

Abstract
Background
There are two, largely autonomous antioxidant pathways in many organisms, one based on thioredoxin and one based on glutathione, with each pathway having a unique flavoprotein oxidoreductase to maintain them in a reduced state. A recently discovered protein, thioredoxin glutathione reductase (TGR) potentially connects these two pathways. In a large group of parasitic worms, responsible for hundreds of millions of infections in humans and animals, untold morbidity and significant mortality, TGR is the sole enzyme present to maintain redox balance.
Scope of Review
In this review, the current understanding of the biochemical properties of TGR enzymes is compared to the related enzymes thioredoxin reductase and glutathione reductase. The role of the rare amino acid selenocysteine is discussed. An overview of the potential to target TGR for drug development against a range of parasitic worms and preliminary results to identify TGR inhibitors for schistosomiasis treatment is presented.
Major Conclusions
TGR has properties that are both unique and common to other flavoprotein oxidoreductases. TGR plays a fundamentally different and essential role in the redox biology of parasitic flatworms. Therefore, TGR is a promising target for drug development for schistosomiasis and other trematode and cestodes infections.
General Significance
TGR may have differing functions in host organisms, but through analyses to understand its ability to reduce both glutathione and thioredoxin we can better understand the reaction mechanisms of an important class of enzymes. The unique properties of TGR in parasitic flatworms provide promising routes to develop new treatments for diseases.
Keywords: drug development, flavoprotein oxidoreductase, glutathione, schistosomiasis, selenocysteine, thioredoxin
1. Introduction
1.1. Glutathione and thioredoxin redox pathways
Most organisms possess two major pathways for regulating cellular redox homeostasis, antioxidant defense and to provide reducing equivalents for a variety of chemical transformations. These are the glutathione/glutathione disulfide (GSH/GSSG) and the thioredoxin (Trx) dependent systems. The mammalian GSH/GSSG system is composed of GSH, a γ-Glu-Cys-Gly tripeptide, the pyridine nucleotide disulfide oxidoreductase GSSG reductase (GR) recycling oxidized glutathione using NADPH, several glutaredoxin (Grx) isoforms acting as thiol-disulfide reductases as well as a number of GSH peroxidases. The Trx system includes Trx, its NADPH dependent reductase Trx reductase (TrxR) as well as peroxiredoxins possessing Trx-dependent peroxidase activities. GR and TrxR are closely related oxidoreductases with a highly similar structure and reaction mechanism belonging to the same protein family – the GR superfamily of dimeric flavoenzymes [1], which also includes lipoamide dehydrogenase, and trypanothione reductase [2, 3]. One intriguing difference between GR and TrxR enzymes is the C-terminal extension of 16 amino acids in TrxR. An additional active site is located here, which in certain organisms, e.g., mammals, contains the rare amino acid selenocysteine (Sec) in the motif Gly-Cys-Sec-Gly that is indispensable for most endogenous functions of TrxR [4, 5]. In mammals there are two genes encoding TrxR, one of that encodes a protein targeted to the cytoplasm (TrxR1) and one that encodes a protein targeted to both the mitochondria and cytosol (TrxR2) [6].
In addition to the GSH/GSSG and Trx pathways, mammals express a fusion protein of TrxR and Grx domains capable of transporting electrons from NADPH to both Trx and GSH systems. Therefore, the enzyme is called thioredoxin glutathione reductase (TGR), also known as TXNRD3/TrxR3. TGR proteins characterized thus far are selenoproteins, with Sec occurring as the penultimate amino acid in the redox center Glys-Cys-Sec-Gly as in mammalian TrxR proteins [7-10]. TGR, mainly expressed in male germ cells and only at low levels in other tissues [9-11], is implicated in spermatogenesis, presumably playing a role in disulfide bond formation and protein isomerization through disulfide isomerase activity present in the Grx domain [11]. It has been proposed that mouse TGR along with phospholipid hydrogenperoxide glutathione peroxidase (or glutathione peroxidase 4) facilitate protein folding during sperm maturation [11]. Under conditions of oxidative stress and in the presence of retinoic acid, mammalian TGR was recently shown to bind the retinoic acid receptor, presumably to stabilize the receptor and allow its continued signaling activity [12].
Redox pathways in other metazoans can vary from the pattern described above for mammals. For instance, TGR appears to be the Trx-reducing enzyme in fishes as TrxR1 and TrxR2 are absent from their genomes [10]. Reduction of GSSG in insects, which lack authentic GR enzymes, is accomplished nonenzymatically by Trx [13], thereby linking the Trx and GSH/GSSG pathways. Further linkage is seen in the parasitic worm Schistosoma mansoni and other parasitic Platyhelminthes (flatworms including trematodes, the flukes, and cestodes, the tapeworms), which lack authentic TrxR and GR enzymes. Instead, TGR is uniquely present resulting in a fusion of the GSH and Trx pathways in a single, essential protein. It is the only antioxidant enzyme with TrxR or GR activities found in these organisms [8, 14] making it indispensable for cell growth and defense against host-induced oxidative stress, an important part of the innate immune response. Recent analysis of the genome of a free-living platyhelminth, Schmidtea mediterranea, indicates that the fusion of GSH/GSSG and Trx pathways does not occur and that Schmidtea has redox pathways with TrxR, GR and TGR enzymes [15].
1.2. Eukaryotic selenoprotein expression
All TGR proteins characterized to date (and many TrxR proteins) contain a C-terminal redox-active site containing a Cys and a Sec residue. Sec, the 21st amino acid, a highly reactive Cys analog with a selenium atom in place of the sulfur, is co-translationally incorporated into proteins. A highly specific protein complex is required to accomplish Sec insertion; justification for presence of selenium rather than sulfur in a small subset of the total proteins of an organism is based on the unique chemical properties of selenium [16]. The role of selenium in Sec proteins is complex. Selenium is more nucleophilic than sulfur. Therefore, the presence of Sec increases the reaction rates of Sec-containing proteins with substrates. In addition, after reacting with substrates, the resultant thiol-selenolate can efficiently accept electrons from other redox-active cysteine pairs because Sec has a significantly lower pKa value than Cys making it a better leaving group [17]. In addition, under oxidative conditions, Sec-containing proteins are more resistant to inactivation by irreversible oxidation than Cys-containing proteins [17]. However, since Sec is mainly deprotonated at physiological pH it is highly reactive towards alkylation by electrophilic agents and for this reason TrxR proteins have been targeted for cancer chemotherapy [1, 3, 18]. Humans have at least 25 selenoproteins found in 17 distinct protein families [19]. In S. mansoni, four selenoproteins have been identified and characterized, namely two GPx, TGR, and selenoprotein T [8, 20-23]. Analysis of the S. mansoni genome indicates that four additional selenoproteins occur in S. mansoni; these are selenoprotein S, selenophosphate synthetase 2, selenoprotein W and the 15 kDa selenoprotein (Table 1). Selenoproteins I, O, and U occur in S. mansoni as Cys variants, while selenoproteins commonly found in vertebrates, H, J, K, M, N, P, R, V and DI, appear to be absent from the S. mansoni genome. Therefore, S. mansoni has seven selenoprotein families and a total of eight selenoproteins. By contrast, nematodes (e.g., Caenorhabditis elegans) have a single selenoprotein (TrxR) and insects appear to have (at most) only three (H, K, and SPS2) [24, 25].
Table 1.
Presence or Absence of Typical Eukaryotic Selenoproteins in Schistosoma mansoni
| Selenoprotein | Present in S. mansoni | Accession No. | Sec in S. mansoni | Sec location in protein (length of protein) | Identity / similarity to human orthologue (%) | SECISa |
|---|---|---|---|---|---|---|
| 15 kDa | YES | TC37305b | YES | 80 (149) | 36 / 59 | YES |
| DI | NO | -- | -- | -- | -- | -- |
| glutathione | ||||||
| peroxidase 1 | YES | XP_002577160 | YES | 43 (169) | 52 / 67 | YES |
| glutathione | ||||||
| peroxidase 2 | YES | XP_002571873 | YES | 62 (179) | 47 / 63 | YES |
| H | NO | -- | -- | -- | -- | -- |
| I | YES | CAX73136d | NO | -- | 39 / 59 | -- |
| J | NO | -- | -- | -- | -- | -- |
| K | NO | -- | -- | -- | -- | -- |
| M | NO | -- | -- | -- | -- | -- |
| N | NO | -- | -- | -- | -- | -- |
| O | YES | AAW26859e | NO | -- | 46 / 61 | -- |
| P | NO | -- | -- | -- | -- | -- |
| R | NOc | -- | -- | -- | -- | -- |
| S | YES | XP_002569582 | YES | 126 (127) | 30 / 42 | ?f |
| SPS2 | YES | XP_002580459 | YES | 35 (392) | 48 / 65 | YES |
| T | YES | XP_002581129 | YES | 56 (204) | 45 / 63 | YES |
| TGR | YES | AAK85233 | YES | 597 (598) | 54 / 71 | YES |
| U | YES | XP_002574167 | NO | -- | 42 / 62 | -- |
| V | NO | -- | -- | -- | -- | -- |
| W | YES | TC38149b | YES | 13 (84) | 61 / 80 | YES |
Selenocysteine insertion sequences (SECIS) identified using SECISearch 2.19 [20] at http://genome.unl.edu/SECISearch.html.
TC37305 and TC38149 are tentative consensus sequences generated from expressed sequence tags of S. mansoni. The sequences are available at http://compbio.dfci.harvard.edu/tgi/cgibin/tgi/gimain.pl?gudb=s_mansoni
SelR is a form of methionine-R-sulfoxide reductase (MsrB). S. mansoni has two MsrB proteins (MsrB2a - AAT77263 and MsrB2b - AAT77264) neither of which contains Sec [110].
S. japonicum GenBank accession for Sel I; the S. mansoni Sel I is incorrectly annotated.
Partial S. japonicum GenBank accession for an ortholog for Sel O. The S. mansoni Sel O is incorrectly annotated.
Although predicted to be a selenoprotein, no SECIS element was identified by SECISearch.
