sub-lethal model of hemorrhagic shock, we have demonstrated that HDACI treatment with valproic acid (VPA) can attenuate ERK1/2 activation in the lung at 1 and 4 hours after injury (11).
In the current study we utilized a sub-lethal model of hemorrhagic shock (40% blood loss, 100% survival) to investigate the molecular mechanisms associated with HDACI treatment over a 20-hour time course following injury. In our experiment, 40% hemorrhage led to activation (phosphorylation) of MAP kinases JNK and p38 in lung tissue at early time points (1 and 4 hours) and increased pulmonary inflammation (myeloperoxidase levels) at 20 hours in control animals. Treatment with HDACI valproic acid (VPA) significantly attenuated these changes. We elected to study lung as it is a frequent site of damage (secondary insult) following a primary injury such as hemorrhagic or septic shock (18, 19, 26).
Injuries, including hemorrhage, trigger the release of DAMPs , which are transduced by several cellular receptors and converge to activate MAP kinase proteins (7). There are several well studied MAP kinase proteins namely the extracellular signaling kinase 1/2 (ERK 1/2), the c-Jun terminal kinase (JNK), and the p38 protein kinase. These three seronine/threonine kinases regulate the cellular stress response through protein modifications and direct targeting of transcription factors. Their diverse role as effector molecules are highly context dependent. The effect of a given MAP kinase differs according to three major parameters: 1) complex interactions with upstream regulators and scaffolding proteins, 2) subcellular localization, and 3) the concentration and duration of the activated MAPK module (27). In the setting of traumatic injury and shock, activation of these proteins has been strongly associated with poor outcomes while inhibition of MAP kinase activity has been associated with survival in animal models of blood loss, ischemia reperfusion injury, sepsis, and neurotrauma. A brief description of these molecules is provided to place our findings in the proper context.
JNK
c-Jun N-terminal kinase (JNK) is a major regulator of apoptosis in response to cellular stress signals. During hemorrhage, tissue hypoperfusion and subsequent oxidative stress can activate JNK via phosphorylation (28, 29). Phosphorylation of JNK leads to a downstream signaling cascade that favors cell death through proteolytic cleavage of anti-apoptotic proteins and activation of pro-apoptotic signals (30). Pharmacologic inhibition of JNK can attenuate inflammation and tissue damage in several contexts. In a model of severe hemorrhage, Relja et al (33) demonstrated that inhibition of JNK after blood loss but prior to resuscitation could reduce hepatic cell death and markers of hepatocellular inflammation. Inhibition of JNK activation also induces a strongly neuroprotective effect and reduces cerebral edema following ischemic stroke in rodents (34). JNK inhibition can also attenuate pulmonary injury following abdominal sepsis (32). These data suggest a role for sustained activation of JNK in the development of ‘second hit’ injury following a primary insult. Likewise, in our experiment, VPA-induced attenuation of JNK activation was associated with a reduction in neutrophil infiltration in the lung, measured by a decrease in pulmonary myeloperoxidase activity.
P38
P38 kinase is known to be a major regulator of macrophage function, inflammatory processes, and apoptosis in response to injury in several organs (35–37). It up regulates the expression of inflammatory cytokines, prostaglandins and inducible nitric oxide synthase at the level of transcription and translation (38). This key role of p38 in the inflammatory response has made it a promising target for pharmacologic inhibition; several p38 inhibitors have been developed to date (39). In the lung, p38 MAP kinase activation has been shown to be increased by hemorrhagic shock and play a key role in chemokine production and development of acute lung injury (40, 41). Thus, the ability of VPA to reduce activation of p38 and pulmonary inflammation in our experiment represents a potential means of improving the inflammatory milieu in the acute phase following hemorrhagic shock.
ERK1/2
ERK1/2 is known to be involved in a variety of biological functions such as cellular proliferation and differentiation (42). As with other MAP kinases, an important role for ERK1/2 signaling during times of stress is emerging. ERK1/2 is activated during ischemic insults such as stroke and HS (43, 44), and its activation may contribute to cellular injury and death. Increased ERK1/2 activation has been demonstrated following cell damage and necrosis (45, 46) whereas specific inhibition of ERK1/2 signaling has been shown to attenuate cell injury and inflammation (47, 48). We recently demonstrated that VPA can inhibit hemorrhage-induced ERK1/2 activation and improve survival in lethal and sublethal models of hemorrhagic shock (11).
