

Abstract
Selenium, in the form of selenocysteine, is a critical component of some major redox-regulating enzymes, including thioredoxin reductase (TrxR) and glutathione peroxidase (Gpx). TrxR has emerged as an anticancer target for drug development due to its elevated expression level in many aggressive human tumors. Acylfulvenes (AFs) are semisynthetic derivatives of the natural product illudin S, and display improved cytotoxic selectivity profiles. AF and illudin S alkylate cellular macromolecules. Compared to AFs, illudin S more readily reacts with thiol-containing small molecules such as cysteine, glutathione and cysteine-containing peptides. However, a previous study indicates the reactivity of AFs and illudin S with glutathione reductase, a thiol-containing enzyme, are inversely correlated with reactivity toward small molecule thiols. In this study, we investigate mechanistic aspects underlying the enzymatic and cellular effects of the AFs and illudin S on thioredoxin reductase. Both AF and HMAF were found to inhibit mammalian TrxR in the low- to sub-micromolar range, but illudin S was significantly less potent. TrxR inhibition by AFs was shown to be irreversible, concentration- and time-dependent, and mediated by alkylation of C-terminus active site Sec/Cys residues. In contrast, neither AFs nor illudin S inhibit Gpx, demonstrating that enzyme structure-specific small molecule interactions have a significant influence over the inherent reactivity of the Sec residue. In human cancer cells, TrxR activity can be inhibited by low micromolar concentrations of all three drugs. Finally, it was demonstrated that preconditioning cells by addition of selenite to the cell culture media results in an enhancement in cell sensitivity towards AFs. These data suggest potential strategies for increasing drug activity by combination treatments that promote selenium enzyme activity.
Keywords: thioredoxin reductase, glutathione peroxidase, illudin S, acylfulvene, redox enzyme inhibition
Introduction
Selenium (Se)1 is a biologically important trace element; low levels are required for cell growth and high concentrations are cytotoxic (1). Human blood Se levels are low to sub μM and vary with regional dietary Se content, as well as health status. Se homeostasis can inhibit tumor cell invasion and decrease the risk of some human cancers, such as prostate, lung, and colon cancer (2, 3). Se is incorporated into proteins in the form of selenocysteine (Sec) (4), the naturally occurring selenium analog of cysteine (Cys), regarded as the 21st amino acid (5, 6). As such, Se acts as an antioxidant (7), and changes in selenoenzyme levels may be an underlying mechanism for Se’s chemopreventive and chemotherapeutic effects (3, 8). Similarly, if the action of a drug is dictated even in part by selenoenzymes, its supplementation may impact drug effectiveness or toxicity.
The cellular antioxidant enzymes thioredoxin reductase (TrxR) and glutathione peroxidase (Gpx) rely on Sec residues, and have both been found to be deregulated in tumor cells, such that TrxR levels have been found to be high and Gpx levels have been found to be low in cancer cell lines and human tumor tissue (9-11). TrxR has a broad range of substrates, including disulfide-containing proteins, such as thioredoxin (Trx), many small molecule thiols, such as 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) (12), and even non-disulfide compounds such as dehydroascorbic acid (13). The Gpxs are a family of peroxidases that contain one essential active site Sec and require glutathione (GSH) as a co-substrate. Gpx 1 is the predominant Gpx isoform. It is found in the cytoplasm of all mammalian tissues and it catalyzes the cellular reduction of inorganic hydrogen peroxide or organic lipid hydrogen peroxides to water or corresponding alcohols (14). The active site Sec of Gpx is highly sensitive to oxidative modification, and nitric oxide-mediated modifications of the Gpx Sec has been shown to inactivate the enzyme (15). In TrxR, the Sec residue is located at the C-terminus active site as part of the tetrapeptide Gly-Cys-Sec-Gly motif (6). The flexible C-terminus of the TrxR active site is exposed to the protein surface and is accessible to substrates and inhibitors. In addition, the Sec residue is more reactive than Cys towards electrophiles due to its lower pKa (5.24 vs 8.25) (16). These features make TrxR highly susceptible to reactions with electrophilic compounds, and previous studies show that some alkylating agents such as the nitrogen mustard compounds chlorambucil and melphalan inhibit TrxR with micromolar IC50s (17, 18). However, the potential role of selenoenzyme inhibition by more selectively toxic non-traditional anticancer agents, such as acylfulvene, in cytotoxicity has not been tested.