Selenoproteins are present in all kingdoms of life [16]. However, the prokaryotic selenocysteine insertion machinery differs strongly from that in archea and eukaryotes. Most studies on eukaryotic selenoprotein production were performed in mammalian systems and little is known on the details in lower eukaryotic organisms such as S. mansoni. Sec is encoded by a UGA codon [26], which normally signals termination of translation. Therefore, a rather complex secondary RNA stem-loop structure present in the 3’ untranslated region of the eukaryotic mRNA of selenoproteins serves as a recoding signal (Figure 1). This structure is called the Sec insertion sequence (SECIS) element and may be located up to several kilobases downstream of the UGA codon [27, 28]. Despite low sequence similarities among the SECIS elements of different selenoproteins their secondary structure is highly conserved with consensus sequences essential for Sec incorporation [29].
Figure 1.

Model for the translational insertion of selenocysteine. A secondary structure present in the 3’ untranslated region of the mRNA called the selenocysteine insertion sequence (SECIS) element interacts with the SECIS binding protein-2 (SBP2). This complex then recruits other factors required for the re-interpretation of the codon TGA, normally a termination codon, for selenocysteine incorporation including selenocysteine elongation factor (EFSec), SECp43, ribosomal protein L30, and the specialized tRNAsec. Other factors, known and unknown, are involved in the process [111].
Selenium is taken up by the organism through the diet and can be metabolized via different pathways [30]. For Sec incorporation selenide (H2Se) is formed from dietary selenium and becomes activated by selenophosphate synthetase 2, itself a selenoprotein, in a reaction with AMP yielding phosphorylated selenide [31]. Unlike the other genetically encoded amino acids, selenocysteine is directly synthesized on its tRNA, the selenocysteyl-tRNA(Ser)Sec. This tRNA is first charged with a serine, which becomes phosphorylated by the phosphoseryl-tRNA kinase. The Sec synthase finally replaces the phosphate with selenide to form the selenocysteyl-tRNA(Ser)Sec [31].
In addition to the SECIS element and the specific tRNA(Ser)Sec a number of trans-acting protein factors are essential for Sec incorporation, i.e. the specific elongation factor EFSec, SECIS binding protein 2, ribosomal protein L30, SECp43, and possibly other yet unidentified factors [30]. However, the exact functions and interactions of each component of this specific translation machinery as well as the sequence of the formation of the supramolecular complex is yet unknown. In some cases a selenocysteine redefinition element, a specific stem-loop RNA structure, is located in the coding sequence and potentially serves in fine-tuning of the UGA read through [32].
2. Functions and properties of TGR enzymes
2.1. TGR gene structure
The human TGR gene spans 49,162 bases at locus 3q21 and is composed of 17 exons. In humans and many other mammals a CUG codon has been shown to be the start codon [33]. However, translation initiation at this codon is inefficient and a second downstream initiation codon is also used resulting in two forms of TGR protein [33]. Both TGR isoforms are thought to be cytoplasmic, but there may be differences in their tissue distribution [33].
S. mansoni TGR is encoded by a single copy gene composed of 17 exons spanning at least 25,000 basepairs. Similar gene structures have been found for S. japonicum, Echinococcus multilocularis, and E. granulosus [15]. Regulatory elements controlling TGR expression have not been studied nor are their chromosomal locations known. It might be expected that, as in mammals, redox pathways would be present in both mitochondrial and cytoplasmic compartments in parasitic helminths and that TGR should play a role in both. It has been found that in E. granulosus (a tapeworm, causative agent of hydatid cyst disease in humans) that alternative splicing of primary TGR transcripts results in transcripts encoding either mitochondrial or cytoplasmic targeted proteins [7]. In S. mansoni two TGR transcripts are also produced by alternative splicing to encode both mitochondrial and cytoplasmic TGR variants [15, 34]. However, unlike in E. granulosus, in which both mRNA variants are trans-spliced, only the mRNA encoding a cytoplasmic protein in S. mansoni is trans-spliced, while the mitochondrial variant is not. Addition of the trans-spliced leader to the pre-mRNA results in a deletion of the exons encoding the mitochondrial targeting sequence (Williams et al, unpublished). Alternative transcription start sites in TGR mRNAs may also contribute to the production of TGR transcripts in S. mansoni. TGR mRNAs contain a consensus type 2 SECIS element located in the 3’noncoding region of their transcripts [8, 15].
2.2. S. mansoni TGR protein structure
The structure of S. mansoni TGR has been determined [35, 36]. Like other members of the family of pyridine nucleotide disulfide oxidoreductases, TGRs are dimeric proteins with a head-tail arrangement; the molecular weight of each monomer is about 65 kDa [8, 9, 37]. Each monomer has a Grx domain at its N-terminus fused to the C-terminal TrxR domain. Figure 2 compares SmTGR domains to its related mammalian proteins. S. mansoni TGR dimer appears as a distorted ‘W’ structure, with the dimerization interface representing the central ‘Λ’ of the structure formed through the interaction of the two TrxR domains (Figure 3). The two Grx domains are at the top of the two outer arms of the ‘W’. The structure of the TrxR domain of TGR proteins is similar to other pyridine nucleotide disulfide oxidoreductases with three subdomains. The N-terminal subdomain contains a flavin adenine dinucleotide (FAD) binding site with a redox active cysteine pair adjacent to FAD in the sequence Cys-Val-Asn-Val-Gly-Cys, which is also found in GR and TrxR proteins (Figure 2). The central subdomain has an NADPH-binding pocket. The C-terminal subdomain acts as the interface between monomers in the dimer and has a 16 amino acid extension found also in TrxR. This extension contains the C-terminal redox pair in the sequence Gly-Cys-Sec-Gly. The dimer interface of TGR presents differences from both TrxR and GR dimer interfaces regions most notably in a hydrophobic cavity known to be a site for noncompetitive inhibitor binding in GR [38]. More polar residues are present in this pocket in TGR than in TrxR and GR and this difference could potentially be used to design TGR-specific inhibitors. The active sites of TGR proteins are composed of residues from both subunits: an FAD, a redox cysteine pair adjacent to FAD, and a redox-active cysteine pair in the Grx domain from one subunit, and a redox-active selenocysteine-cysteine pair from the other subunit.
Figure 2.

Comparison of the domain structure of pyridine nucleotide disulfide oxidoreductases. Glutathione reductase (GR), thioredoxin reductase (TrxR) and thioredoxin glutathione reductase (TGR) all contain a common domain characteristic of pyridine nucleotide disulfide oxidoreductases (yellow) with N-terminal redox active cysteines (CVNVGC), FAD- and NADPH-binding pockets and a interface domain that is involved in protein dimerization. TrxR and TGR proteins both contain a C-terminal extension (blue) with redox active cysteine and selenocysteine (U) (or cysteine). TGR proteins have an additional N-terminal domain with sequence and structural similarities to glutaredoxin (purple). This domain has a redox active cysteine pair (CPYC) as in Schistosoma mansoni TGR or single cysteine (CPHS) as in human TGR.
Figure 3.

Comparison of the proposed reaction mechanisms of pyridine nucleotide disulfide oxidoreductases. The active forms of all proteins are dimmers of identical subunits. (A) and (B) Schistosoma mansoni thioredoxin glutathione reductase. In (A) reducing equivalents are shown being transferred from NADPH to the FAD and then to the adjacent cysteine pair in the sequence (CVNVGC). This reduced cysteine pair then transfers the hydride to the C-terminal cysteineselenocysteine in the other peptide chain. (B) Reduction of the C-terminal cysteine-selenocysteine pair produces a structural change in the flexible C-terminal arm allowing it to be repositioned to either react with the glutaredoxin domain active site (CPYC) or directly with oxidized thioredoxin (and potentially other substrates). The reduced glutaredoxin domain can then interact with its substrates, glutathione disulfide (shown) or other substrates such as glutathionylated peptides. In (C), electrons from NADPH pass to the flavin and then to the redox active cysteine pair (CVNVGC) and subsequently to glutathione disulfide in glutathione reductase proteins. In (D), electrons from NADPH pass to the flavin, then to the redox active cysteine pair (CVNVGC) and subsequently to the C-terminal cysteine-selenocysteine couple in the other subunit in thioredoxin reductases. The reduced C-terminal active site can then transfer reducing equivalents to substrates (e.g., oxidized thioredoxin).
2.3. TGR Enzymology - Functions of S. mansoni TGR
The catalytic mechanism of GR proteins has been extensively studied. The flow of electrons during GR catalysis is from NADPH to FAD to the active center disulfide (CDistalVNVGCProximal (to the flavin)) to GSSG (Figure 3). Although the enzyme can accept four electrons per subunit (two at the flavin and two at the disulfide), the principal catalytically active forms are the two-electron reduced EH2 and the oxidized Eox forms [39-43]. The catalytic mechanisms of the high Mr TrxR proteins from eukaryotes are similar to that of GR [44-48]. High Mr TrxR is a dimer with each monomer containing an FAD and two pairs of redox active cysteines/selenocysteine. Thus, it resembles GR with an extra domain containing a C-terminal redox active selenol-thiol or dithiol pair that communicates with substrates (Figure 2, Figure 3). Anaerobic reductive titrations and presteady state kinetic analyses have shown that the C-terminal redox pair of cysteines (GlyCysSecGly in mammalian TrxR, CysXaaXaaXaaXaaCys in TrxR from Plasmodium falciparum, or SerCysCysSer in TrxR from dipteran insects) is redox active and communicates between the N-terminal active site dithiols of the other TrxR subunit and the substrates [48-50] (Figure 3).
