

Conspectus
Catalysis in living cells is carried out by both proteins and RNA. Protein enzymes have been known for over 200 years, but RNA enzymes, or ‘ribozymes,’ were discovered only 30 years ago. Developing insight into RNA enzyme mechanisms is invaluable for better understanding both extant biological catalysis as well as the primitive catalysis envisioned in an early RNA-catalyzed life. Natural ribozymes include large RNAs such as the group I and II introns; small RNAs such as the hepatitis D virus and the hairpin, hammerhead, VS, and glmS ribozymes; and the RNA portion of the ribosome and spliceosome. RNA enzymes use many of the same catalytic strategies as protein enzymes, but do so with much simpler sidechains. Among these strategies are metal ion, general acid–base, and electrostatic catalysis. In this Account, we examine evidence for participation of charged nucleobases in RNA catalysis. Our overall approach is to integrate direct measurements on catalytic RNAs with thermodynamic studies on oligonucleotide model systems.
The charged amino acids make critical contributions to the mechanisms of nearly all protein enzymes. Ionized nucleobases should be critical for RNA catalysis as well. Indeed, charged nucleobases have been implicated in RNA catalysis as general acid–bases and oxyanion holes. We provide an overview of ribozyme studies involving nucleobase catalysis and the complications involved in developing these mechanisms. We also consider driving forces for perturbation of the pKa values of the bases. Mechanisms for pKa values shifting towards neutrality involve electrostatic stabilization and the addition of hydrogen bonding. Both mechanisms couple protonation with RNA folding, which we treat with a thermodynamic formalism and conceptual models. Furthermore, ribozyme reaction mechanisms can be multichannel, which demonstrates the versatility of ribozymes but makes analysis of experimental data challenging.
We examine advances in measuring and analyzing perturbed pKa values in RNA. Raman crystallography and fluorescence spectroscopy have been especially important for pKa measurement. These methods reveal pKa values for the nucleobases A or C equal to or greater than neutrality, conferring potential histidine-, lysine/arginine-like behavior on them. Structural support for ionization of the nucleobases also exists: an analysis of RNA structures in the databases conducted herein suggests that charging of the bases is neither especially uncommon nor difficult to achieve under cellular conditions. Our major conclusions are that cationic and anionic charge states of the nucleobases occur in RNA enzymes and that these states make important catalytic contributions to ribozyme activity. We conclude by considering outstanding questions and possible experimental and theoretical approaches for further advances.
Introduction
A paradigm shift in biological catalysis occurred in the 1980s when Cech and Altman discovered that certain RNAs are enzymes.1 This was remarkable for elucidating new mechanisms of enzyme catalysis, and for insights into possible mechanisms of primitive catalysis in an RNA World. Since these landmark discoveries much has been learned about ribozymes: additional classes have been discovered, numerous ribozymes and DNA enzymes (‘DNAzymes’) have been reported, high-resolution structures of ribozymes have been solved, and theoretical approaches to understanding ribozyme reactions have made headway.2
The focus of this Account is the direct participation of nucleobases in RNA catalysis. We begin by considering roles for ionized bases in RNA catalysis. Structural support for ionization is provided via analysis of databases. This is followed by discussion of experimental and conceptual advances for measuring and interpreting pKa values. Next, we examine select cases wherein ionized bases have been implicated in RNA catalysis. Lastly, driving forces for shifting pKa values are considered. Our approach is to bridge functional study on ribozymes with thermodynamic measurements on model oligonucleotides.
Charged Nucleobases and Their Potential for Catalysis
A major difference between RNA and protein enzymes is the diversity of the sidechains. Proteins are comprised of 20 chemically diverse amino acids, whereas RNA has just four similar heterocycles.3 It is well established that the charged amino acids play key roles in protein enzyme catalysis: lysine and arginine act as oxyanion holes to stabilize charge development in the transition state, whereas histidine cycles between neutral and positive when it transfers protons. Thus, one might inquire whether the nucleobases play similar roles.
In contrast to the amino acids, the unmodified nucleosides in RNA are typically neutral at biological pH.3 The pKa values closest to neutrality on adenosine and cytidine are ~3.5 and ~4.2, respectively, while those on guanosine and uridine are ~9.2 (Figure 1).3 A and C become cationic when protonated, while G and U become anionic when deprotonated. However, lacking pKa perturbation, the bases are only ~0.1–1% in their charged forms at neutrality. On the one hand, these pKa’s help nucleic acids store genetic information by virtue of complementary hydrogen bonding. However, these pKa’s limit potential contributions to catalysis by electrostatics and proton transfer.
Figure 1.
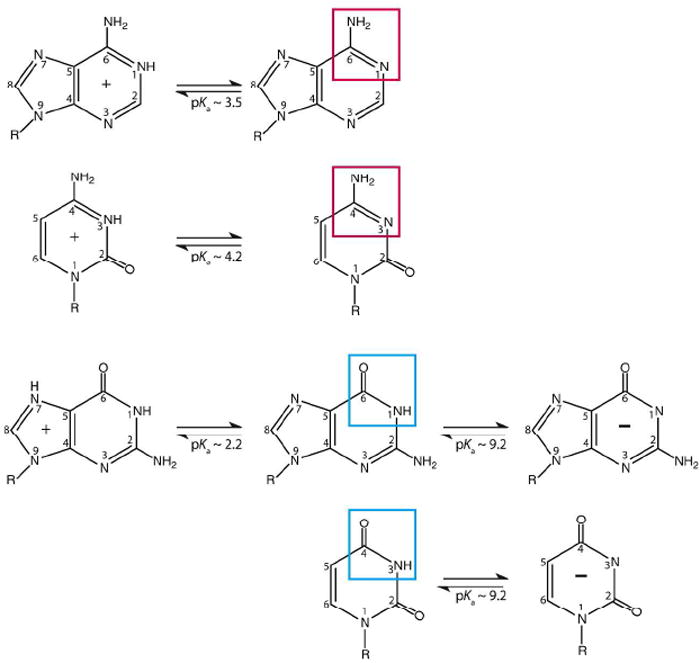
Charged nucleobases in RNA. Adenosine and cytidine can protonate on N1 and N3, with pKa’s near 3.5 and 4.2. Guanosine and uridine can deprotonate on N1 and N3 with pKa’s near 9.2; guanine has protonation possible at N7. Boxes indicate functional groups responsible for these groupings. These pKa values are often perturbed in natural RNAs.