HDACIs and MAP Kinases
While the mechanism by which HDACI VPA attenuates MAP kinase activation remains undefined, recent study has suggested several putative links between acetylation and regulation of the MAP kinase cascade. Interestingly, JNK kinase activity can be regulated by NFkB (31). NFkB activation requires the formation of a multi-protein complex. Interestingly, one of the key co-activator proteins in this complex, p300/CBP–associated factor has histone acetyltransferase activity and is required for NFkB -activated transcription (49). Acetylation of the RelA subunit of NFkB promotes the stability and long term of activity of NFkB in the nucleus. (50, 51) As an HDACI, VPA may have an activating and stabilizing effect on acetylation of NFkB, thereby promoting transcription of pro-survival genes and attenuating the deleterious effects of the JNK/caspase 3 pro-apoptotic pathway (52, 53).
MAP kinase activation (phosphorylation) is also regulated by mitogen activated protein kinase phoshphotases (MKPs). MKPs are regulated post-translationally by phosphorylation, oxidation, and acetylation (54). Cao et al (55) demonstrated that exposure to LPS and/or HDACI treatment can increase MKP-1 acetylation. Moreover, actetylation of MKP-1 via HDACIs increases MKP-1’s interaction with p38 and decreases phosphorylation (activation) of p38 in alveolar macrophages and in mice treated with LPS. Therefore, in our model of hemorrhagic shock, VPA may attenuate the acute phase increase in activated p38 through acetylation of MKP-1.
Current treatment for hemorrhagic shock focuses on pathophysiology at the level of organ systems, and emphasis is placed on hemorrhage control along with adequate resuscitation. Unfortunately, this resource intensive protocol remains difficult to administer, particularly in austere environments, where advanced medical management is not immediately available (56). Moreover, this macroscopic approach fails to address much of the damage that takes place at the cellular level as a result of hypoperfusion during hemorrhage and reperfusion during treatment (57). Since ERK, JNK, and p38 serve as ‘signaling hubs’ in the cellular stress response, therapies that target these convergent kinases possess great potential for success as therapies in shock. The MAP kinases’ proven role in hemorrhagic shock and in the ‘second hit’ pathophysiology of ALI and MODS makes treatments that decrease their activation useful not only in treating primary injury but in preventing subsequent morbidity and mortality. Hence, HDACI such as VPA constitute promising adjuncts to standard trauma care, especially in austere environments.
It should be noted, that this study is limited in several ways. Firstly, VPA exerts its pro-survival and anti-inflammatory phenotype through many signaling pathways and mechanisms including HDACI, GABAnergic neurotransmitter inhibition, mediation of sodium and potassium ion flux. As an HDACI, VPA promotes the acetylation of multiple histone and non-histone proteins. VPA also has several intra and extracellular effects independent of acetylation that may contribute to treated animals’ overall physiologic and molecular phenotype. Interestingly, VPA has been shown to have pro-apoptotic actions in in vitro and in vivo models of cancer (24, 58) when administered lower doses (25–50 mg/kg) for several cycles. These actions are incompletely understood but several pathways including the extrinsic caspase and Fas ligand pathways as well as the alterations in JNK dependent cell cycle signaling pathways (59). Further studies are required to explain the seemingly paradoxical, anti-apoptotic action of HDACI in trauma and pro-apoptotic action in cancer and the relevance of the differences in HDACI dosing for these indications. One obvious difference between these conditions is that ischemic cells are essentially normal cells undergoing a stressful insult, whereas neoplastic cells have an abnormal expression of genes/proteins that regulate normal cell cycle. Given VPA’s multiple effects, we have used a second HDACI in many of our prior studies to confirm that the observed effects are indeed due to an inhibition of HDAC (and increased acetylation). Treatment with a more specific (and more potent) HDADI such as suberoylanilide hydroxamic acid (SAHA) results in nearly identical benefits. These mechanistic studies have been summarized in a recent review (60), and taken together data so far suggests that the beneficial effects of VPA are primarily due to its HDACI properties. Other mechanisms cannot be completely ruled out, however. Thus, the pathways discussed here likely describe some but not all of the mechanisms underlying VPA’s effects in the pathophysiology of hemorrhagic shock.