Because the active sites of TGR proteins from different organisms are virtually identical, the catalytic mechanisms of all TGR proteins are thought to be similar (Figure 3). It has been proposed that a hydride ion from NADPH is passed to FAD that in turn reduces the redox-active cysteine pair (Cys154/Cys159 in S. mansoni TGR). The nascent dithiol reduces the redox-active selenylsulfide (Cys596/Sec597 in S. mansoni TGR). Then, electrons from the reduced redox-active selenyldithiol can be passed either to substrates, e.g., Trx, or to the redox-active disulfide in the Grx domain (Cys28/Cys31 in S. mansoni TGR) [9, 10]. The C-terminal tail, where the redox-active selenocysteine-cysteine couple is located, should be highly mobile in order to meet the proposed electron flow in TGRs and the available structures of TGRs have not been able to visualize the C-terminal tail [35, 36]. Via their redox-active cysteine pairs and the selenocysteine-cysteine couple, TGRs are able to reduce a wide variety of substrates [9, 34]. However, the specific binding sites on TGR proteins of different substrates remain to be determined.
TGR is the sole enzyme in S. mansoni possessing TrxR and GR activities. It also appears to contribute to the majority of Grx activity [8]. S. mansoni TGR reduces the typical TrxR substrates, such as Trx, DTNB, as well as GSSG and some mixed disulfides (e.g., that formed between 2-hydroxyethyl disulfide and GSH) using NADPH as an electron donor [34]. S. mansoni TGR has been shown to be able to reduce various low molecular weight compounds, such as sodium selenite, tert-butyl hydroperoxide, hydrogen peroxide, alloxan, lipoic acid, and lipoamide, which also can be reduced by mammalian TrxR. In contrast, S. mansoni TGR has very weak activity toward dehydroascrobic acid and ubiquinone, both of which are substrates of mammalian TrxR [34, 51, 52].
The Grx activity of TGR proteins may function independently of the Sec residue. Significant Grx activity could still be found in a recombinant S. mansoni TGR lacking Sec [8, 36]. This indicates that the N-terminal Grx domain could be functionally dissociated from the Sec-containing C-terminal domain provided there is an external source of GSH. In addition, the Grx domain of mammalian TGR was active when expressed alone [10].
2.4. The TrxR domain and selenocysteine in TGRs
It has been shown that Sec is important to TrxR and GR activities of TGR proteins. The DTNB, TrxR, and GR activities of a mouse TGR variant, Sec614Ser, decreased to 2, 1.8, and 1.4% of wild-type activities, respectively [10]. In addition, a truncated form of mouse TGR where Sec614 and Gly615 are missing has very low TrxR and GR activities [10]. A cysteine variant of S. mansoni TGR, Sec597Cys, also displayed significantly decreased DTNB, GR and TrxR activities compared to its wild-type enzyme, corroborating that Sec is crucial but not essential to TGR activity [53]. Similar to selenoprotein TrxRs, the redox-active selenosulfide is thought to confer broad substrate specificity to TGRs [9, 51]. However, selenocysteine is not absolutely essential for the broad substrate specificity of S. mansoni TGR: the TGR variant, Sec597Cys, is also able to reduce low molecular weight compounds even though its activities against these substrates were low compared to those of wild-type S. mansoni TGR [53].
The highly reactive Sec in selenoprotein TrxRs is susceptible to inhibition by several gold compounds, quinones and nitroso compounds [3, 18, 54-57]. By analogy, it is thought that Sec-dependent activity of TGR is important for inhibitory activity of a number of compounds. Many of these compounds are reduced by the redox-active selenocysteine of TGRs and are converted into TGR inhibitors. However, research by Angelucci et al. [35] showed that auranofin, a gold compound and a potent TGR inhibitor, was able to inhibit a truncated form of S. mansoni TGR as well as yeast GR (which lacks Sec), but that the inhibition was very slow. The activity of auranofin against GR and truncated S. mansoni TGR could be accelerated by supplying an exogenous source of selenium [36]. Our recent observations indicate that the S. mansoni TGR Sec597Cys mutant is also rapidly inhibited by auranofin [53]. Therefore, the presence of Sec is not essential for activity of TGR inhibitors, but Sec or a solvent accessible Cys in the S. mansoni TGR Sec597Cys mutant, could accelerate the inhibitory effects of auranofin [36, 53].
2.5. The Grx activity of TGR proteins
The additional Grx domain found in TGR proteins relative to TrxR and GR proteins allows them to catalyze deglutathionylation reactions. Glutathionylation is an important protective mechanism to prevent further, irreversible oxidation of amino acids in proteins, especially cysteines, when cells are challenged by oxidative stress [59]. As the oxidative stress is removed, deglutathionylation of amino acid residues can restore normal functions to proteins. In addition, glutathionylation/deglutathionylation has been shown to regulate multiple factors, e.g., signaling mediators involved in several cellular processes under non-stress conditions [60]. It should be noted that in TGR proteins from a wide range of eukaryotes including Schistosoma, a Cys-Xaa-Xaa-Cys motif is present in the Grx domain, whereas a Cys-Xaa-Xaa-Ser motif is found in the Grx domain of most mammalian TGR proteins [8-11, 34, 38, 61]. At high GSH concentrations, Grx catalyze deglutathionylation via a monothiol mechanism [62-64]. The N-terminal cysteine residue in Grx is responsible for attacking glutathionylated mixed disulfides, leading to Grx-GSH mixed disulfides. Then, Grx-GSH mixed disulfides can be resolved by the second molecule of GSH, resulting in the formation of GSSG and reduced Cys in Grx. In contrast, at low concentrations of GSH, the formation of intramolecular disulfides in Grx is more favorable, leading to oxidized Grx; the oxidized Grx can interact with GSH to form Grx-GSH mixed disulfides again [63, 64]. The latter scenario is thought to be futile because no glutathionylated mixed disulfides can be turned over. Mutation of the C-terminal cysteine residue in the Cys-Xaa-Xaa-Cys motif resulted in increased activity of Grx1 from human and Grx1 and Grx2 from Saccharomyces cerevisiae [65, 66], though the same mutation in Escherichia coli Grx decreased activity to 25% of wild-type Grx activity and slightly decreased the activity of human Grx2 [62, 64, 65].
Although it is known that TGRs are able to catalyze deglutathionylation, the catalytic mechanisms remain to be explored [8, 9, 37, 61]. Recently, a mechanism for the Grx deglutathionylation activity of TGR from Echinococcus granulosus (EgTGR) has been suggested [67]. It was shown that the Grx activity of EgTGR requires the Grx domain and the selenocysteine residue. When the Grx domain of EgTGR undergoes deglutathionylation reactions, the resultant mixed disulfide between GSH and Cys-31 (equivalent to Cys-28 of SmTGR) is resolved by the redox-active cysteine-selenocysteine pair from the TrxR domain instead of Cys-34 (equivalent to Cys-31 of SmTGR). However, the deglutathionylation reactions catalyzed by S. mansoni TGR display different reaction mechanisms. Studies on S. mansoni TGR have found that the N-terminal cysteine residue (Cys28) in the Grx domain is responsible for the Grx activity; mutation of this particular cysteine residue led to the loss of Grx activity [53]. In addition, Cys31 variants (Cys31Ala and Cys31Ser) have decreased Grx activity [53]. Thus, we propose that the deglutathionylation catalyzed by S. mansoni TGR appears to be accomplished by two mechanisms. One is the conventional monothiol mechanism in which only Cys28 is involved. The other mechanism uses Cys31 to form an intramolecular disulfide with Cys28 to deglutathionylate this residue and in the process release GSH. The resulting disulfide in the Grx domain would then be reduced by NADPH via the redox–active C-terminal Cys-Sec pair or by GSH [35, 53]. The intra-molecular transfer of reducing equivalents from the TrxR domain to the Grx domain of TGR is reminiscent of the inter-molecular transfer from TrxR to Grx2 in the mammalian mitochondrial redox system [65].
2.6. The unique properties of GR activity of TGRs
In GR proteins, GSSG receives reducing equivalents from the redox-active dithiol that is itself reduced by FAD [2, 40]. Although the active sites and catalytic mechanisms of high Mr TrxRs are similar to those of GRs, high Mr TrxRs cannot catalyze the reduction of GSSG [47, 50]. In order to explain this phenomenon, two mechanisms have been proposed. The GSSG binding site in GR proteins is found buried far from the protein surface; access by GSSG is through a narrow cleft [42]. One argument developed by Sandalova et al., [69] suggests that GSSG is excluded from binding to the N-terminal redox active site near FAD in high Mr TrxRs by the C-terminal extension. However, C-truncated TrxR also cannot reduce GSSG [70]. Another proposal by Urig et al., [70] suggests the electrostatic distribution of amino acids in the GSSG binding pocket interacting with the redox-active dithiol adjacent to FAD of GRs is essential for binding GSSG. The amino acids in the same region of high Mr TrxRs is not favorable for GSSG binding. Although significant similarity exists between high Mr TrxR and TGR, much more so than between GR and TGR on a sequence identity level, TGR exhibits GR activity. Thus, it is of interest to explore the catalytic mechanism of reduction of GSSG by TGR.
It has been shown that both Sec and the Grx domain are important to the GR activity of TGR from E.granulosus [61]. Sec is thought to be essential for the GR activity of S. mansoni TGR [8]. However, Angelucci et al. [35] could measure very low GR activity (~0.8 %) in a truncated form of S. mansoni TGR where Sec-597 and Gly-598 are missing. This argument is supported by structural and modeling data study, which indicate that the putative GSSG binding pocket of TGR contains many positively charged residues and thus is more similar to GR than to TrxR suggesting that GSSG could receive electrons from the redox-active cysteine pair adjacent to FAD (Cys154/Cys159) in addition to the Cys28/Cys31 found in the Grx domain of TGR [35, 71]. Thus, it was proposed that whereas the majority of GSSG reduction occurs as the result of electrons transferred from the FAD to the neighboring Cys154/Cys159, then to the C-terminal Cys596/Sec597 of the other subunit and finally to Cys28/Cys31 in the N-terminal Grx domain of the first subunit, a small fraction of electrons might be directly transferred from the FAD site to GSSG [35, 71]. This is supported by recent analysis of a Cys28Ala/Cys31Ala mutant that retains ~4% of wild-type-TGR activity [53]. Others have found that in E. granulosus TGR lacking the Grx domain [61] and mammalian TGR with Sec614Ser mutation or truncated TGR Cys613Stop [10] lack any residual GR activity suggesting that GSSG reduction occurs exclusively by the Grx domain in these orthologs.