The ability of charged and near-neutral functionalities to enhance nucleic acid enzyme activity has been elegantly demonstrated by Perrin and co-workers through studies on chimeric DNAzymes containing amine and imidazole functionalities.4 We have asserted that poor population of the functional charged forms of the bases is the greatest limitation to RNA catalysis, because once properly charged RNA catalyzes reactions with rates estimated to equal those of protein enzymes.5 Acidic and basic amino acids often have pKa values shifted 2 or more units in proteins. The pKa ’s of RNA sidechains shift as well. Indeed, every time a Watson-Crick base pair forms, linkage to folding causes the pKa to shift further away from neutrality by a few units.6
Interestingly, examples are also known where A and C shift towards neutrality. We divide such pKa shifts into ‘Class I’ and ‘Class II’, depending on whether the loaded proton is sequestered in base pairing (Figure 2).7 Both classes have potential to participate in catalysis: Class I primarily by serving as an oxyanion hole (Figure 3, Interaction ‘3’), and Class II by participating primarily in proton transfer (Figure 3, Interactions ‘1’ and ‘2’), although these roles are not exclusive owing to acidification of external amino protons upon protonation of an imino proton.8 As described below, pKa values for Class I motifs from 7 to 8 have been reported in model systems, indicating potential for populating the protonated state at neutrality, and lysine/arginine-like behavior. Also, pKa values for Class II motifs near 7 have been reported for A and C, indicating potential for histidine-like behavior.
Figure 2.
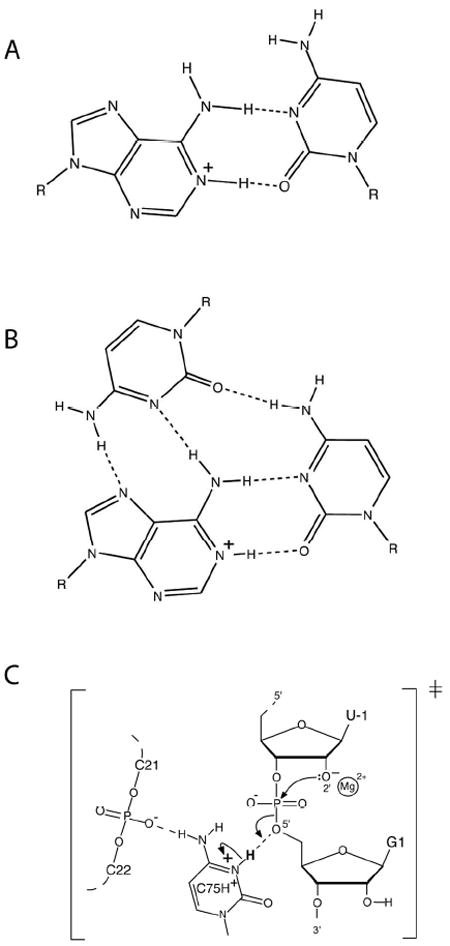
Examples of Class I and II protonated base interactions. (A) Structure of a two-hydrogen bond A+•C base pair. (Class I base pair.) (B) Structure of a base triple involving a two-hydrogen bond A+•C base pair and a third base. (Class I base pair.) (C) Model for active site of the HDV ribozyme. (Class II interaction.)24
Figure 3.
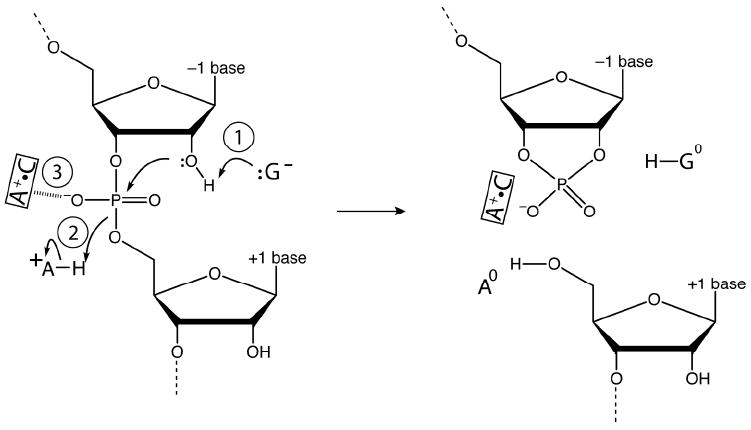
Involvement of charged bases in RNA catalysis. Shown are three potential interactions of charged nucleobases in the mechanism of small ribozymes. Interaction 1 involves an anionic G acting as a general base. Interaction 2 involves a cationic A acting as a general acid. Interaction 3 involves a cationic A+•C base pair acting as an oxyanion hole.
Structural Support for Ionization of the Nucleobases
Given that enzymes need a compact tertiary structure with a cleft for conducting catalysis, a major issue in RNA folding is neutralization of the backbone. Catalytic RNAs achieve compact structures primarily through counterion condensation: cations neutralize the backbone, which allows compaction.9 Another strategy for attaining electrostatic stabilization is protonation of A or C, which affords a two-hydrogen bond cationic base. We focus on A+•C wobbles because these are common and compatible with an A-form helix, making them amenable to experiments.10
In an effort to assess structural support for protonated bases in RNA, we surveyed databases for Class I protonated base pairs. We inspected the Fox lab RNA database, as well as a database from the Major lab.11,12 A base motif was inferred as protonated if two hydrogen-bonding heteroatoms were properly positioned (2.5–3.5 Å between heteroatoms), and maintained an angle (X-H---Y) of 140-180°. These criteria were verified for both of the hydrogen bonds in each A+•C wobble. We found 57 unique occurrences in 37 crystal or NMR structures consistent with these criteria for protonation of the A. Of these, 54 motifs were isolated A+•C pairs, and three were base triples involving an A+•C base pair. For those structures that reported experimental pH, half had a pH in the range of 6.5 to 7.5, suggesting a pKa near biological pH. This analysis suggests that charging of the bases into a cationic state is not uncommon.
Anionic base formation has less direct structural support. Given that RNA is a polyanion, electrostatic stabilization of a negative charge might seem improbable. Supporting this notion, molecular dynamics (MD) trajectories on the hairpin and glmS ribozymes suggest that an anionic G near the active site is disfavored due to electrostatic repulsion.13,14 On the other hand, experimental data show that RNA is fully capable of binding anions. Anion sites were first described by Auffinger and co-workers,15 while Golden and Kieft recently described sites for sulfate and selenate in RNA.16 They proposed that anion binding can be achieved through two motifs: consecutive helical C’s, whose major groove amines provide a positive potential, or positioning of one or two metal ions near the anion. A third possible motif is through-space electrostatic interaction with a cationic base, while a fourth is hydrogen bonding of a deprotonated functionality, in analogy to protonated functionalities. For example, an anionic guanosine could hydrogen bond with a 2’-OH nucleophile (Figure 3), which is supported structurally from hairpin ribozyme precleaved, product, and transition state structures.17,18
Measuring and Interpreting pKa Values in RNA
There has been growing interest in measuring pKa values of RNA. The two basic approaches are monitoring the fraction of RNA in the protonated state as a function of pH as reported by (1) spectroscopic or (2) kinetics measurements. First, we consider spectroscopic approaches. Importantly, pKa shifts have to occur in the ground state, rather than the transition state, to affect reactive populations;7,19 this means such shifts should be measurable by non-kinetics means such as spectroscopy. A two-state model of protonated-to-unprotonated states is typically invoked to interpret these data. A spectroscopic signal (e.g. UV, fluorescence, Raman, and NMR) is fit to a two-state Henderson-Hasselbalch equation to yield a pKa and a Hill coefficient to allow for multiple linked protonations.