Secondly, in this hemorrhage model we aimed to establish a delicate balance between model severity and survivability. While our sublethal model eliminates survival bias, it may have been too mild to detect some of the pathophysiologic effects of hemorrhage and HDACI therapy. Finally, as with any time course study, the power and precision of our study were limited by using only 3 or 4 animals per group and harvesting organs at only three time points.
In summary, we have demonstrated that VPA can attenuate acute ERK, JNK and p38 MAP kinase activation in the lung in a sub-lethal model of hemorrhagic shock. We have also shown that VPA can reduce pulmonary neutrophil infiltration 20 hours after blood loss. Further understanding of these mechanisms is key to providing the most effective combined cellular and systemic treatment for hemorrhage control and prevention of ‘second hit’ morbidity and mortality.
Acknowledgments
Some of these data were presented at the annual meeting of the American College of Surgeons (Surgical Forum; October, 2010). Supported by NIH RO1 GM084127 (HBA).
Abbreviations
- ALI
acute lung injury
- ERK1/2
extracellular signal related protein 1/2
- HDACI
Histone deacetylase inhibitor
- IL
Interleukin
- JNK
cJun N-terminal kinase
- MAPK
mitogen activated protein kinase
- MKP
mitogen activated protein kinase phoshphotase
- MODS
multi-organ system
- MPO
myeloperoxidase
- NFkB
nuclear factor kappa b
- VPA
valproic acid
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Murphy TJ, Paterson HM, Mannick JM, Lederer JA. Injury sepsis, and the regulation of Toll-like receptor responses. J Leukoc Biol. 2004;75:400–407. doi: 10.1189/jlb.0503233. [DOI] [PubMed] [Google Scholar]
- 2.Partrick DA, Moore FA, Moore EE, Barnett CC, Jr, Silliman CC. Neutrophil priming and activation in the pathogenesis of postinjury multiple organ failure. New Horiz. 1996;4:194–210. [PubMed] [Google Scholar]
- 3.Botha AJ, Moore FA, Moore EE, Kim FJ, Banerjee A, Peterson VM. Postinjury neutrophil priming and activation: an early vulnerable window. Surgery. 1995;118:358–364. doi: 10.1016/s0039-6060(05)80345-9. [DOI] [PubMed] [Google Scholar]
- 4.Abraham E, Aracaroli J, Shenkar R. Activation of Extracellular Signal Related Kinases, NF-kappa B and Cyclic Adenosine 5′ Monophosphate Response Element Binding Protein in Lung Neutrophils Occurs by Differing Mechanisms After Hemorrhage or Endotoxemia. J Immunol. 2001;166:522–530. doi: 10.4049/jimmunol.166.1.522. [DOI] [PubMed] [Google Scholar]
- 5.Chen H, Alam HB, Querol RI, Rhee P, Li Y, Koustova E. Identification of expression patterns associated with hemorrhage and resuscitation: integrated approach to data analysis. J Trauma. 2006;60:701–723. doi: 10.1097/01.ta.0000203699.91475.f6. [DOI] [PubMed] [Google Scholar]
- 6.Lin T, Alam HB, Chen H, Britten-Webb J, Rhee P, Kirkpatrick J, Koustova E. Cardiac histones are substrates of histone deacetylase activity in hemorrhagic shock and resuscitation. Surgery. 2006;139:365–376. doi: 10.1016/j.surg.2005.08.022. [DOI] [PubMed] [Google Scholar]
- 7.Winter-Vann AM, Johnson GL. Integrated activation of MAP3Ks balances cell fate in response to stress. J Cell Biochem. 2007;102:848–858. doi: 10.1002/jcb.21522. [DOI] [PubMed] [Google Scholar]