Hysteresis, the slow response of an enzyme to rapid changes in substrate or modifier concentrations, was first found in the GR activity of TGR from Taenia crassiceps (TcTGR) [37]. Later, hysteretic behavior was observed in E. granulosus TGR [61]. Hysteresis was initially found in enzymes associating with metabolic regulation; this effect can be caused by different mechanisms (e.g., isomerization of enzymes induced by ligand binding) [72, 73]. In the case of TGRs, at higher concentrations of GSSG (>100 μM), slow initial rates of GSSG reduction are followed by faster rates in the kinetic traces. The hysteretic effect in TGRs can be abolished by the addition of low concentrations of GSH, decreasing the concentrations of GSSG or increasing concentrations of enzymes [37, 61]. It has been suggested that the hysteretic effect is caused by glutathionylation by GSSG on two cysteine residues of E. granulosus TGR: Cys88 and Cys354. These cysteine residues appear to be involved in maintenance of structural integrity rather than catalysis but are not conserved in all TGR proteins known to be affected by hysteresis. Glutathionylation at Cys88 might prevent conformational changes associated with the transfer of reducing equivalents in the protein as this residue is close to the mobile linker of Grx-TrxR domains. However, neither glutathionylated Cys residue identified in E. granulosus TGR is conserved in S. mansoni TGR, which also displays hysteresis at high GSSG concentrations [34, 53], suggesting that either different Cys residues are involved in glutathionylation in orthologous proteins or that as yet unidentified, conserved Cys residues are modified in TGRs with hysteretic behavior. Studies on E. granulosus TGR could not exclude the possibility that the redox-active cysteine (and selenocysteine) residues were glutathionylated because these modified (seleno)cysteine residues may undergo deglutathionylation via E. granulosus TGR itself and were not identified by MS analysis [61]. Recent studies strongly suggest that glutathionylation of Cys31 in the Grx domain of S. mansoni TGR is responsible for hysteresis: a Cys31Ser variant of S. mansoni TGR has full GR activity (equal to wild-type S. mansoni TGR) but displays no hysteretic affects at high GSSG concentrations [53]. The ratio of GSH:GSSG in S. mansoni worms has been found to be between 18:1 [34] and 28:1 [74]. Under these conditions minimal if any hysteretic effects on TGR by GSSG would occur. Therefore, although it is possible that S. mansoni worms occasionally experience extreme oxidative stress, the physiological/in vivo relevance of the hysteretic behavior of TGR observed in vitro is unknown.
3. TGR as a drug target: Schistosomiasis
3.1. Schistosomiasis
Human schistosomiasis is caused by five species of schistosomes, of which S. mansoni and S. japonicum (infections resulting in intestinal schistosomiasis) and S. haematobium (infections resulting in urinary schistosomiasis) are most common. The disease remains a major neglected, poverty-related disease in many tropical areas. Schistosomes have a complex life cycle involving both freshwater snails and vertebrate hosts. Asexual reproduction in the snail produces cercariae, which emerge from infected snails and swim in water until they can penetrate the skin of people in contact with the water. Once in the body, the parasites turn into larvae and migrate to the liver where they become juvenile worms. They eventually develop into 1 to 2 cm-long adult male and female parasites, which pair and live together in human blood vessels for up to 30 years [75]. The female parasites release 200 to 2000 eggs per day over an average of 5 years. Many of the eggs are excreted in the feces or urine of the host, but some eggs remain trapped in body tissues causing disease symptoms such as bleeding and damage to organs as a result of the inflammatory response to the eggs. It is estimated that more than 200 million people are afflicted with schistosomiasis, with 779 million at risk of infection [76]. Schistosome infections result in 280,000 deaths annually and more than 20 million infected individuals experience high morbidity [77, 78]. Bladder cancer is common in advanced cases of urinary schistosomiasis. Recent analyses indicate that the disease burden due to schistosomiasis is much more significant in many individuals, even those with light infections, than previously estimated and is associated with significant anemia, chronic pain, diarrhea, reduced exercise tolerance, and malnutrition and to undermine productivity of farmers and impact children's growth and school performance in endemic areas [79]. Schistosome infections lead to the development of altered immune responses that may change susceptibility to other infections, including HIV/AIDS, malaria and tuberculosis, and vaccinations [80]. Furthermore, many individuals infected with schistosome are also co-infected with other worms (e.g., hook worms, Ascaris, filarial) and these co-infections can result in a synergistic effect on morbidity [81].
3.2 Schistosomiasis treatment
Current treatment of schistosomiasis relies exclusively on one drug, praziquantel (PZQ), an effective drug that is active against all schistosome species. By the mid-1980s, PZQ was widely used, replacing previous treatments including the highly toxic antimonials and oxamniquine [82]. In nearly 30 years of use in the field, it has been shown to be safe and effective, a single oral dose of 40–60 mg/kg is sufficient to achieve cure rates of 60–90% [82]. Unfortunately, people get reinfected and need to be retreated regularly; millions of people are treated annually with PZQ [83]. One notable failing of PZQ is its reduced efficacy against immature parasites relative to adult worms [84]. Although PZQ is effective, reliance on a single drug is a major concern because of the potential clinical development and spread of PZQ-resistant parasites. Even now patients who are not cured by multiple doses of PZQ have been identified from various locales, suggesting that resistance to the drug may already be present in the field [85-89]. Additionally, several in vitro and animal model studies have demonstrated resistance to PZQ [90]. There is neither a large body of field evidence that schistosomes are becoming less sensitive to PZQ nor sufficient research in this area to identify clinical resistance. However, given the wide clinical use of PZQ, drug-resistant parasites will eventually evolve. Other drugs are available, but they are more expensive, less effective, have unacceptable side effects and/or are effective on only one schistosome species [82]. Therefore, it is imperative to identify alternative drugs to ensure that PZQ resistance does not become a major health concern. Although the target of PZQ is not known with certainty [91, 92], TGR is not inhibited by PZQ [34] and therefore TGR inhibitors could potentially be used in combination therapies with PZQ.
3.3. Consequences of TGR inhibition
An important consideration in the rational design of new drugs is that the target should be required for pathogen persistence; several studies have determined that S. mansoni TGR is an essential enzyme for the survival of schistosome worms. Using double-stranded RNA to specifically silence S. mansoni TGR expression in schistosomula, the larval parasites, TGR activity was significantly reduced after three days and parasite survival was approximately eight percent after one additional day compared to control parasites that showed >95% survival [34]. Auranofin is a very potent inhibitor of mammalian TrxR [49] and S. mansoni TGR [8, 34] as well as TGR from the related parasites E. granulosus [61] and T. crassiceps [37]. A concentration of 10 μM auranofin led to a nearly 100% inhibition of TrxR and GR activities in S. mansoni worm homogenates after only 1 h and a 100% mortality of male and female worms after 9 h [34]. In addition, 5 μM auranofin also killed larval, skin and juvenile liver stage parasites within 10 h, whereas the mammalian myeloma cell line SP2/0 tolerated high concentrations of auranofin (100 μM) for up to 5 days [34]. Administration of a non-toxic dose of 6 mg/kg auranofin twice daily for 9 days to mice infected with S. mansoni resulted in an approximately 60% decrease in worm burden compared to control infected mice [34]. These results demonstrate the importance of S. mansoni TGR for parasite survival and efficient TGR inhibition is a promising strategy in the development of new antischistosome drugs. Differences in redox pathways between mammals and helminths [14] as well as biochemical differences between host and parasite pyridine nucleotide disulfide oxidoreductases [34] suggest that chemotherapy for flatworm infections based on selective inhibition of worm TGR should be feasible. In fact, two compounds, potassium antimonyl tartrate and oltipraz, which have previously been used in antischistosomal therapy have been shown to inhibit S. mansoni TGR in vitro [14]. However, these drugs are obsolete today due to severe adverse side effects [93, 94]. Nevertheless, high throughput screening, establishment of structure activity relationships and lead optimization in order to develop highly specific inhibitors for TGR has great potential for improving treatment of schistosome infections and perhaps infections caused by E.granulosus, T. crassiceps and other trematode and cestodes infections [61, 95, 96].
3.4. Identification of TGR inhibitors
Using resources available through the NIH Chemical Genomics Center Roadmap Molecular Libraries Initiative a quantitative high throughput screen [97] for TGR inhibitors was conducted. The Molecular Libraries Initiative addresses the problem of the limited interest of the pharmaceutical industry in the development of new therapies for rare and neglected tropical diseases by providing pharma-scale infrastructure and technologies to academic researchers searching for novel drug leads [98]. Williams, Simeonov and co-workers developed an assay for S. mansoni TGR suitable for a quantitative high throughput screen to identify novel and potent inhibitors of the S. mansoni redox cascade [99]. Monitoring NADPH consumption as a decrease in fluorescence was used to follow reaction progression (Scheme 1).
Scheme 1.

Of 71,028 compounds tested as 7- to 15-point concentrations in 1536-well plates, 39 compounds were active exhibiting clear concentration responses with IC50 values below 10 μM. The most interesting groups of compounds, oxadiazole-2-oxides and phosphinic amides, were analyzed in more detail for inhibition of TGR both in vitro and in S. mansoni worm extracts, as well as for effectiveness towards the parasite in culture and in a mouse model [100]. Furoxan (4-phenyl-1,2,5-oxadiazole-3-carbonitrile-2-oxide; Figure 4) was a potent inhibitor of TrxR and GR activities of TGR in worm homogenates and the most effective compound in killing cultured adult worms.
Figure 4.