Acylfulvenes (AFs) are a class of semisynthetic antitumor agents derived from the natural sesquiterpene cytotoxin illudin S, isolated from the Jack O’Lantern mushroom (Chart 1) (19-22). Illudin S kills various drug-resistant cancer cells, but exhibits low malignant cell versus normal cell selectivity and therefore has no practical potential in cancer therapy (23). On the other hand, newer semisynthetic acylfulvene (AF) analogues, including hydroxymethylacylfulvene (HMAF), have improved cancer cell selectivity (24, 25). Studies regarding their cellular accumulation suggest that illudin S and AFs covalently bind to cellular DNA, RNA, and protein (26, 27). There is evidence that cytotoxicity is associated with bioactivation to an unstable cyclohexadiene intermediate that more potently reacts with DNA and interrupts DNA synthesis/repair (28-33). The specificity and efficiency with which tumor-associated reductase enzymes activate AFs versus illudins appear to contribute in part to the selectivity of acylfulvene analogs compared to illudin S. However, this process does not account for the full magnitude of cytotoxicity differences between these structurally related small molecules.
Chart 1.
Structure of illudin S and acylfulvene derivatives AF and HMAF.
Differences in amino acid alkylation proficiencies between the illudin and acylfulvene analogs, presumed to extend to the protein level, have been hypothesized to additionally contribute to toxicity. Thus, illudin S reacts with cysteine under mild conditions (aqueous buffer at pH 6) more readily than acylfulvenes; no data is available for Sec reactivity, but it should be expected to be more reactive toward the electrophilic compounds (21, 34-36). Despite the increased reactivity of illudin S toward Cys, however, we have previously found that for glutathione reductase (GR), an enzyme with a redox-active disulfide at its active site, acylfulvenes are more reactive as inhibitors. They disrupt enzyme structure and inhibit GSSG reductase activity, and in particular, HMAF covalently binds active site Cys residues (18).
In a continued effort to understand potential contributions of reactive redox enzyme-drug interactions in dictating selective toxicities of alkylating agents, in this study, we aimed to test the hypothesis that the high nucleophilicity and accessibility of the Sec-containing enzymes TrxR and Gpx may render them susceptible to illudin S and/or AFs. If so, we wished to determine whether there could be any basis for chemical selection in this process. Thus, we have characterized the reactivities of illudin S and AFs toward isolated TrxR and Gpx enzymes, and the results reveal differences in inhibition potencies and enzyme alkylation profiles as a function of active site residue/accessibility and drug structure. A further compelling finding is that in cancer cells there exists a positive correlation between selenium availability and drug sensitivity. These data suggest that the more selectively cytotoxic AF derivatives may act in part by interacting with reactive Sec or Cys protein residues more abundant in sensitive cells, and inhibit these cellular redox enzymes.
Experimental procedures
Chemicals and reagents
Illudin S from the mature fungal Lampteromyces japonicas was provided by MGI Pharma (Bloomington, MN) (37). Acylfulvene and HMAF were synthesized according to the published procedure with illudin S as the starting material (21, 22). Purified rat TrxR, Gpx from bovine erythrocytes, Tris base, 5,5′-Dithiobis(2-nitrobenzoic acid) (DTNB), hydrogen peroxide (H2O2), reduced glutathione (GSH), iodoacetamide, and EDTA were obtained from Sigma Chemical. Biotin-conjugated iodoacetamide (BIAM) was purchased from Invitrogen. Reduced nicotinamide adenine dinucleotide phosphate (NADPH) was purchase from EMD chemicals. Dulbecco’s modified Eagle’s medium (DMEM) was purchased from Mediatech (Herndon, VA). Fetal bovine serum (FBS) was purchased from Atlanta Biologicals (Lawrenceville, GA). Phosphate-buffered saline (PBS), 0.25% trypsin-EDTA, penicillin-streptomycin were obtained from Invitrogen. Tris-buffered saline was purchased from Bio-Rad. Glutathione reductase was purchased from MP Biomedicals (Solon, OH). Drug stock solutions were prepared in DMSO.