- 8.Chen Z, Gibson TB, Robinson F, Silvestro L, Pearson G, Wright A, Vanderbilt C, Cobb MH. MAP Kinases. Chemical Reviews. 2001;101:2449–2476. doi: 10.1021/cr000241p. [DOI] [PubMed] [Google Scholar]
- 9.Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocrinol Rev. 2001;22:153–183. doi: 10.1210/edrv.22.2.0428. [DOI] [PubMed] [Google Scholar]
- 10.Shults C, Sailhamer EA, Li Y, Liu B, Tabbara M, Butt MU, Shuja F, Demoya M, Velmahos G, Alam HB. Surviving blood loss without fluid resuscitation. J Trauma. 2008;64:629–638. doi: 10.1097/TA.0b013e3181650ff3. [DOI] [PubMed] [Google Scholar]
- 11.Fukudome EY, Kochanek AR, Li Y, Smith EJ, Liu B, Kheirbek T, Lu J, Kim K, Hamwi K, Velmahos GC, Alam HB. Pharmacologic Resuscitation Promotes Survival and Attenuates Hemorrhage-Induced Activation of Extracellular Signal-Regulated Kinase 1/2. J Surg Res. doi: 10.1016/j.jss.2010.04.013. Epub 2010 May 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Alam HB, Shuja F, Butt MU, Duggan M, Li Y, Zacharias N, Fukudome EY, Liu B, Demoya M, Velmahos GC. Surviving blood loss without blood transfusion in a swine poly-trauma model. Surgery. 2009;146:325–333. doi: 10.1016/j.surg.2009.04.007. [DOI] [PubMed] [Google Scholar]
- 13.Li Y, Liu B, Zhao H, Sailhamer EA, Fukudome EY, Zhang X, Kheirbek T, Finkelstein RA, Velmahos GC, deMoya M, Hales CA, Alam HB. Protective effect of suberoylanilide hydroxamic acid against LPS-induced septic shock in rodents. Shock. 2009;32:517–523. doi: 10.1097/SHK.0b013e3181a44c79. [DOI] [PubMed] [Google Scholar]
- 14.Li Y, Liu B, Fukudome EY, Kochanek AR, Finkelstein RA, Chong W, Jin G, Lu J, deMoya MA, Velmahos GC, Alam HB. Surviving lethal septic shock without fluid resuscitation in a rodent model. Surgery. 2010;148:246–254. doi: 10.1016/j.surg.2010.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li Y, Liu B, Sailhamer EA, Yuan Z, Shults C, Velmahos GC, deMoya M, Shuja F, Butt MU, Alam HB. Cell protective mechanism of valproic acid in lethal hemorrhagic shock. Surgery. 2008;144:217–224. doi: 10.1016/j.surg.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 16.Finkelstein RA, Li Y, Liu B, Shuja F, Fukudome E, Velmahos GC, Demoya M, Alam HB. Treatment with Histone Deacetylase Inhibitor Attenuates MAP Kinase Mediated Liver Injury in a Lethal Model of Septic Shock. J Surg Res. doi: 10.1016/j.jss.2010.04.024. Epub 2010 May 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lheureux PE, Penaloza A, Zahir S, Gris M. Science review: carnitine in the treatment of valproic acid-induced toxicity - what is the evidence? Crit Care. 2005;9:431–40. doi: 10.1186/cc3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Abraham E, Carmody A, Shenkar R, Arcaroli J. Neutrophils as early immunologic effectors in hemorrhage or endotoxemia-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2000;279:1137–1145. doi: 10.1152/ajplung.2000.279.6.L1137. [DOI] [PubMed] [Google Scholar]
- 19.Song Y, Ao L, Raeburn CD, et al. A low level of TNF-alpha mediates hemorrhage-induced acute lung injury via p55 TNF receptor. Am J Physiol Lung Cell Mol Physiol. 2001;281:L677–684. doi: 10.1152/ajplung.2001.281.3.L677. [DOI] [PubMed] [Google Scholar]
- 20.Lin T, Chen H, Koustova E, Sailhamer EA, Li Y, Shults C. Histone deacetylase as therapeutic target in a rodent model of hemorrhagic shock: effect of different resuscitation strategies on lung and liver. Surgery. 2007;141:784–794. doi: 10.1016/j.surg.2007.01.014. [DOI] [PubMed] [Google Scholar]