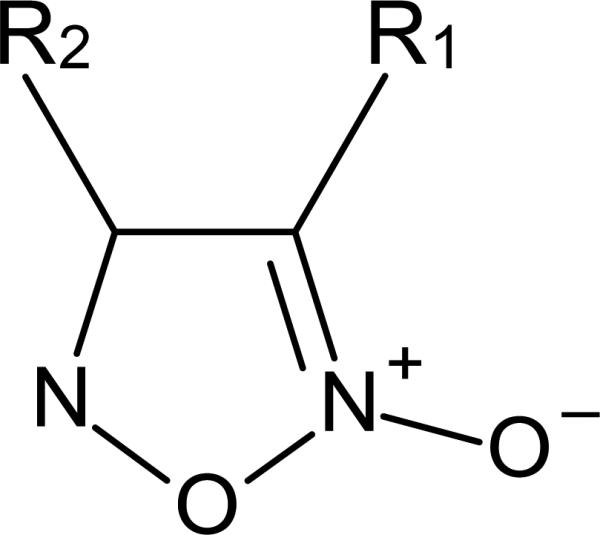
The oxadiazole-oxide pharmacophore. In furoxan (4-phenyl-1,2,5-oxadiazole-3-carbonitrile-2-oxide) R1 = carbonitrile (C≡N) and R2 = phenyl.
In addition, furoxan was toxic to all parasite stages, i.e. skin larvae, lung-stage schistosomula, juvenile liver worms as well as adult worms, including S. japonicum and S. haematobium. A mouse myeloma cell line (SP2/0) tolerated much higher concentrations of furoxan than the parasites. Treating S.mansoni-infected mice with 10 mg/kg furoxan i.p. once daily resulted in a tremendous decrease in worm burden and clearly reduced pathology for both early and late onset of treatment. Thus, this new drug lead was equal to or more effective than PZQ and artemether. Moreover, it surpasses all criteria of the World Health Organization set for new anti-schistosomal drug leads [101].
Intriguingly, the oxadiazole-2-oxide core of furoxan has been demonstrated to be an efficient NO donor [102, 103]. Thus, furoxan and other oxadiazole-2-oxides used in the study mentioned above were evaluated for the capacity of NO donation. NO can have antiparasitic effects [104-106] and therefore might be important for the worm killing activities. Only furoxan, the most potent anti-schistosomal analogue tested, showed significant NO production in the presence of TGR and NADPH. Hence, NO release was dependent on the catalytic activity of TGR and also essential for worm killing as furoxan in the presence of the NO scavenger C-PTIO was less effective [100].
Additional structure-activity relationship studies were performed using furoxan as a lead [107]. Analogues that lacked or regiochemically transposed the N-oxide did not release NO nor were they active against TGR or in worm killing assays. Analogues with a substitution of the carbonitrile group (R1 in Figure 4; -CH3, -CH2OH, -CHO, -COOH, or -CONH2) were all inactive in worm killing and most did not inhibit TGR. Thus, the 3-cyano-1,2,5-oxadiazole-2-oxide core was identified as the pharmacophore necessary for potent TGR inhibition and worm killing efficacy. Substitutions at the phenyl ring (R2 in Figure 4) revealed that electron-withdrawing moieties resulted in slight enhancements of potency. A similar effect was found when the phenyl ring was replaced by a furan or thiophene. Compounds containing two oxadiazole 2-oxide rings joined to a common phenyl or thiophene ring also had increased activity in TGR inhibition and worm killing. However, these ‘bis’ compounds also have increased cellular toxicity (Williams unpublished). Based on these studies it is clear that both effective inhibition of TGR and the ability to release NO were important for worm killing. Using a biotin switch assay [108, 109], it was shown that TGR catalysis of these compounds resulted in S- or Se-nitrosylation due to the liberated NO, which might eventually cause the loss of TGR activity. In addition, NO produced by TGR may result in the inactivation of bystander proteins increasing cellular stress. With a selection of promising furoxan analogues, specificity towards inhibition of TGR was tested by establishing the IC50 values of these compounds for S. mansoni TGR, human GR and rat TrxR1. Out of 31 compounds only three displayed considerable inhibition of GR and TrxR, thus demonstrating a highly selective behavior for most analogues towards TGR and the related important host enzymes. Evaluations of absorption, distribution, metabolism, excretion, and toxicity drug properties demonstrated a high variation of solubility, membrane permeability, microsomal stability and potential heart muscle toxicity among the compounds tested. Although none of the analogues displayed an excellent overall ADMET profile, this high variation within the group of compounds that all derived from the same pharmacophore promises the potential to modulate these properties by further substitution while maintaining the biological activity. Thus, through continued research a new, effective and orally available anti-schistosomal drug targeting TGR will hopefully be in our hands in several years.
Acknowledgements
This work was supported by National Institute of Allergy and Infectious Diseases Grant AI065622 (D.L.W.).
Abbreviations
- FAD
flavin adenine dinucleotide
- Grx
glutaredoxin
- GSH
glutathione
- GSSG
glutathione disulfide
- GR
glutathione disulfide reductase
- PZQ
praziquantel
- Sec
selenocysteine
- SECIS
Sec insertion sequence
- Trx
thioredoxin
- TrxR
thioredoxin reductase
- TGR
thioredoxin-glutathione reductase
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Arner ES. Focus on mammalian thioredoxin reductases--important selenoproteins with versatile functions, Biochim. Biophys. Acta. 2009;1790:495–526. doi: 10.1016/j.bbagen.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 2.Williams CH. Lipoamide Dehydrogenase, Glutathione Reductase, Thioredoxin reductase, and Mercuric ion reductase-A family of Flavoenzyme Transhydrogenases. CRC press, Inc; Boca Raton, FL: 1992. [Google Scholar]
- 3.Gromer S, Urig S, Becker K. The thioredoxin system--from science to clinic. Med. Res. Rev. 2004;24:40–89. doi: 10.1002/med.10051. [DOI] [PubMed] [Google Scholar]
- 4.Tamura T, Stadtman TC. A new selenoprotein from human lung adenocarcinoma cells: purification, properties, and thioredoxin reductase activity. Proc. Natl. Acad. Sci. U. S. A. 1996;93:1006–1011. doi: 10.1073/pnas.93.3.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gladyshev VN, Jeang KT, Stadtman TC. Selenocysteine, identified as the penultimate C-terminal residue in human T-cell thioredoxin reductase, corresponds to TGA in the human placental gene. Proc. Natl. Acad. Sci. U. S. A. 1996;93:6146–6151. doi: 10.1073/pnas.93.12.6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turanov AA, Su D, Gladyshev VN. Characterization of alternative cytosolic forms and cellular targets of mouse mitochondrial thioredoxin reductase. J. Biol. Chem. 2006;281:22953–22963. doi: 10.1074/jbc.M604326200. [DOI] [PubMed] [Google Scholar]
- 7.Agorio A, Chalar C, Cardozo S, Salinas G. Alternative mRNAs arising from trans-splicing code for mitochondrial and cytosolic variants of Echinococcus granulosus thioredoxin Glutathione reductase. J. Biol. Chem. 2003;278:12920–12928. doi: 10.1074/jbc.M209266200. [DOI] [PubMed] [Google Scholar]
- 8.Alger HM, Williams DL. The disulfide redox system of Schistosoma mansoni and the importance of a multifunctional enzyme, thioredoxin glutathione reductase. Mol. Biochem. Parasitol. 2002;121:129–139. doi: 10.1016/s0166-6851(02)00031-2. [DOI] [PubMed] [Google Scholar]
- 9.Sun QA, Kirnarsky L, Sherman S, Gladyshev VN. Selenoprotein oxidoreductase with specificity for thioredoxin and glutathione systems. Proc. Natl. Acad. Sci. U. S. A. 2001;98:3673–3678. doi: 10.1073/pnas.051454398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sun QA, Su D, Novoselov SV, Carlson BA, Hatfield DL, Gladyshev VN. Reaction mechanism and regulation of mammalian thioredoxin/glutathione reductase. Biochemistry. 2005;44:14528–14537. doi: 10.1021/bi051321w. [DOI] [PubMed] [Google Scholar]
- 11.Su D, Novoselov SV, Sun QA, Moustafa ME, Zhou Y, Oko R, Hatfield DL, Gladyshev VN. Mammalian selenoprotein thioredoxin-glutathione reductase. Roles in disulfide bond formation and sperm maturation. J. Biol. Chem. 2005;280:26491–26498. doi: 10.1074/jbc.M503638200. [DOI] [PubMed] [Google Scholar]
- 12.Park UH, Han HS, Um E, An XH, Kim EJ, Um SJ. Redox regulation of transcriptional activity of retinoic acid receptor by thioredoxin glutathione reductase (TGR) Biochem. Biophys. Res. Commun. 2009;390:241–246. doi: 10.1016/j.bbrc.2009.09.097. [DOI] [PubMed] [Google Scholar]
- 13.Kanzok SM, Fechner A, Bauer H, Ulschmid JK, Muller HM, Botella-Munoz J, Schneuwly S, Schirmer R, Becker K. Substitution of the thioredoxin system for glutathione reductase in Drosophila melanogaster. Science. 2001;291:643–646. doi: 10.1126/science.291.5504.643. [DOI] [PubMed] [Google Scholar]
- 14.Salinas G, Selkirk ME, Chalar C, Maizels RM, Fernandez C. Linked thioredoxin-glutathione systems in platyhelminths. Trends Parasitol. 2004;20:340–346. doi: 10.1016/j.pt.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 15.Otero L, Bonilla M, Protasio AV, Fernandez C, Gladyshev VN, Salinas G. Thioredoxin and glutathione systems differ in parasitic and free-living platyhelminths. BMC Genomics. 2010;11:237. doi: 10.1186/1471-2164-11-237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Johansson L, Gafvelin G, Arner ES. Selenocysteine in proteins-properties and biotechnological use. Biochim. Biophys. Acta. 2005;1726:1–13. doi: 10.1016/j.bbagen.2005.05.010. [DOI] [PubMed] [Google Scholar]
- 17.Hondal RJ, Ruggles EL. Differing views of the role of selenium in thioredoxin reductase. Amino Acids. 2010 doi: 10.1007/s00726-010-0494-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Witte AB, Anestal K, Jerremalm E, Ehrsson H, Arner ES. Inhibition of thioredoxin reductase but not of glutathione reductase by the major classes of alkylating and platinum-containing anticancer compounds. Free Radic. Biol. Med. 2005;39:696–703. doi: 10.1016/j.freeradbiomed.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 19.Kryukov GV, Castellano S, Novoselov SV, Lobanov AV, Zehtab O, Guigo R, Gladyshev VN. Characterization of mammalian selenoproteomes. Science. 2003;300:1439–1443. doi: 10.1126/science.1083516. [DOI] [PubMed] [Google Scholar]