The pKa values of larger RNAs have been determined by NMR spectroscopy, especially by changes in 13C chemical shift. Legault and Pardi showed, using the lead-dependent ribozyme, that nucleobase pKa’s cluster into three classes: unstructured loops have pKa’s that are unshifted, Watson-Crick base pairs have pKa’s perturbed away from neutrality, and wobble A+•C pairs have pKa’s perturbed towards neutrality.6
NMR was also applied to the HDV ribozyme (structure and mechanism provided in Figure 4) to measure the pKa of the catalytic nucleobase C75 using 13C spectroscopy.20 In the cleaved form, the pKa was found to be largely unperturbed, while line broadening and splitting precluded pKa determination in the precleaved state. More recently, we collaborated with the Carey and Golden labs to measured a pKa for the precleaved state of the HDV ribozyme using Raman crystallography.21 The interested reader is directed to a review article on the technique.22 We designed non-cleavable forms of the HDV ribozyme in which the 2’-hydroxyl of U-1 was modified. A pKa of 6.15 was found in 20 mM Mg2+, which shifted to 6.4 in 2 mM Mg2+ (Figure 5). These pKa’s and their anticooperative coupling to Mg2+ were in agreement with intrinsic pKa’s from kinetics of 5.9 and 7.25 for Mg2+-bound and -free states, respectively.23 This anticooperative coupling is likely due to through-space repulsion between protonated C75 and bound Mg2+, as suggested by our crystal structure of the precleaved HDV ribozyme.24 Remarkably, these near-neutral pKa values, confer a histidine-like pKa on cytosine, which is optimal for general acid-base chemistry.19 Ability of Raman crystallography of RNA, which is conducted at room temperature,22 to measure these pKa’s may be because crystallography selects for one state of the ribozyme, thus preventing misfolding. Indeed, we have shown that the HDV ribozyme is especially prone to misfolding.25 Also, our crystallographic constructs were based upon sequences engineered to have fast and monophasic folding.
Figure 4.
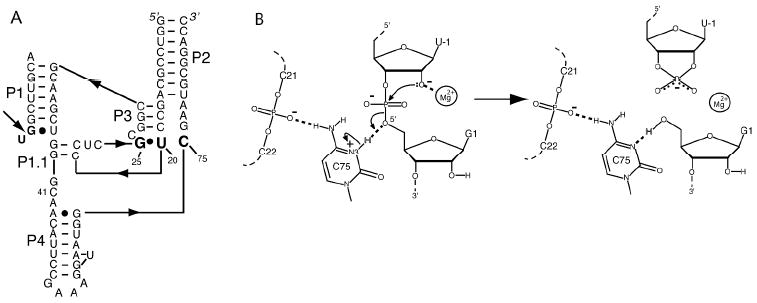
Structure and proposed mechanism of the HDV ribozyme. (A) Secondary structure of two-piece ribozyme. Arrow depicts cleavage site. (B) Proposed mechanism with Mg2+ acting as a Lewis Acid and protonated C75 as a general acid. Reproduced with permission 49. Copyright 2011 American Chemical Society.
Figure 5.
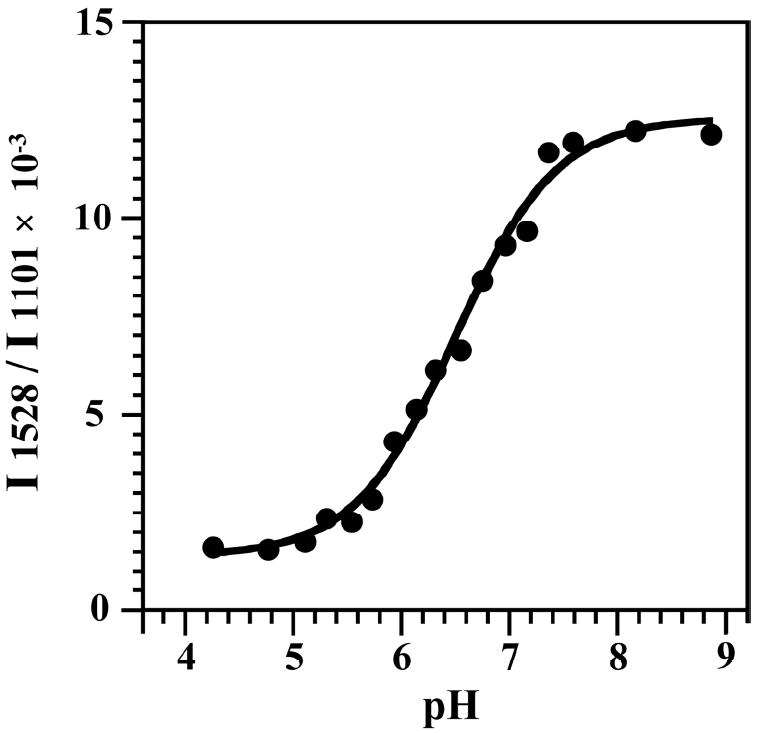
pH titration of HDV ribozyme crystals, using construct shown in Figure 4A. The y-axis is the relative intensity ratio of 1528 cm-1 band (neutral cytosine) to the intensity of the band at 1101 cm-1 (PO2−, internal standard). A pKa of 6.40 ± 0.05 at 2 mM Mg2+ was obtained. Data were fit to a Henderson-Hasselbalch equation with a Hill constant of unity. Adapted with permission 21. Copyright 2007 American Chemical Society.