- 21.Lenz A, Franklin GA, Cheadle WG. Systemic inflammation after trauma. Injury. 2007;38:1336–1345. doi: 10.1016/j.injury.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 22.Rotstein OD. Modeling the two-hit hypothesis for evaluating strategies to prevent organ injury after shock/resuscitation. J Trauma. 2003;54:203–206. doi: 10.1097/01.TA.0000064512.62949.92. [DOI] [PubMed] [Google Scholar]
- 23.Sailhamer EA, Li Y, Smith EJ, Liu B, Shuja F, Soupir CP, DeMoya MA, Velmahos GC, Alam HB. Hypoxic "second hit" in leukocytes from trauma patients: Modulation of the immune response by histone deacetylase inhibition. Cytokine. 2010;49:303–311. doi: 10.1016/j.cyto.2009.11.013. [DOI] [PubMed] [Google Scholar]
- 24.Bolden JE, Peart MJ, Johnstone RW. Anticancer activities of histone deacetylase inhibitors. Nat Rev Drug Discov. 2006;5:769–84. doi: 10.1038/nrd2133. [DOI] [PubMed] [Google Scholar]
- 25.Lee HB, Blaufox MD. Blood volume in the rat. J Nucl Med. 1985;26:72–76. [PubMed] [Google Scholar]
- 26.Paterson H, Murphy T, Purcell E. Injury primes the innate immune system for enhanced toll-like receptor reactivity. J Immunol. 2003;171:1473–1483. doi: 10.4049/jimmunol.171.3.1473. [DOI] [PubMed] [Google Scholar]
- 27.Johnson GL, Dohlman HG, Graves LM. MAPK kinase kinases (MKKKs) as a target class for small molecule inhibition to modulate signaling networks and gene expression. Curr Opin Chem Biol. 2005;9:325–331. doi: 10.1016/j.cbpa.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 28.Mollen KP, McCloskey CA, Tanaka H, et al. Hypoxia activates c-Jun N-terminal kinase via Rac1-dependent reactive oxygen species production in hepatocytes. Shock. 2007;28:270–276. doi: 10.1097/shk.0b013e3180485acd. [DOI] [PubMed] [Google Scholar]
- 29.Shen HM, Liu ZG. JNK signaling pathway is a key modulator in cell death mediated by reactive oxygen and nitrogen species. Free Radic Biol Med. 2006;40:928–939. doi: 10.1016/j.freeradbiomed.2005.10.056. [DOI] [PubMed] [Google Scholar]
- 30.Budd RC. Activation-induced cell death. Curr Opin Immunol. 2001;13:356–362. doi: 10.1016/s0952-7915(00)00227-2. [DOI] [PubMed] [Google Scholar]
- 31.Bubici C, Papa S, Dean K. Mutual cross-talk between reactive oxygen species and nuclear factor-kB: Molecular basis and biological significance. Oncogene. 2006;25:6731–6748. doi: 10.1038/sj.onc.1209936. [DOI] [PubMed] [Google Scholar]
- 32.Chen LW, Tseng HT, Chen PH, Hsu CM. Peritonitis induced peroxynitrite and lung damage depends on c-Jun NH2-terminal kinase signaling of hematopoietic cells. Crit Care Med. 2010;38:1168–1178. doi: 10.1097/CCM.0b013e3181d44e06. [DOI] [PubMed] [Google Scholar]
- 33.Relja B, Schwestka B, Lee VS, Henrich D, Czerny C, Borsello T, Marzi I, Lehnert M. Inhibition of c-Jun N-terminal kinase after hemorrhage but before resuscitation mitigates hepatic damage and inflammatory response in male rats. Shock. 2009;32:509–516. doi: 10.1097/SHK.0b013e3181a2530d. [DOI] [PubMed] [Google Scholar]
- 34.Michel-Monigadon D, Bonny C, Hirt L. c-Jun N-Terminal Kinase Pathway Inhibition in Intracerebral Hemorrhage. Cerebrovasc Dis. 2010;29:564–570. doi: 10.1159/000306643. [DOI] [PubMed] [Google Scholar]