- 20.Kryukov GV, Kryukov VM, Gladyshev VN. New mammalian selenocysteine-containing proteins identified with an algorithm that searches for selenocysteine insertion sequence elements. J. Biol. Chem. 1999;274:33888–33897. doi: 10.1074/jbc.274.48.33888. [DOI] [PubMed] [Google Scholar]
- 21.Maiorino M, Pierce R, Flohe L. Product of the Schistosoma mansoni glutathione peroxidase gene is a selenium containing phospholipid hydroperoxide glutathione peroxidase (PHGPx) sharing molecular weight and substrate specificity with its mammalian counterpart. Biomed. Environ. Sci. 1997;10:209–213. [PubMed] [Google Scholar]
- 22.Maiorino M, Roche C, Kiess M, Koenig K, Gawlik D, Matthes M, Naldini E, Pierce R, Flohe L. A selenium-containing phospholipid-hydroperoxide glutathione peroxidase in Schistosoma mansoni. Eur. J. Biochem. 1996;238:838–844. doi: 10.1111/j.1432-1033.1996.0838w.x. [DOI] [PubMed] [Google Scholar]
- 23.Williams DL, Pierce RJ, Cookson E, Capron A. Molecular cloning and sequencing of glutathione peroxidase from Schistosoma mansoni. Mol. Biochem. Parasitol. 1992;52:127–130. doi: 10.1016/0166-6851(92)90042-i. [DOI] [PubMed] [Google Scholar]
- 24.Taskov K, Chapple C, Kryukov GV, Castellano S, Lobanov AV, Korotkov KV, Guigo R, Gladyshev VN. Nematode selenoproteome: the use of the selenocysteine insertion system to decode one codon in an animal genome? Nucleic Acids Res. 2005;33:2227–2238. doi: 10.1093/nar/gki507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lobanov AV, Hatfield DL, Gladyshev VN. Selenoproteinless animals: selenophosphate synthetase SPS1 functions in a pathway unrelated to selenocysteine biosynthesis. Protein Sci. 2008;17:176–182. doi: 10.1110/ps.073261508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee BJ, Worland PJ, Davis JN, Stadtman TC, Hatfield DL. Identification of a selenocysteyl-tRNA(Ser) in mammalian cells that recognizes the nonsense codon. UGA, J. Biol. Chem. 1989;264:9724–9727. [PubMed] [Google Scholar]
- 27.Berry MJ, Banu L, Chen YY, Mandel SJ, Kieffer JD, Harney JW, Larsen PR. Recognition of UGA as a selenocysteine codon in type I deiodinase requires sequences in the 3' untranslated region. Nature. 1991;353:273–276. doi: 10.1038/353273a0. [DOI] [PubMed] [Google Scholar]
- 28.Berry MJ, Banu L, Harney JW, Larsen PR. Functional characterization of the eukaryotic SECIS elements which direct selenocysteine insertion at UGA codons. EMBO J. 1993;12:3315–3322. doi: 10.1002/j.1460-2075.1993.tb06001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krol A. Evolutionarily different RNA motifs and RNA-protein complexes to achieve selenoprotein synthesis. Biochimie. 2002;84:765–774. doi: 10.1016/s0300-9084(02)01405-0. [DOI] [PubMed] [Google Scholar]
- 30.Papp LV, Lu J, Holmgren A, Khanna KK. From selenium to selenoproteins: synthesis, identity, and their role in human health. Antioxid. Redox Signal. 2007;9:775–806. doi: 10.1089/ars.2007.1528. [DOI] [PubMed] [Google Scholar]
- 31.Xu XM, Carlson BA, Mix H, Zhang Y, Saira K, Glass RS, Berry MJ, Gladyshev VN, Hatfield DL. Biosynthesis of selenocysteine on its tRNA in eukaryotes. PLoS Biol. 2007;5:e4. doi: 10.1371/journal.pbio.0050004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Howard MT, Aggarwal G, Anderson CB, Khatri S, Flanigan KM, Atkins JF. Recoding elements located adjacent to a subset of eukaryal selenocysteine-specifying UGA codons. EMBO J. 2005;24:1596–1607. doi: 10.1038/sj.emboj.7600642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gerashchenko MV, Su D, Gladyshev VN. CUG start codon generates thioredoxin/glutathione reductase isoforms in mouse testes. J. Biol. Chem. 2010;285:4595–4602. doi: 10.1074/jbc.M109.070532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuntz AN, Davioud-Charvet E, Sayed AA, Califf LL, Dessolin J, Arner ES, Williams DL. Thioredoxin glutathione reductase from Schistosoma mansoni: an essential parasite enzyme and a key drug target. PLoS Med. 2007;4:e206. doi: 10.1371/journal.pmed.0040206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Angelucci F, Miele AE, Boumis G, Dimastrogiovanni D, Brunori M, Bellelli A. Glutathione reductase and thioredoxin reductase at the crossroad: the structure of Schistosoma mansoni thioredoxin glutathione reductase. Proteins. 2008;72:936–945. doi: 10.1002/prot.21986. [DOI] [PubMed] [Google Scholar]
- 36.Angelucci F, Sayed AA, Williams DL, Boumis G, Brunori M, Dimastrogiovanni D, Miele AE, Pauly F, Bellelli A. Inhibition of Schistosoma mansoni thioredoxin-glutathione reductase by auranofin: structural and kinetic aspects. J. Biol. Chem. 2009;284:28977–28985. doi: 10.1074/jbc.M109.020701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rendon JL, del Arenal IP, Guevara-Flores A, Uribe A, Plancarte A, Mendoza-Hernandez G. Purification, characterization and kinetic properties of the multifunctional thioredoxin-glutathione reductase from Taenia crassiceps metacestode (cysticerci) Mol. Biochem. Parasitol. 2004;133:61–69. doi: 10.1016/j.molbiopara.2003.09.003. [DOI] [PubMed] [Google Scholar]
- 38.Savvides SN, Karplus PA. Kinetics and crystallographic analysis of human glutathione reductase in complex with a xanthene inhibitor. J. Biol. Chem. 1996;271:8101–8107. doi: 10.1074/jbc.271.14.8101. [DOI] [PubMed] [Google Scholar]
- 39.Arscott LD, Veine DM, Williams CH., Jr Mixed disulfide with glutathione as an intermediate in the reaction catalyzed by glutathione reductase from yeast and as a major form of the enzyme in the cell. Biochemistry. 2000;39:4711–4721. doi: 10.1021/bi9926431. [DOI] [PubMed] [Google Scholar]
- 40.Rietveld P, Arscott LD, Berry A, Scrutton NS, Deonarain MP, Perham RN, Williams CH., Jr Reductive and oxidative half-reactions of glutathione reductase from Escherichia coli. Biochemistry. 1994;33:13888–13895. doi: 10.1021/bi00250a043. [DOI] [PubMed] [Google Scholar]
- 41.Karplus PA, Pai EF, Schulz GE. A crystallographic study of the glutathione binding site of glutathione reductase at 0.3-nm resolution. Eur. J. Biochem. 1989;178:693–703. doi: 10.1111/j.1432-1033.1989.tb14500.x. [DOI] [PubMed] [Google Scholar]
- 42.Pai EF, Schulz GE. The catalytic mechanism of glutathione reductase as derived from x-ray diffraction analyses of reaction intermediates. J. Biol. Chem. 1983;258:1752–1757. [PubMed] [Google Scholar]
- 43.Arscott LD, Thorpe C, Williams CH., Jr Glutathione reductase from yeast. Differential reactivity of the nascent thiols in two-electron reduced enzyme and properties of a monoalkylated derivative. Biochemistry. 1981;20:1513–1520. doi: 10.1021/bi00509a016. [DOI] [PubMed] [Google Scholar]
- 44.Gromer S, Johansson L, Bauer H, Arscott LD, Rauch S, Ballou DP, Williams CH, Jr, Schirmer RH, Arner ES. Active sites of thioredoxin reductases: why selenoproteins? Proc. Natl. Acad. Sci. U. S. A. 2003;100:12618–12623. doi: 10.1073/pnas.2134510100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bauer H, Massey V, Arscott LD, Schirmer RH, Ballou DP, Williams CH., Jr The mechanism of high Mr thioredoxin reductase from Drosophila melanogaster. J. Biol. Chem. 2003;278:33020–33028. doi: 10.1074/jbc.M303762200. [DOI] [PubMed] [Google Scholar]
- 46.Zhong L, Arner ES, Holmgren A. Structure and mechanism of mammalian thioredoxin reductase: the active site is a redox-active selenolthiol/selenenylsulfide formed from the conserved cysteine-selenocysteine sequence. Proc. Natl. Acad. Sci. U. S. A. 2000;97:5854–5859. doi: 10.1073/pnas.100114897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Williams CH, Arscott LD, Muller S, Lennon BW, Ludwig ML, Wang PF, Veine DM, Becker K, Schirmer RH. Thioredoxin reductase two modes of catalysis have evolved. Eur. J. Biochem. 2000;267:6110–6117. doi: 10.1046/j.1432-1327.2000.01702.x. [DOI] [PubMed] [Google Scholar]
- 48.Wang PF, Arscott LD, Gilberger TW, Muller S, Williams CH., Jr Thioredoxin reductase from Plasmodium falciparum: evidence for interaction between the C-terminal cysteine residues and the active site disulfide-dithiol. Biochemistry. 1999;38:3187–3196. doi: 10.1021/bi982674g. [DOI] [PubMed] [Google Scholar]
- 49.Gromer S, Arscott LD, Williams CH, Jr, Schirmer RH, Becker K. Human placenta thioredoxin reductase. Isolation of the selenoenzyme, steady state kinetics, and inhibition by therapeutic gold compounds. J. Biol. Chem. 1998;273:20096–20101. doi: 10.1074/jbc.273.32.20096. [DOI] [PubMed] [Google Scholar]
- 50.Arscott LD, Gromer S, Schirmer RH, Becker K, Williams CH., Jr The mechanism of thioredoxin reductase from human placenta is similar to the mechanisms of lipoamide dehydrogenase and glutathione reductase and is distinct from the mechanism of thioredoxin reductase from Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 1997;94:3621–3626. doi: 10.1073/pnas.94.8.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Arner ES, Zhong L, Holmgren A. Preparation and assay of mammalian thioredoxin and thioredoxin reductase. Methods Enzymol. 1999;300:226–239. doi: 10.1016/s0076-6879(99)00129-9. [DOI] [PubMed] [Google Scholar]
- 52.Xia L, Nordman T, Olsson JM, Damdimopoulos A, Bjorkhem-Bergman L, Nalvarte I, Eriksson LC, Arner ES, Spyrou G, Bjornstedt M. The mammalian cytosolic selenoenzyme thioredoxin reductase reduces ubiquinone. A novel mechanism for defense against oxidative stress. J. Biol. Chem. 2003;278:2141–2146. doi: 10.1074/jbc.M210456200. [DOI] [PubMed] [Google Scholar]
- 53.Huang HH, Day L, Cass CL, Ballou DP, Williams CH, Jr., Williams DL. Investigations of the catalytic mechanism of thioredoxin glutathione reductase (TGR) from Schistosoma mansoni. Biochemistry. doi: 10.1021/bi200107n., Just Accepted Manuscript. PMID: 21630672; DOI: 10.1021/bi200107n Publication Date (Web): June 1, 2011.