Measurements of pKa’s of the general acid and base of the hairpin ribozyme have been made recently. Wedekind, Carey and co-workers used Raman crystallography to determine a pKa of 5.5 for A38, the putative general acid in the hairpin ribozyme,26 and a similar value of 5.9 was extrapolated by Fedor and colleagues using fluorescence spectroscopy;27 moreover these pKa values agree with analyses of rate-pH studies. In addition, Fedor and colleagues used fluorescence spectroscopy to obtain a pKa of 10.6 for G8, the putative general base, which was extrapolated from a value of 9.5 measured with the fluorescent base 8-azaguanine.28 There appears to be agreement that A38 serves as the general acid, but the role of G8 is unsettled: rate-pH profiles can be interpreted with G8 as the general base in the reaction,19,29 but hydroxide could deprotonate the 2’OH and G8 could simply hydrogen bond in the transition state.28,30
We have also measured pKa values for model hairpin oligonucleotides using 31P NMR and UV-vis spectroscopy.10,31,32 For Class I oligonucleotides with an A+•C base pair, ionization was measured by placing a thiophosphate near the site of protonation.31 The resulting mixture of diastereomers leads to two peaks in the 31P NMR that are separated from other phosphorus resonances and can be tracked as a function of pH. We measured effects of position, temperature, and ionic strength for an A+•C wobble in DNA using this method.33 Under the most favorable conditions—low temperature, low ionic strength, and central positioning of the wobble—a pKa of 8.0 was found, suggesting that nucleobases can be cationic under biological conditions.
In addition, pKa values of unstructured oligonucleotides with a single ionizable group, such as UUCUU and UUAUU, have been determined using UV-detected pH titrations.10,32 These measurements provide unfolded state pKa estimates (see below).10 Also, the ionic strength dependence of these systems was used to interpret effects of ionic strength on the HDV ribozyme as lack of ion penetration.32
Lastly, chemical modification, such as methylation, as a function of pH has been used to determine pKa’s in large RNAs, such as the ribosome. These have led to measurement of a pKa near 7.34 One problem with this method is potential for nonequivalence in sites of deprotonation and methylation, which is because complex RNA structures can refold in a way that can expose distal, non-ionizing sites to methylation.
Next we consider kinetics approaches of attaining pKa’s. Most often kinetics data are plotted linear in free energy, i.e. log kobs vs pH (Figure 6). This is doable because decreasingly small amounts of RNA in the functional protonation state remain detectable by gathering longer time points. Kinetic assays thus offer the ability to detect poorly populated (e.g. 1 % or less) general acids and bases; typically this is not afforded by spectroscopy, which often has detection limits of ~5% bound. By analyzing such ‘log-linear’ regions of rate-pH profiles, one can attain additional information, such as reaction channels35,36 and non-productive roles for protons, such as misfolding the RNA.10,37 The slope affords the net number of protons ionizing in the step.
Figure 6.
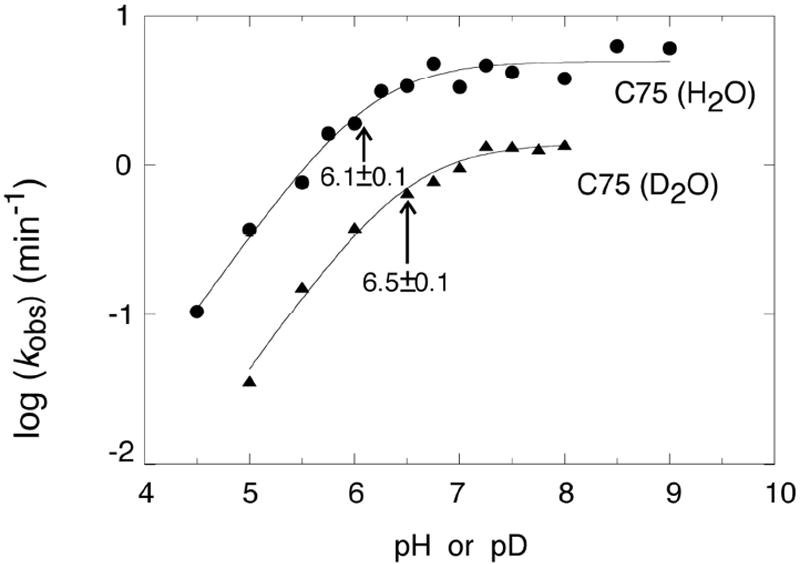
Rate-pH profile for the HDV ribozyme in H2O or D2O and 10 mM Mg2+. Note that the pKa is near the rounding or flex point of the curve and that data can be obtained over a wide range of rates. The slope of the log-linear region is ~1 consistent with one proton ionizing in this region. The offset in the pKa and the difference in the plateau values between H2O and D2O are consistent with chemistry as the rate-limiting step. Adapted with permission 5. Copyright 2000 American Association for the Advancement of Science.
Extraction of pKa values from kinetics can be very complex. First, data can fit well to the Henderson-Hasselbalch equation but have nothing to do with charging of bases; for example, apparent pKa’s can be due to a change in the rate-limiting step from chemistry to a conformational change, or another reaction channel. Second, because there is typically ionization of both a general acid and a general base (along with any other ionization related to structure), the shape of the rate-pH profile is not a reliable guide as to whether the observed pKa comes from the general acid or base. We have developed a partition function analysis to aid interpreting rate-pH profiles, which can provide insight, but not proof of mechanism.19,29 Third, extremes of pH chemically denature RNA and DNA by rupturing hydrogen bonding. Moreover, because ionization of any number of bases can unfold the RNA, an apparent pKa for loss of cleavage activity shifted towards neutrality can occur owing to statistical effects, as first described by Knitt and Herschlag.10,37
Because of the above hazards in interpreting rate-pH profiles, it is critical to have complementary measurements of the pKa, such as the aforementioned spectroscopic approaches. Calculations can help as well, and we recently used MD simulations to gain insight into how protonation of a non-catalytic base triple affects the active site.38 These simulations were consistent with complex kinetic rate-pH profiles.23 In addition, deconvoluting pKa’s from rate-pH profiles can be aided by studies with nucleobase analogs with altered pKa’s.39,40
Involvement of Charged Bases in RNA Catalysis
The reaction pathways of RNA enzymes vary in terms of contributions of metal ion, general acid-base and electrostatic catalysis. In general, large ribozymes prefer metal ion catalysis, while smaller ribozymes prefer general acid-base catalysis. Large ribozymes have been extensively studied biochemically and structurally.1 With the exception of the spliceosome, high-resolution structures of these RNAs have been solved, often in several catalytic states. A major contribution to the catalytic mechanism is stabilization of charge development by divalent ions, which act as Lewis acids.41 Smaller ribozymes such as the HDV, hairpin, hammerhead, VS, and glm S have also been intensively studied by biochemical and structural approaches.1 With the exception of the VS ribozyme, there are high-resolution structures of these RNAs as well, including reactant states, product states, and transition state-mimics. A major catalytic theme for these reactions has been the direct involvement of nucleobases in self-cleavage.42
One can associate the termini produced in RNA catalysis with the chemical roles that the nucleobases play. Small ribozymes perform phosphodiester bond cleavage in which a nucleophilic 2’-OH attacks the adjoining phosphorus, with the bridging 5’-oxygen as the leaving group (Figure 3). This leaving group has no vicinal source of labile protons, which might serve as a general acid. In contrast, in large ribozymes the bridging 3’-oxygen leaving group has the 2’-OH to assist in its departure. In addition, metal ion and nucleobase catalysis are not exclusive within these ribozyme classes: there is strong evidence that the HDV ribozyme employs a Mg2+ ion as a Lewis acid (Figure 4B),24 and it is possible that large ribozymes use nucleobases in catalysis.