- 35.Guo X, Gerl RE, Schrader JW. Defining the involvement of p38a MAPK in the production of anti- and proinflammatory cytokines using an SB 203580-resistant form of the kinase. J Biol Chem. 2003;278:22237–22242. doi: 10.1074/jbc.M300847200. [DOI] [PubMed] [Google Scholar]
- 36.Tourian L, Jr, Zhao H, Srikant CB. p38alpha, but not p38beta, inhibits the phosphorylation and presence of c-FLIPS in DISC to potentiate Fas-mediated caspase-8 activation and type I apoptotic signaling. J Cell Sci. 2004;15:6459–6471. doi: 10.1242/jcs.01573. [DOI] [PubMed] [Google Scholar]
- 37.Bassi R, Heads R, Marber MS, Clark JE. Targeting p38-MAPK in the ischaemic heart: kill or cure? Curr Opin Pharmacol. 2008;8:141–146. doi: 10.1016/j.coph.2008.01.002. [DOI] [PubMed] [Google Scholar]
- 38.Kumar S, Boehm J, Lee JC. p38 MAP kinases: key signalling molecules as therapeutic targets for inflammatory diseases. Nat Rev Drug Discov. 2003;2:717–726. doi: 10.1038/nrd1177. [DOI] [PubMed] [Google Scholar]
- 39.Adams JL, Badge AM, Kumar S, Lee JC. p38 MAP kinase: molecular target for the inhibition of proinflammatory cytokines. Prog Med Chem. 2001;38:1–60. doi: 10.1016/s0079-6468(08)70091-2. [DOI] [PubMed] [Google Scholar]
- 40.Chen C, Wang Y, Zhang Z, Wang C, Peng M. Toll-like receptor 4 regulates heme oxygenase-1 expression after hemorrhagic shock induced acute lung injury in mice: requirement of p38 mitogen-activated protein kinase activation. Shock. 2009;31:486–492. doi: 10.1097/SHK.0b013e318188f7e1. [DOI] [PubMed] [Google Scholar]
- 41.Wang YX, Xu XY, Su WL, Wang Q, Zhu WX, Chen F, Jin G, Liu YJ, Li YD, Sun YP, Gao WC, Ruan CP. Activation and Clinical Significance of p38 MAPK Signaling Pathway in Patients with Severe Trauma. J Surg Res. 2010;161:119–125. doi: 10.1016/j.jss.2008.10.030. [DOI] [PubMed] [Google Scholar]
- 42.Irving EA, Barone FC, Reith AD, Hadingham SJ, Parsons AA. Differential activation of MAPK/ERK and p38/SAPK in neurons and glia following focal cerebral ischaemia in the rat. Brain Res Mol Brain Res. 2000;77:65–75. doi: 10.1016/s0169-328x(00)00043-7. [DOI] [PubMed] [Google Scholar]
- 43.Namura S, Iihara K, Takami S, Nagata I, Kikuchi H, Matsushita K, Moskowitz MA, Bonventre JV, Alessandrini A. Intravenous administration of MEK inhibitor U0126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc Natl Acad Sci U S A. 2001;98:11569–11574. doi: 10.1073/pnas.181213498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hsu JT, Kan WH, Hsieh CH. Role of extracellular signal-regulated protein kinase (ERK) in 17β-estradiol-mediated attenuation of lung injury after trauma-hemorrhage. Surgery. 2009;145:226–234. doi: 10.1016/j.surg.2008.10.008. [DOI] [PubMed] [Google Scholar]
- 45.Chanteux H, Guisset AC, Pilette C, Sibille Y. LPS induces IL-10 production by human alveolar macrophages via MAPKinases- and Sp1-dependent mechanisms. Respir Res. 2007;8:71. doi: 10.1186/1465-9921-8-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ramachandiran S, Huang Q, Dong J, Lau SS, Monks TJ. Mitogen-activated protein kinases contribute to reactive oxygen species-induced cell death in renal proximal tubule epithelial cells. Chem Res Toxicol. 2002;15:1635–1642. doi: 10.1021/tx0200663. [DOI] [PubMed] [Google Scholar]