Instrumentation
LC/MS analysis of drug-treated enzymes were performed on an Agilent 1100 capillary HPLC in line with an Agilent 1100 ion trap mass spectrometer (Agilent Technologies, Santa Clara, CA) operated in positive ion mode. For drug-treated TrxR peptide mixtures, an Agilent Zorbax SB-C18 column (150 mm × 0.5 mm, 5 μm) was used. Analytes were eluted with a gradient of solvent A (0.5% formic acid/0.01% TFA in water (v/v)) and solvent B (0.5% formic acid/0.01% TFA in acetonitrile (v/v)) at a flow rate of 15 μL/min: initial conditions, 3:97 B:A, were held constant for 3 min, and then increased to 5:95 B:A in 7 min and held for 10 min followed by linear increase to 35:65 B:A over a course of 95 min, and finally to 75:25 B:A in 10 min. For drug-treated Gpx, a Zorbax 300 SB-C3 column (150 mm × 0.5 mm, 5 μm) was used for chromatography. Analytes were eluted with a solvent gradient of 0.05% TFA in water (A) and 0.05% TFA in acetonitrile (B), at a flow rate of 15 μL/min: initial conditions, 30:70 B:A, were held 3 min followed by a linear increase to 80:20 B:A over a course of 20 min. Absorbance measurements for enzyme assays were determined using a Varian Cary UV 100 UV/visible spectrophotometer (Varian, Inc., Palo Alto, CA).
TrxR enzyme activity assays
Drug stock solutions were prepared in DMSO. All other solutions used in the assay were prepared in TE buffer (50 mM Tris-Cl, 1 mM EDTA) at pH 7.2. TrxR activity was determined at 25 °C with a UV/visible spectrophotometer (Varian Cary-100). TrxR (80 nM) was first reduced by addition of excess NADPH (100 μM) to result in a total volume of 0.1 mL. After 10 min at 25 °C, varying amounts of drug were added to the pre-reduced TrxR followed by incubating at 25 °C for the time indicated. Negative control runs were conducted by adding the same amounts of DMSO. The enzyme activities were measured by DTNB reducing assay in which at the end of incubation, 0.4 mL of assay solution (2 mM DTNB and 200 μM NADPH in TE buffer) was added and the absorbance at 420 nm was monitored for 3 min. Initial data points were fit to a straight line to obtain relative inhibition concentrations under the conditions of each experiment. To determine the reversibility of inhibition, TrxR was allowed to react with AFs as described above. After the 2 h reaction period, unbound drug was removed by gel-filtration with a size-exclusion Micro Bio-Spin™ P-6 pre-packed column (containing 10 mM Tris-HCl buffer, pH 7.4, with 0.02% sodium azide (w/v)) according to the Bio-Rad’s protocols. Briefly, the column was placed in 2 mL centrifuge tube to drain the excess packing buffer by gravity. After discarding the drained buffer, the column was placed back into the tube and centrifuged to remove any remaining packing buffer (2 min, 1000 g). Afterwards, the column was placed in a clean 1.5 mL centrifuge tube, and the reaction solution was loaded into two columns (100 μL/each) followed by centrifugation for 4 min at 1000 g. The resulting solutions were combined, and the activity was determined following the procedure for the DTNB reducing assay described above. Each data point represents an average of at least three measurements.