We favor general acid-base chemistry as a predominant mechanism for small ribozymes for three reasons: (1) rate-pH profiles are consistent with general acid-base chemistry,5,19,29,32,35 (2) hyperactivated leaving group studies implicate nucleobases as general acids,43,44 and (3) Brønsted plots based on small molecule rescue show nucleobase acidity contributes to catalysis.45 Participation in general acid-base chemistry does not eliminate structural or electrostatic roles for the bases—in fact, these are likely given that the general acid and base are charged for part of the reaction cycle. Indeed, RNase A, which catalyzes the same reaction, uses a lysine to stabilize charge development.46 Kinetically equivalent modes of catalysis in which nucleobases hydrogen bond to the active site and solvent species transfer protons, remain possibilities in ribozyme reactions where Brønsted and activated leaving group studies have not been performed.
An emerging theme in the general acid-base mechanisms for most small ribozymes is a reactant state in which both the general base is anionic and general acid is cationic (Figure 3). Anionic guanine has been implicated as a general base for the hairpin, VS, glm S, and hammerhead ribozymes. At the general acid position, cationic species are generally found: cytosine for the HDV ribozyme, adenine for the hairpin and VS ribozymes, and GlcN6pP for the glm S ribozyme. All of these species cycle from charged to neutral as they accept and donate their protons (Figure 3). The observation that the general acid and base are charged together in the reactant state of many ribozymes suggests the possibility for through-space electrostatic stabilization.
Mechanisms used by small ribozymes can be remarkably complex. We described the reaction of the HDV ribozymes with a multichannel mechanism, in which each channel leads to the same products (Scheme 1),32,35,36 Channel 1 operates without participation of Mg2+, Channel 2 with structural Mg2+, and Channel 3 with catalytic Mg2+, and thus the dominant reaction mechanism is a function of Mg2+ concentration. Ions were classified as structural or catalytic on the basis of effects of ion size and affinity.35,36 The presence of multiple channels was inferred from the complexity of rate-pH and rate-Mg2+ profiles over wide pH and Mg2+ ranges.32,35,36 Channels 1 and 2 have rate-pH profiles inverted from Channel 3. Moreover, Channels 1 and 2 have subchannels, in which water or hydroxide participates as the base, depending on pH.32 One theme to emerge is that C75 is cationic and serves as the general acid in all three channels, indicating robustness and importance of its shifted pKa to function.32
Scheme 1.

Other ribozymes likely have multiple reaction channels.47 On the one hand, multichannel mechanisms have the potential to confound model-driven interpretation of rate-pH profiles; indeed, careful tuning of experimental conditions is needed to isolate a single channel. On the other hand, these mechanisms reveal the versatility of ribozymes—that they can employ metal ions, nucleobases, and solvent to catalyze the same reaction in multiple ways.
Driving Forces for Ionizing the Bases
We have considered thermodynamic driving forces for pKa perturbation.10,48 Two types of molecular mechanisms for these driving forces are (1) gain of hydrogen bonding and (2) electrostatic stabilization, both of which involve coupling of protonation and RNA folding.
Coupling between protonation of A or C and gain of hydrogen bonding can lead to new base pairs (Figure 2).10 We previously developed a formalism in which the driving force for nucleobase protonation in the folded state is the stabilization in RNA folding offered by protonation, ΔGx (Figure 7A, right side green box).
| (1) |
Given that pKa shifts of 3 or 4 units are known, ΔGx can be a remarkably large, −4 to −6 kcal/mol. This arises from strengthened stacking, hydrogen bonding, and electrostatics.
Figure 7.
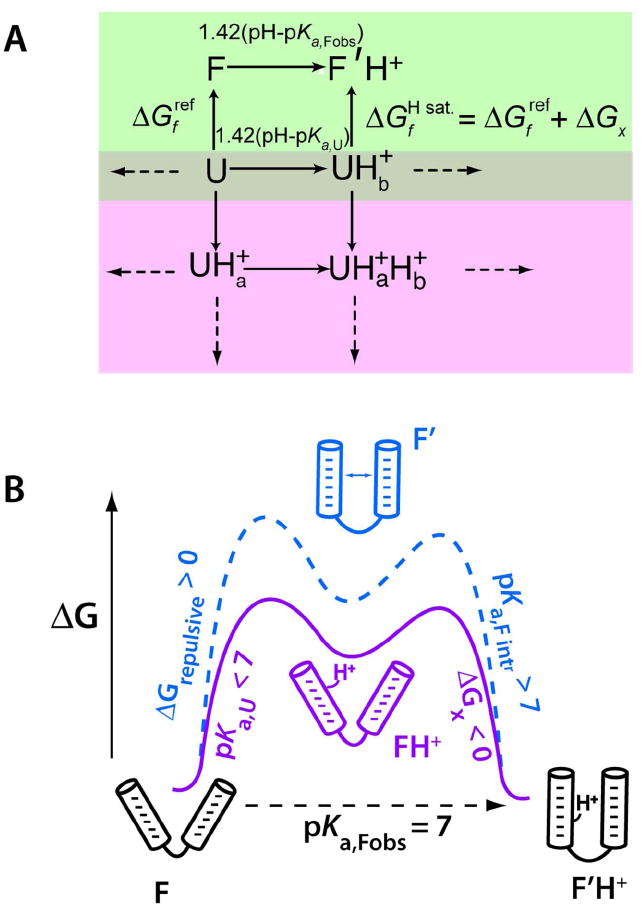
Linkage between folding and pKa shifting.10 (A) Thermodynamic model linking RNA folding (green box, vertical lines) and protonation of the folded (F) and unfolded (U) states (green box, horizontal lines; pink boxes, all lines). This model assumes a single shifted pKa in the folded state. Values are free energies in kcal/mol. We measure the folded and unfolded state pKa’s, as well as the reference free energy , and calculate ΔGx. (B) Free energy diagram showing model-driven interpretation of pKa values for electrostatic stabilization at pH 7. The model is conceptual and the difference in the free energy of the two paths is unknown; the two paths are offset to allow adequate visualization. Protonation is three-state, with either path involving a high-energy intermediate. Panel A is adapted with permission 10. Copyright 2005 RNA Society.