- 54.Smith AD, Guidry CA, Morris VC, Levander OA. Aurothioglucose inhibits murine thioredoxin reductase activity in vivo. J. Nutr. 1999;129:194–198. doi: 10.1093/jn/129.1.194. [DOI] [PubMed] [Google Scholar]
- 55.Rigobello MP, Scutari G, Folda A, Bindoli A. Mitochondrial thioredoxin reductase inhibition by gold(I) compounds and concurrent stimulation of permeability transition and release of cytochrome c. Biochem. Pharmacol. 2004;67:689–696. doi: 10.1016/j.bcp.2003.09.038. [DOI] [PubMed] [Google Scholar]
- 56.Lu J, Chew EH, Holmgren A. Targeting thioredoxin reductase is a basis for cancer therapy by arsenic trioxide. Proc. Natl. Acad. Sci. U. S. A. 2007;104:12288–12293. doi: 10.1073/pnas.0701549104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Cenas N, Nivinskas H, Anusevicius Z, Sarlauskas J, Lederer F, Arner ES. Interactions of quinones with thioredoxin reductase: a challenge to the antioxidant role of the mammalian selenoprotein. J. Biol. Chem. 2004;279:2583–2592. doi: 10.1074/jbc.M310292200. [DOI] [PubMed] [Google Scholar]
- 58.Cenas N, Prast S, Nivinskas H, Sarlauskas J, Arner ES. Interactions of nitroaromatic compounds with the mammalian selenoprotein thioredoxin reductase and the relation to induction of apoptosis in human cancer cells. J. Biol. Chem. 2006;281:5593–5603. doi: 10.1074/jbc.M511972200. [DOI] [PubMed] [Google Scholar]
- 59.Mieyal JJ, Gallogly MM, Qanungo S, Sabens EA, Shelton MD. Molecular mechanisms and clinical implications of reversible protein S-glutathionylation. Antioxid. Redox Signal. 2008;10:1941–1988. doi: 10.1089/ars.2008.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Protein S-glutathionylation: a regulatory device from bacteria to humans. Trends Biochem. Sci. 2009;34:85–96. doi: 10.1016/j.tibs.2008.11.002. [DOI] [PubMed] [Google Scholar]
- 61.Bonilla M, Denicola A, Novoselov SV, Turanov AA, Protasio A, Izmendi D, Gladyshev VN, Salinas G. Platyhelminth mitochondrial and cytosolic redox homeostasis is controlled by a single thioredoxin glutathione reductase and dependent on selenium and glutathione. J. Biol. Chem. 2008;283:17898–17907. doi: 10.1074/jbc.M710609200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bushweller JH, Aslund F, Wuthrich K, Holmgren A. Structural and functional characterization of the mutant Escherichia coli glutaredoxin (C14----S) and its mixed disulfide with glutathione. Biochemistry. 1992;31:9288–9293. doi: 10.1021/bi00153a023. [DOI] [PubMed] [Google Scholar]
- 63.Fernandes AP, Holmgren A. Glutaredoxins: glutathione-dependent redox enzymes with functions far beyond a simple thioredoxin backup system. Antioxid. Redox Signal. 2004;6:63–74. doi: 10.1089/152308604771978354. [DOI] [PubMed] [Google Scholar]
- 64.Saaranen MJ, Salo KE, Latva-Ranta MK, Kinnula VL, Ruddock LW. The C-terminal active site cysteine of Escherichia coli glutaredoxin 1 determines the glutathione specificity of the second step of peptide deglutathionylation. Antioxid. Redox Signal. 2009;11:1819–1828. doi: 10.1089/ars.2008.2387. [DOI] [PubMed] [Google Scholar]
- 65.Johansson C, Lillig CH, Holmgren A. Human mitochondrial glutaredoxin reduces S-glutathionylated proteins with high affinity accepting electrons from either glutathione or thioredoxin reductase. J. Biol. Chem. 2004;279:7537–7543. doi: 10.1074/jbc.M312719200. [DOI] [PubMed] [Google Scholar]
- 66.Yang Y, Jao S, Nanduri S, Starke DW, Mieyal JJ, Qin J. Reactivity of the human thioltransferase (glutaredoxin) C7S, C25S, C78S, C82S mutant and NMR solution structure of its glutathionyl mixed disulfide intermediate reflect catalytic specificity. Biochemistry. 1998;37:17145–17156. doi: 10.1021/bi9806504. [DOI] [PubMed] [Google Scholar]
- 67.Bonilla M, Denicola A, Marino SM, Gladyshev VN, Salinas G. Linked thioredoxinglutathione systems in platyhelminth parasites: Alternative pathways for glutathione reduction and deglutathionylation. J. Biol. Chem. 2011;286:4959–4967. doi: 10.1074/jbc.M110.170761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Discola KF, de Oliveira MA, Rosa Cussiol JR, Monteiro G, Barcena JA, Porras P, Padilla CA, Guimaraes BG, Netto LE. Structural aspects of the distinct biochemical properties of glutaredoxin 1 and glutaredoxin 2 from Saccharomyces cerevisiae. J. Mol. Biol. 2009;385:889–901. doi: 10.1016/j.jmb.2008.10.055. [DOI] [PubMed] [Google Scholar]
- 69.Sandalova T, Zhong L, Lindqvist Y, Holmgren A, Schneider G. Three-dimensional structure of a mammalian thioredoxin reductase: implications for mechanism and evolution of a selenocysteine-dependent enzyme. Proc. Natl. Acad. Sci. U. S. A. 2001;98:9533–9538. doi: 10.1073/pnas.171178698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Urig S, Lieske J, Fritz-Wolf K, Irmler A, Becker K. Truncated mutants of human thioredoxin reductase 1 do not exhibit glutathione reductase activity. FEBS Lett. 2006;580:3595–3600. doi: 10.1016/j.febslet.2006.05.038. [DOI] [PubMed] [Google Scholar]
- 71.Sharma M, Khanna S, Bulusu G, Mitra A. Comparative modeling of thioredoxin glutathione reductase from Schistosoma mansoni: a multifunctional target for antischistosomal therapy. J. Mol. Graph. Model. 2009;27:665–675. doi: 10.1016/j.jmgm.2008.10.009. [DOI] [PubMed] [Google Scholar]
- 72.Frieden C. Slow transitions and hysteretic behavior in enzymes. Annu. Rev. Biochem. 1979;48:471–489. doi: 10.1146/annurev.bi.48.070179.002351. [DOI] [PubMed] [Google Scholar]
- 73.Frieden C. Kinetic aspects of regulation of metabolic processes. The hysteretic enzyme concept. J. Biol. Chem. 1970;245:5788–5799. [PubMed] [Google Scholar]
- 74.Mkoji GM, Smith JM, Prichard RK. Glutathione redox state, lipid peroxide levels, and activities of glutathione enzymes in oltipraz-treated adult Schistosoma mansoni. Biochem. Pharmacol. 1989;38:4307–4313. doi: 10.1016/0006-2952(89)90530-3. [DOI] [PubMed] [Google Scholar]
- 75.Vermund SH, Bradley DJ, Ruiz-Tiben E. Survival of Schistosoma mansoni in the human host: estimates from a community-based prospective study in Puerto Rico. Am. J. Trop. Med. Hyg. 1983;32:1040–1048. doi: 10.4269/ajtmh.1983.32.1040. [DOI] [PubMed] [Google Scholar]
- 76.Steinmann P, Keiser J, Bos R, Tanner M, Utzinger J. Schistosomiasis and water resources development: systematic review, meta-analysis, and estimates of people at risk. Lancet Infect. Dis. 2006;6:411–425. doi: 10.1016/S1473-3099(06)70521-7. [DOI] [PubMed] [Google Scholar]
- 77.van der Werf MJ, de Vlas SJ, Brooker S, Looman CW, Nagelkerke NJ, Habbema JD, Engels D. Quantification of clinical morbidity associated with schistosome infection in sub-Saharan Africa. Acta Trop. 2003;86:125–139. doi: 10.1016/s0001-706x(03)00029-9. [DOI] [PubMed] [Google Scholar]
- 78.King CH, Dangerfield-Cha M. The unacknowledged impact of chronic schistosomiasis. Chronic Illn. 2008;4:65–79. doi: 10.1177/1742395307084407. [DOI] [PubMed] [Google Scholar]
- 79.King CH. Parasites and poverty: the case of schistosomiasis. Acta Trop. 2010;113:95–104. doi: 10.1016/j.actatropica.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hotez PJ, Fenwick A, Savioli L, Molyneux DH. Rescuing the bottom billion through control of neglected tropical diseases. Lancet. 2009;373:1570–1575. doi: 10.1016/S0140-6736(09)60233-6. [DOI] [PubMed] [Google Scholar]