Cell culture
HeLa cells were maintained as monolayers in Dulbecco’s Modified Eagle Medium (DMEM) medium supplemented with 10% Fetal Bovine Serum (FBS) and 1% penicillin-streptomycin in a humidified, 5% CO2 atmosphere at 37 °C. HeLa cells were subcultured in the medium described above for three days to reach 80% confluence (100 mm2 plate, 2×106 cells/plate) evaluated with microscope. Cells were treated with test compounds diluted with medium (0.1% final concentration of DMSO) for 4 h. After treatment cells were washed with 5 mL phosphate buffered saline (PBS) twice. Cells were collected as follows: 2 mL of 0.25% trypsin-ethylenediaminetetraacetic acid (EDTA) was added and the cells were incubated for 5 min at 25 °C. Cells were scratched off from the plates and divided evenly into three centrifuge tubes (1.5 mL), and centrifuged (5 min, 1000g). The supernatant was removed by pipette. Cells were resuspended in 0.2 mL of lysis buffer (50 mM Tris-HCl, pH 7.5; 1 mM EDTA; 0.1% Triton X-100; 1 mM phenylmethanesulfonyl fluoride; 1 mM benzamidine; 1.4 μM pepstatin A; and 2.0 μM leupeptin) and sonicated at 4 °C (5 s bursts). The resulting cell lysate was centrifuged for 10 min (8000 g, 4 °C), and the supernatant was withdrawn for analysis. Cell cytosol containing 50 μg protein as determined by the bicinchoninic acid (BCA) protein assay reagent (Pierce, Rockford, IL) was incubated with an 80 μL mixture of insulin (1 mg/mL) and NADPH (200 μM) in TE buffer at 25 °C for 10 min. Trx (50 μL, 1 mg/mL) was added, and the rate of NADPH consumption was determined by monitoring changes in absorbance at 340 nm for 5 min at 25 °C.
Immunoassay of TrxR levels in HeLa cells
Cell cytosol containing 50 μg protein was separated by 4-12% SDS-PAGE (Invitrogen) and transferred onto a PVDF membrane (Invitrogen) for 1 h at 33 V, 4 °C. Membranes were blocked with 5% (w/v) nonfat milk powder in tris-buffered saline (TBS)/Tween 20 (0.05%) overnight at 4 °C. The membrane was incubated with primary anti-TrxR (ABR, Rockford, IL) and anti-actin (Invitrogen) antibodies diluted 2000 times in TBS/Tween 20 (5 mL) for 1 h. The membrane was washed three times with TBS/Tween 20 (5 mL, 5 min) and subject to incubation with 5 mL of secondary conjugated antibody (goat anti-rabbit IgG horseradish peroxidase, Bio-Rad, 1:2000 dilution in TBS/Tween 20,) for 1 h. After four 5 min washes with TBS/Tween 20 (5 mL/each), the Peroxide Solution and the Luminol Enhancer Solution from the ECL Western Blotting Substrate analysis system (Pierce, Rockford, IL) were mixed in a 1:1 ratio in a tray and rinsed over the membrane. The membrane was exposed to a film, developed, and the chemiluminescence was quantified.
Cell viability assay
Drug cytotoxicity toward HeLa cells was determined by Promega CellTiter 96 Aqueous One Solution Cell Proliferation Assay, where viable cells yield a colored, quantifiable product via bioreduction of 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium, inner salt (MTS) (38). Briefly, cells were seeded on 96-well plate with a starting density of 1000 cells/well in either regular medium or medium containing 1 μM sodium selenite for 3 days. The exponentially growing cells were then exposed to drugs in varying concentrations for 12 h. The drug-containing medium was exchanged with fresh regular medium. Cell viability was determined by exposing cells to an MTS-containing solution for 2 h at 37 °C, and measuring formazan formation, i.e. absorbance at 490 nm, with a microplate reader. All experiments were carried out in triplicate.
Chemical labeling and assay of AF-modified TrxR
NADPH-reduced TrxR (0.9 μM, prepared as described above) and AFs at varying concentrations in DMSO were allowed to react at 37 °C for 2 h. The samples containing equivalent amounts of DMSO only were used for control experiments. After incubation, 1 μl of the reaction mixture was taken out and added to new tubes containing 19 μL of BIAM (100 mM, pH 6.5 and 8.5 in TE buffer) further incubated at 37 °C for another 30 min to allow the alkylation at the remaining free –SeH and –SH groups in the enzyme. Fifteen microliters of BIAM-modified enzyme were mixed with 5 μL of loading buffer (Invitrogen), 20 μL of the samples were subjected to SDS-PAGE on a 4-12% gel, and the separated proteins were transferred to nitrocellulose membrane. Proteins labeled with BIAM were detected with horseradish peroxidase-conjugated streptavidin and enhanced chemiluminescence detection.