The large value of ΔGx is never fully observed, however. This is because directly measuring it would require starting in the protonated unfolded state (Figure 7A, UH+ state), but this is impossible because lowering the pH enough to fully populate it leads to protonation of other sites in the unfolded state (Figure 7A, pink box), which drives acid denaturation of the helix. Practically speaking, the minimum in folding free energy for most RNAs occurs around pH 5.5. We thus typically calculate ΔGx, using the thermodynamic cycle in Figure 7A (green box) and experimental values of pKa,Fobs, pKa,U, and neutral state stability ΔGf,ref .
Regarding electrostatic stabilization as a driving force for ionization, Pyle and Honig have reported non-linear Poisson-Boltzmann (NLPB) electrostatics calculations in which regions of high negative electrostatic potential drive protonation of A and C,48 and we have provided qualitatively similar NLPB arguments on the HDV ribozyme for upwards shifting of the pKa of C75 (Figure 8).35,49 These types of calculations confirm which regions of the RNA contribute to shifting of ionization potentials, but are presently limited in predicting pKa values.
Figure 8.
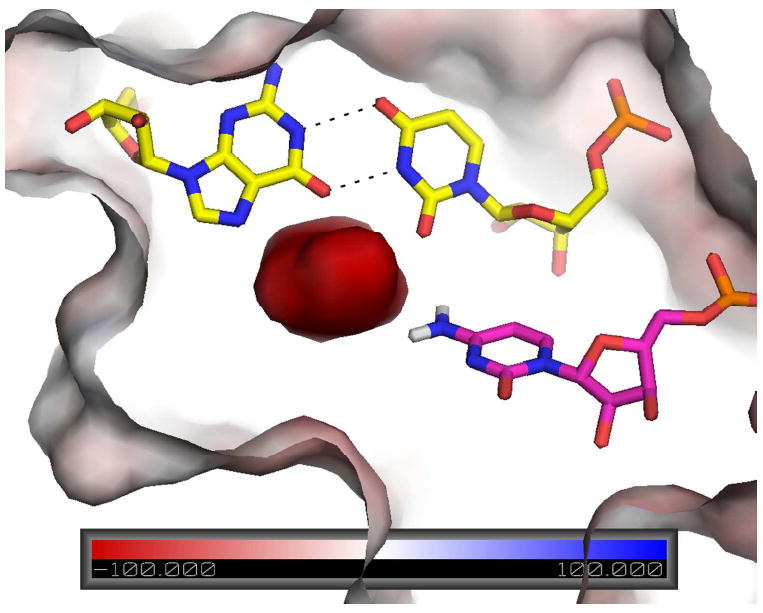
Surface electrostatic potential of precleaved HDV ribozyme. Reverse G•U wobble is in yellow and C75 is in magenta. Note the extreme negative potential near the N4 of C75. This potential helps stabilize the positive charge of protonated C75, which delocalizes to N4 according to RESP calculations, and orient N3 for proton transfer to the leaving group. Reproduced with permission 49. Copyright 2011 American Chemical Society.
As mentioned, mechanisms for pKa shifting invoke coupling between proton loading and RNA folding. This coupling can be understood in terms of the simplified conceptual free energy diagram in Figure 7B. Coupling arises because the proton-binding site has too much negative repulsion to be stable without a cation (state F’) and too little affinity to bind proton without folding (state FH+). A minimal model for coupling between folding and protonation is thus three-state. Because the intermediate is high in energy, it is generally hidden from experimental observation, and the system behaves cooperatively. It is therefore customary to treat the system as two-state, with a single pKa in the folded state (Figure 7A). Nonetheless, it is conceptually useful to consider pKa,Fobs, as arising from two terms.
Along path 1 (Figure 7B, solid line), we consider the system protonating with unperturbed pKa,U to a high energy state followed by a folding bonus (i.e. negative) of ΔGx,
| (2) |
Thus, pKa,Fobs could be raised by interactions upon folding of the protonated state. Along path 2 (Figure 7B, dashed line), we consider the system folding to a high energy state with a repulsive (i.e. positive) free energy ΔGrep followed by protonating with an intrinsic pKa,Fintr given by
| (3) |
where pKa,Fintr is even more basic than pKFobs. If there are extensive interactions in the RNA that help it remain folded despite repulsive interactions, then the repulsive state might become the reference state (i.e. F’ and F’H+ both lowered in energy), and the observed pKa could become pKa,Fintr and be exceptionally basic. One might envision such a mechanism in a large RNA with extensive pre-organization, such as the ribosome. Coupling of folding to molecular crowding could further favor protonation, as supported by recent studies on the i-motif.50
At present, almost nothing is known about coupling between folding and deprotonation of the nucleobases. However, if new interactions are gained upon deprotonation, there will also be a driving force. New interactions could be offered by metal ions or protonated bases, the cationic functionalities on the bases, or hydrogen bonding to the 2’OH. Moreover, we note that the unperturbed pKa’s of G and U start closer to biological pH than do the unperturbed pKa of A and C, meaning less driving force is needed.
Concluding Remarks
Nucleobases can have pKa’s near neutrality that contribute to catalysis. This functional diversity is remarkable when one considers that it arises solely from reversible, non-covalent interactions of functionally bereft monomers. Roles of cationic bases in RNA catalysis seem well-established. At the same time, much remains controversial or unknown in the field: Do negatively charged bases populate and do they contribute to catalysis? Do the bases tautomerize? Are current ribozyme structures catalytically competent? In RNAs with measurable pKa’s, which bases are ionizing? Are the ionizing bases communicating with each other? Do nucleobases contribute to catalysis in large ribozymes? Do in vivo conditions alter pKa shifting? Answering these questions requires collaborations between chemists, biologists, crystallographers, spectroscopists, and theorists. Theory will be critical because charged bases often populate poorly and thus remain hidden from many structural and spectroscopic studies.
Acknowledgments
This work was supported by NIH grant GM095923. We thank Paul Carey and Sharon Hammes- Schiffer for comments.
Biographies
Jennifer L. Wilcox received her B.S. in chemistry from The University of Scranton in 2007 where she completed undergraduate research in organic geochemistry. She is now a graduate student at The Pennsylvania State University pursuing her Ph.D. in chemistry. Her research focuses on determining the driving forces for pKa shifting in RNA.
Amarpreet Ahluwalia is an undergraduate student pursuing a B.S. in biochemistry and molecular biology at The Pennsylvania State University in the Schreyer Honors College. Her research interests include identifying protonated bases in RNA through computational and experimental approaches.
Philip C. Bevilacqua received his B.S. in chemistry and physics from John Carroll University in 1987. He obtained the Ph.D. from The University of Rochester in 1993 from Douglas Turner, and was a post-doctoral fellow at the University of Colorado, Boulder with Thomas Cech. He is currently Professor of Chemistry at The Pennsylvania State University. His research interests include the structure and function of catalytic and regulatory RNA.