- 81.Ezeamama AE, McGarvey ST, Acosta LP, Zierler S, Manalo DL, Wu HW, Kurtis JD, Mor V, Olveda RM, Friedman JF. The synergistic effect of concomitant schistosomiasis, hookworm, and trichuris infections on children's anemia burden. PLoS Negl Trop. Dis. 2008;2:e245. doi: 10.1371/journal.pntd.0000245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Caffrey CR. Chemotherapy of schistosomiasis: present and future. Curr. Opin. Chem. Biol. 2007;11:433–439. doi: 10.1016/j.cbpa.2007.05.031. [DOI] [PubMed] [Google Scholar]
- 83.Fenwick A, Webster JP, Bosque-Oliva E, Blair L, Fleming FM, Zhang Y, Garba A, Stothard JR, Gabrielli AF, Clements AC, Kabatereine NB, Toure S, Dembele R, Nyandindi U, Mwansa J, Koukounari A. The Schistosomiasis Control Initiative (SCI): rationale, development and implementation from 2002-2008. Parasitology. 2009;136:1719–1730. doi: 10.1017/S0031182009990400. [DOI] [PubMed] [Google Scholar]
- 84.Sabah AA, Fletcher C, Webbe G, Doenhoff MJ. Schistosoma mansoni: chemotherapy of infections of different ages. Exp. Parasitol. 1986;61:294–303. doi: 10.1016/0014-4894(86)90184-0. [DOI] [PubMed] [Google Scholar]
- 85.Fallon PG, Sturrock RF, Niang AC, Doenhoff MJ. Short report: diminished susceptibility to praziquantel in a Senegal isolate of Schistosoma mansoni. Am. J. Trop. Med. Hyg. 1995;53:61–62. [PubMed] [Google Scholar]
- 86.Stelma FF, Talla I, Sow S, Kongs A, Niang M, Polman K, Deelder AM, Gryseels B. Efficacy and side effects of praziquantel in an epidemic focus of Schistosoma mansoni. Am. J. Trop. Med. Hyg. 1995;53:167–170. doi: 10.4269/ajtmh.1995.53.167. [DOI] [PubMed] [Google Scholar]
- 87.Ismail M, Metwally A, Farghaly A, Bruce J, Tao LF, Bennett JL. Characterization of isolates of Schistosoma mansoni from Egyptian villagers that tolerate high doses of praziquantel. Am. J. Trop. Med. Hyg. 1996;55:214–218. doi: 10.4269/ajtmh.1996.55.214. [DOI] [PubMed] [Google Scholar]
- 88.Guisse F, Polman K, Stelma FF, Mbaye A, Talla I, Niang M, Deelder AM, Ndir O, Gryseels B. Therapeutic evaluation of two different dose regimens of praziquantel in a recent Schistosoma mansoni focus in Northern Senegal. Am. J. Trop. Med. Hyg. 1997;56:511–514. doi: 10.4269/ajtmh.1997.56.511. [DOI] [PubMed] [Google Scholar]
- 89.Melman SD, Steinauer ML, Cunningham C, Kubatko LS, Mwangi IN, Wynn NB, Mutuku MW, Karanja DM, Colley DG, Black CL, Secor WE, Mkoji GM, Loker ES. Reduced susceptibility to praziquantel among naturally occurring Kenyan isolates of Schistosoma mansoni. PLoS Negl Trop. Dis. 2009;3:e504. doi: 10.1371/journal.pntd.0000504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Botros SS, Bennett JL. Praziquantel resistance. Expert Opinion on Drug Discovery. 2007;2:S35–S40. doi: 10.1517/17460441.2.S1.S35. [DOI] [PubMed] [Google Scholar]
- 91.Doenhoff MJ, Cioli D, Utzinger J. Praziquantel: mechanisms of action, resistance and new derivatives for schistosomiasis. Curr. Opin. Infect. Dis. 2008;21:659–667. doi: 10.1097/QCO.0b013e328318978f. [DOI] [PubMed] [Google Scholar]
- 92.Jeziorski MC, Greenberg RM. Voltage-gated calcium channel subunits from platyhelminths: potential role in praziquantel action. Int. J. Parasitol. 2006;36:625–632. doi: 10.1016/j.ijpara.2006.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Davis A. Recent advances in schistosomiasis. Q. J. Med. 1986;58:95–110. [PubMed] [Google Scholar]
- 94.Katz N, Coelho PM. Clinical therapy of schistosomiasis mansoni: the Brazilian contribution. Acta Trop. 2008;108:72–78. doi: 10.1016/j.actatropica.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 95.Cioli D, Valle C, Angelucci F, Miele AE. Will new antischistosomal drugs finally emerge? Trends Parasitol. 2008;24:379–382. doi: 10.1016/j.pt.2008.05.006. [DOI] [PubMed] [Google Scholar]
- 96.Martinez-Gonzalez JJ, Guevara-Flores A, Alvarez G, Rendon-Gomez JL, Del Arenal IP. In vitro killing action of auranofin on Taenia crassiceps metacestode (cysticerci) and inactivation of thioredoxin-glutathione reductase (TGR) Parasitol. Res. 2010 doi: 10.1007/s00436-010-1867-1. [DOI] [PubMed] [Google Scholar]
- 97.Inglese J, Auld DS, Jadhav A, Johnson RL, Simeonov A, Yasgar A, Zheng W, Austin CP. Quantitative high-throughput screening: a titration-based approach that efficiently identifies biological activities in large chemical libraries. Proc. Natl. Acad. Sci. U. S. A. 2006;103:11473–11478. doi: 10.1073/pnas.0604348103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Austin CP, Brady LS, Insel TR, Collins FS. NIH Molecular Libraries Initiative. Science. 2004;306:1138–1139. doi: 10.1126/science.1105511. [DOI] [PubMed] [Google Scholar]
- 99.Simeonov A, Jadhav A, Sayed AA, Wang Y, Nelson ME, Thomas CJ, Inglese J, Williams DL, Austin CP. Quantitative high-throughput screen identifies inhibitors of the Schistosoma mansoni redox cascade. PLoS Negl Trop. Dis. 2008;2:e127. doi: 10.1371/journal.pntd.0000127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Sayed AA, Simeonov A, Thomas CJ, Inglese J, Austin CP, Williams DL. Identification of oxadiazoles as new drug leads for the control of schistosomiasis. Nat. Med. 2008;14:407–412. doi: 10.1038/nm1737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Nwaka S, Hudson A. Innovative lead discovery strategies for tropical diseases. Nat. Rev. Drug Discov. 2006;5:941–955. doi: 10.1038/nrd2144. [DOI] [PubMed] [Google Scholar]
- 102.Medana C, Ermondi G, Fruttero R, Di Stilo A, Ferretti C, Gasco A. Furoxans as nitric oxide donors. 4-Phenyl-3-furoxancarbonitrile: thiol-mediated nitric oxide release and biological evaluation. J. Med. Chem. 1994;37:4412–4416. doi: 10.1021/jm00051a020. [DOI] [PubMed] [Google Scholar]
- 103.Gasco A, Fruttero R, Sorba G, Di Stilo A, Calvino R. NO donors: Focus on furoxan derivatives. Pure and Applied Chemistry. 2004;76:973–981. [Google Scholar]
- 104.Colasanti M, Gradoni L, Mattu M, Persichini T, Salvati L, Venturini G, Ascenzi P. Molecular bases for the anti-parasitic effect of NO (Review) Int. J. Mol. Med. 2002;9:131–134. [PubMed] [Google Scholar]
- 105.Brunet LR. Nitric oxide in parasitic infections. Int. Immunopharmacol. 2001;1:1457–1467. doi: 10.1016/s1567-5769(01)00090-x. [DOI] [PubMed] [Google Scholar]
- 106.Rivero A. Nitric oxide: an antiparasitic molecule of invertebrates. Trends Parasitol. 2006;22:219–225. doi: 10.1016/j.pt.2006.02.014. [DOI] [PubMed] [Google Scholar]
- 107.Rai G, Sayed AA, Lea WA, Luecke HF, Chakrapani H, Prast-Nielsen S, Jadhav A, Leister W, Shen M, Inglese J, Austin CP, Keefer L, Arner ES, Simeonov A, Maloney DJ, Williams DL, Thomas CJ. Structure mechanism insights and the role of nitric oxide donation guide the development of oxadiazole-2-oxides as therapeutic agents against schistosomiasis. J. Med. Chem. 2009;52:6474–6483. doi: 10.1021/jm901021k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jaffrey SR, Fang M, Snyder SH. Nitrosopeptide mapping: a novel methodology reveals snitrosylation of dexras1 on a single cysteine residue. Chem. Biol. 2002;9:1329–1335. doi: 10.1016/s1074-5521(02)00293-4. [DOI] [PubMed] [Google Scholar]
- 109.Jaffrey SR, Snyder SH. The biotin switch method for the detection of S-nitrosylated proteins. Sci. STKE. 2001:pl1. doi: 10.1126/stke.2001.86.pl1. 2001. [DOI] [PubMed] [Google Scholar]
- 110.Oke TT, Moskovitz J, Williams DL. Characterization of the methionine sulfoxide reductases of Schistosoma mansoni. J. Parasitol. 2009;95:1421–1428. doi: 10.1645/GE-2062.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Squires JE, Berry MJ. Eukaryotic selenoprotein synthesis: mechanistic insight incorporating new factors and new functions for old factors. IUBMB Life. 2008;60:232–235. doi: 10.1002/iub.38. [DOI] [PubMed] [Google Scholar]