- 47.Bhat NR, Zhang P. Hydrogen peroxide activation of multiple mitogen-activated protein kinases in an oligodendrocyte cell line: role of extracellular signal-regulated kinase in hydrogen peroxide-induced cell death. J Neurochem. 1999;72:112–119. doi: 10.1046/j.1471-4159.1999.0720112.x. [DOI] [PubMed] [Google Scholar]
- 48.Jarrar D, Kuebler JF, Rue LW, 3rd, Matalon S, Wang P, Bland KI, Chaudry IH. Alveolar macrophage activation after trauma-hemorrhage and sepsis is dependent on NF-kappaB and MAPK/ERK mechanisms. Am J Physiol Lung Cell Mol Physiol. 2002;283:L799–805. doi: 10.1152/ajplung.00465.2001. [DOI] [PubMed] [Google Scholar]
- 49.Sheppard KA, Rose DW, Haque ZK. Transcriptional activation by NF-kB requires multiple coactivators. Mol Cell Biol. 1999;19:6367–6378. doi: 10.1128/mcb.19.9.6367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang XD, Tajkhorshid E, Chen LF. Functional interplay between acetylation and methylation of the RelA subunit of NF-kappaB. Mol Cell Biol. 2010;30:2170–2180. doi: 10.1128/MCB.01343-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen L, Fischle W, Verdin E. Duration of nuclear NF-kB action regulated by reversible acetylation. Science. 2001;293:1653. doi: 10.1126/science.1062374. [DOI] [PubMed] [Google Scholar]
- 52.Wullaert A, Heyninck K, Beyaert R. Mechanisms of crosstalk between TNF-induced NF-kappaB and JNK activation in hepatocytes. Biochem Pharmacol. 2006;72:1090–101. doi: 10.1016/j.bcp.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 53.Butt MU, Sailhamer EA, Li Y, Liu B, Shuja F, Velmahos GC, DeMoya M, King DR, Alam HB. Pharmacologic resuscitation: cell protective mechanisms of histone deacetylase inhibition in lethal hemorrhagic shock. J Surg Res. 2009;156:290–296. doi: 10.1016/j.jss.2009.04.012. [DOI] [PubMed] [Google Scholar]
- 54.Chi H, Flavell RA. Acetylation of MKP-1 and the Control of Inflammation. Sci Signal. 2009;1:141–144. doi: 10.1126/scisignal.141pe44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cao W, Bao C, Padalko E, Lowenstein CJ. Acetylation of mitogen-activated protein kinase phosphatase-1inhibits Toll-like receptor signaling. J Exp Med. 2008;205:1491–1503. doi: 10.1084/jem.20071728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Champion HR, Bellamy RF, Roberts CP, Leppaniemi A. A profile of combat injury. J Trauma. 2003;5:S13–S19. doi: 10.1097/01.TA.0000057151.02906.27. [DOI] [PubMed] [Google Scholar]
- 57.Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 2007;39:44–84. doi: 10.1016/j.biocel.2006.07.001. [DOI] [PubMed] [Google Scholar]
- 58.Su JM, Li XN, Thompson P, Ou CN, Ingle AM, Russell H, Lau CC, Adamson PC, Blaney SM. Phase 1 Study of Valproic Acid in Pediatric Patients with Refractory Solid or CNS Tumors: A Children’s Oncology Group Report. Clin Cancer Res. 2011;17:589–597. doi: 10.1158/1078-0432.CCR-10-0738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chateauvieux S, Morceau F, Dicato M, Diederich M. Molecular and Therapeutic Potential and Toxicity of Valproic Acid. J Biomed Biotech. 2010;2010:1–18. doi: 10.1155/2010/479364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li Y, Alam HB. Modulation of Acetylation: Creating a Pro-survival and Anti-Inflammatory Phenotype in Lethal Hemorrhagic and Septic Shock. J Biomed Biotechnol. 2011:523481. doi: 10.1155/2011/523481. Epub 2011 Feb 15. [DOI] [PMC free article] [PubMed] [Google Scholar]