References
- 1.Lilley DM, Eckstein F, editors. Ribozymes and RNA Catalysis. Royal Society of Chemistry; Cambridge: 2008. pp. 1–318. [Google Scholar]
- 2.Ferre-D’Amare AR, Scott WG. Small self-cleaving ribozymes. Cold Spring Harb Perspect Biol. 2010:a003574. doi: 10.1101/cshperspect.a003574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Saenger W. In: Principles of Nucleic Acid Structure. Cantor CR, editor. Springer-Verlag; New York: 1984. pp. 1–556. [Google Scholar]
- 4.Thomas JM, Yoon JK, Perrin DM. Investigation of the catalytic mechanism of a synthetic DNAzyme with protein-like functionality: an RNaseA mimic? J Am Chem Soc. 2009;131:5648–5658. doi: 10.1021/ja900125n. [DOI] [PubMed] [Google Scholar]
- 5.Nakano S, Chadalavada DM, Bevilacqua PC. General acid-base catalysis in the mechanism of a hepatitis delta virus ribozyme. Science. 2000;287:1493–1497. doi: 10.1126/science.287.5457.1493. [DOI] [PubMed] [Google Scholar]
- 6.Legault P, Pardi A. Unusual dynamics and pKa shift at the active site of a lead-dependent ribozyme. J Am Chem Soc. 1997;119:6621–6628. [Google Scholar]
- 7.Bevilacqua PC, Brown TS, Nakano S, Yajima R. Catalytic roles for proton transfer and protonation in ribozymes. Biopolymers. 2004;73:90–109. doi: 10.1002/bip.10519. [DOI] [PubMed] [Google Scholar]
- 8.Gueron M, Leroy JL. Studies of base pair kinetics by NMR measurement of proton exchange. Methods Enzymol. 1995;261:383–413. doi: 10.1016/s0076-6879(95)61018-9. [DOI] [PubMed] [Google Scholar]
- 9.Draper DE. A guide to ions and RNA structure. RNA. 2004;10:335–343. doi: 10.1261/rna.5205404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Moody EM, Lecomte JT, Bevilacqua PC. Linkage between proton binding and folding in RNA: A thermodynamic framework and its experimental application for investigating pKa shifting. RNA. 2005;11:157–172. doi: 10.1261/rna.7177505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nagaswamy U, Larios-Sanz M, Hury J, Collins S, Zhang Z, Zhao Q, Fox GE. NCIR: a database of non-canonical interactions in known RNA structures. Nucleic Acids Res. 2002;30:395–397. doi: 10.1093/nar/30.1.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lemieux S, Major F. Automated extraction and classification of RNA tertiary structure cyclic motifs. Nucleic Acids Res. 2006;34:2340–2346. doi: 10.1093/nar/gkl120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Banas P, Walter NG, Sponer J, Otyepka M. Protonation states of the key active site residues and structural dynamics of glmS riboswitch as reveled by molecular dynamics. J Phys Chem B. 2010;114:8701–8712. doi: 10.1021/jp9109699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mlynsky V, Banas P, Hollas D, Reblova K, Walter NG, Sponer J, Otyepka M. Extensive molecular dynamics simulations showing that canonical G8 and protonated A38H+ forms are most consistent with crystal structures of hairpin ribozyme. J Phys Chem B. 2010;114:6642–6652. doi: 10.1021/jp1001258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Auffinger P, Bielecki L, Westhof E. Anion binding to nucleic acids. Structure. 2004;12:379–388. doi: 10.1016/j.str.2004.02.015. [DOI] [PubMed] [Google Scholar]
- 16.Kieft JS, Chase E, Costantino DA, Golden BL. Identification and characterization of anion binding sites in RNA. RNA. 2010;16:1118–1123. doi: 10.1261/rna.2072710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Rupert PB, Ferre-D’Amare AR. Crystal structure of a hairpin ribozyme-inhibitor complex with implications for catalysis. Nature. 2001;410:780–786. doi: 10.1038/35071009. [DOI] [PubMed] [Google Scholar]
- 18.Rupert PB, Massey AP, Sigurdsson ST, Ferre-D’Amare AR. Transition state stabilization by a catalytic RNA. Science. 2002;298:1421–1424. doi: 10.1126/science.1076093. [DOI] [PubMed] [Google Scholar]
- 19.Bevilacqua PC. Mechanistic considerations for general acid-base catalysis by RNA: Revisiting the mechanism of the hairpin ribozyme. Biochemistry. 2003;42:2259–2265. doi: 10.1021/bi027273m. [DOI] [PubMed] [Google Scholar]
- 20.Luptak A, Ferre-D’Amare AR, Zhou K, Zilm KW, Doudna JA. Direct pK(a) measurement of the active-site cytosine in a genomic hepatitis delta virus ribozyme. J Am Chem Soc. 2001;123:8447–8452. doi: 10.1021/ja016091x. [DOI] [PubMed] [Google Scholar]
- 21.Gong B, Chen JH, Chase E, Chadalavada DM, Yajima R, Golden BL, Bevilacqua PC, Carey PR. Direct measurement of a pK(a) near neutrality for the catalytic cytosine in the genomic HDV ribozyme using Raman crystallography. J Am Chem Soc. 2007;129:13335–13342. doi: 10.1021/ja0743893. [DOI] [PubMed] [Google Scholar]
- 22.Gong B, Chen JH, Yajima R, Chen Y, Chase E, Chadalavada DM, Golden BL, Carey PR, Bevilacqua PC. Raman crystallography of RNA. Methods. 2009;49:101–111. doi: 10.1016/j.ymeth.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nakano S, Bevilacqua PC. Mechanistic characterization of the HDV genomic ribozyme: a mutant of the C41 motif provides insight into the positioning and thermodynamic linkage of metal ions and protons. Biochemistry. 2007;46:3001–3012. doi: 10.1021/bi061732s. [DOI] [PubMed] [Google Scholar]
- 24.Chen JH, Yajima R, Chadalavada DM, Chase E, Bevilacqua PC, Golden BL. A 1.9 Å crystal structure of the HDV ribozyme pre-cleavage suggests both Lewis acid and general acid mechanisms contribute to phosphodiester bond cleavage. Biochemistry. 2010;49:6508–6518. doi: 10.1021/bi100670p. [DOI] [PubMed] [Google Scholar]
- 25.Brown TS, Chadalavada DM, Bevilacqua PC. Design of a highly reactive HDV ribozyme sequence uncovers facilitation of RNA folding by alternative pairings and physiological ionic strength. J Mol Biol. 2004;341:695–712. doi: 10.1016/j.jmb.2004.05.071. [DOI] [PubMed] [Google Scholar]
- 26.Guo M, Spitale RC, Volpini R, Krucinska J, Cristalli G, Carey PR, Wedekind JE. Direct Raman measurement of an elevated base pK(a) in the active site of a small ribozyme in a precatalytic conformation. J Am Chem Soc. 2009;131:12908–12909. doi: 10.1021/ja9060883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cottrell JW, Scott LG, Fedor MJ. The pH dependence of hairpin ribozyme catalysis reflects ionization of an active site adenine. J Biol Chem. 2011;286:17658–17664. doi: 10.1074/jbc.M111.234906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Liu L, Cottrell JW, Scott LG, Fedor MJ. Direct measurement of the ionization state of an essential guanine in the hairpin ribozyme. Nat Chem Biol. 2009;5:351–357. doi: 10.1038/nchembio.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson TJ, Lilley DM. Do the hairpin and VS ribozymes share a common catalytic mechanism based on general acid-base catalysis? A critical assessment of available experimental data. RNA. 2011;17:213–221. doi: 10.1261/rna.2473711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kuzmin YI, Da Costa CP, Fedor MJ. Role of an active site guanine in hairpin ribozyme catalysis probed by exogenous nucleobase rescue. J Mol Biol. 2004;340:233–251. doi: 10.1016/j.jmb.2004.04.067. [DOI] [PubMed] [Google Scholar]
- 31.Moody EM, Brown TS, Bevilacqua PC. Simple method for determining nucleobase pKa values by indirect labeling and demonstration of a pKa of neutrality in dsDNA. J Am Chem Soc. 2004;126:10200–10201. doi: 10.1021/ja047362h. [DOI] [PubMed] [Google Scholar]
- 32.Cerrone-Szakal AL, Siegfried NA, Bevilacqua PC. Mechanistic characterization of the HDV genomic ribozyme: Solvent isotope effects and proton inventories in the absence of divalent metal ions support C75 as the general acid. J Am Chem Soc. 2008;130:14504–14520. doi: 10.1021/ja801816k. [DOI] [PubMed] [Google Scholar]
- 33.Siegfried NA, O’Hare B, Bevilacqua PC. Driving forces for nucleic acid pK(a) shifting in an A(+).C wobble: effects of helix position, temperature, and ionic strength. Biochemistry. 2010;49:3225–3236. doi: 10.1021/bi901920g. [DOI] [PubMed] [Google Scholar]
- 34.Muth GW, Ortoleva-Donnelly L, Strobel SA. A single adenosine with a neutral pKa in the ribosomal peptidyl transferase center. Science. 2000;289:947–950. doi: 10.1126/science.289.5481.947. [DOI] [PubMed] [Google Scholar]
- 35.Nakano S, Proctor DJ, Bevilacqua PC. Mechanistic characterization of the HDV genomic ribozyme: assessing the catalytic and structural contributions of divalent metal ions within a multichannel reaction mechanism. Biochemistry. 2001;40:12022–12038. doi: 10.1021/bi011253n. [DOI] [PubMed] [Google Scholar]
- 36.Nakano S, Cerrone AL, Bevilacqua PC. Mechanistic characterization of the HDV genomic ribozyme: classifying the catalytic and structural metal ion sites within a multichannel reaction mechanism. Biochemistry. 2003;42:2982–2994. doi: 10.1021/bi026815x. [DOI] [PubMed] [Google Scholar]
- 37.Knitt DS, Herschlag D. pH dependencies of the Tetrahymena ribozyme reveal an unconventional origin of an apparent pKa. Biochemistry. 1996;35:1560–1570. doi: 10.1021/bi9521147. [DOI] [PubMed] [Google Scholar]
- 38.Veeraraghavan N, Bevilacqua PC, Hammes-Schiffer S. Long distance communication in the HDV ribozyme: Insights from molecular dynamics and experiments. J Mol Biol. 2010;402:278–291. doi: 10.1016/j.jmb.2010.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Das SR, Fong R, Piccirilli JA. Nucleotide analogues to investigate RNA structure and function. Curr Opin Chem Biol. 2005;9:585–593. doi: 10.1016/j.cbpa.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 40.Suydam IT, Levandoski SD, Strobel SA. Catalytic importance of a protonated adenosine in the hairpin ribozyme active site. Biochemistry. 2010;49:3723–3732. doi: 10.1021/bi100234v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Frederiksen JK, Piccirilli JA. Identification of catalytic metal ion ligands in ribozymes. Methods. 2009;49:148–166. doi: 10.1016/j.ymeth.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bevilacqua PC, Yajima R. Nucleobase catalysis in ribozyme mechanism. Curr Opin Chem Biol. 2006;10:455–464. doi: 10.1016/j.cbpa.2006.08.014. [DOI] [PubMed] [Google Scholar]
- 43.Das SR, Piccirilli JA. General acid catalysis by the hepatitis delta virus ribozyme. Nat Chem Biol. 2005;1:45–52. doi: 10.1038/nchembio703. [DOI] [PubMed] [Google Scholar]
- 44.Wilson TJ, Li NS, Lu J, Frederiksen JK, Piccirilli JA, Lilley DM. Nucleobase-mediated general acid-base catalysis in the Varkud satellite ribozyme. Proc Natl Acad Sci U S A. 2010;107:11751–11756. doi: 10.1073/pnas.1004255107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Perrotta AT, Wadkins TS, Been MD. Chemical rescue, multiple ionizable groups, and general acid–base catalysis in the HDV genomic ribozyme. RNA. 2006;12:1282–1291. doi: 10.1261/rna.14106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Raines RT, Ribonuclease A. Chem Rev. 1998;98:1045–1065. doi: 10.1021/cr960427h. [DOI] [PubMed] [Google Scholar]
- 47.Zhou JM, Zhou DM, Takagi Y, Kasai Y, Inoue A, Baba T, Taira K. Existence of efficient divalent metal ion-catalyzed and inefficient divalent metal ion-independent channels in reactions catalyzed by a hammerhead ribozyme. Nucleic Acids Res. 2002;30:2374–2382. doi: 10.1093/nar/30.11.2374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tang CL, Alexov E, Pyle AM, Honig B. Calculation of pK(a)s in RNA: on the structural origins and functional roles of protonated nucleotides. J Mol Biol. 2007;366:1475–1496. doi: 10.1016/j.jmb.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 49.Veeraraghavan N, Ganguly A, Chen JH, Bevilacqua PC, Hammes-Schiffer S, Golden BL. Metal binding motif in the active site of the HDV ribozyme binds divalent and monovalent ions. Biochemistry. 2011;50:2672–2682. doi: 10.1021/bi2000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rajendran A, Nakano S, Sugimoto N. Molecular crowding of the cosolutes induces an intramolecular i-motif structure of triplet repeat DNA oligomers at neutral pH. Chem Commun (Camb) 2010;46:1299–1301. doi: 10.1039/b922050j. [DOI] [PubMed] [Google Scholar]