Trypsin-mediated digestion and LC/MS analysis of AF-treated TrxR
TrxR (50 μg, 1 nmol) was allowed to react with AFs (2 μL, 125 mM) or 2 μL of DMSO as control in a total volume of 0.2 mL TE buffer containing NADPH (1 mM) for 2 h at 25 °C with reaction with 10 μL of DMSO as control. An aliquot (1 μL) of reaction solution was withdrawn and diluted to a total volume of 300 μL; enzyme activity was determined as described above for measurement of TrxR activity. After 2 h, TrxR activity was inhibited by AFs completely. The resulting solutions were diluted to a volume of 1 mL by adding 0.8 mL TE buffer followed by trypsin (0.2 μg/ μL, 10 μL) in 0.1 M HCl. Proteolytic digestion was allowed to occur at 37 °C for 24 h. The resulting mixture was divided into two equal portions, placed into centrifuge tubes, concentrated to dryness on a centrifugal vacuum concentrator, and reconstituted by adding 20 μL of solvent A (0.5% formic acid/0.01% TFA in water (v/v)) to each tube. The resulting samples were analyzed by LC/MS.
Mass analysis of Gpx modification by AFs
Gpx (50 μg, 1.6 nmol) was allowed to react with AFs (10 μL, 125 mM) or 10 μL of DMSO as control in a total volume of 1 mL TE buffer for 2 h at 25 °C. A Microcon® Y-10 centrifuge filter (NMWL 10,000) was used to remove unbound compound and concentrate Gpx to a final volume of 30 μL. Samples (8 μL) were analyzed by LC/MS with the general analytical protocol. Spectra were obtained by full scan data acquisition performed within m/z 100-1500. Mass deconvolution was performed with the Agilent ion trap analysis software (Charge Deconvolution for Data Analysis for LC/MSD Trap, version 3.2).
Gpx enzyme activity assay
The influence of test compounds on Gpx was evaluated by adaptation of a published assay (39). In a microcentrifuge tube (1.5 mL), 0.02 U Gpx (5 μL, 80 nM in phosphate buffer) was allowed to react with varying concentrations of test compound in a total volume of 0.5 mL phosphate buffer (0.1 M, 1 mM EDTA, pH 7.0) at 37 °C. After 40 min, GSH (0.1 mL, 10 mM aqueous) and glutathione reductase (0.1 mL, 0.33 μM in phosphate buffer) were added and vortex mixed. The resulting mixture was allowed to react at 37 °C for an additional 10 min, and then transferred to a disposable cuvette. NADPH (0.1 mL, 1.5 mM in 0.1% NaHCO3 (w/v)) was added to the cuvette. The H2O2-independent consumption of NADPH was determined by monitoring the change in UV absorption of the sample at 340 nm, at 25 °C. After 2-3 minutes, H2O2 (0.1 mL, 1.5 mM aqueous) was added to the cuvette to initiate the H2O2-dependent reaction. The change in UV absorbance was monitored for an additional 7 min, and the slope of the plot of absorption vs. time was normalized to 100% activity as determined by the blank (10 μL DMSO) control. As positive control, Gpx was treated with 1.87 mM iodoacetamide. Data was fit to a linear model for the first 5 data points.
Results
Illudin S and AFs inhibit TrxR in a dose- and time-dependent manner
When NADPH-reduced TrxR was treated with illudin S and AFs, its DTNB reduction activity was inhibited in a dose-dependent manner (Figure 1). The IC50 values were determined to be 260 μM for illudin S, 7.3 μM for AF, and 0.38 μM for HMAF respectively. Without pre-reducing TrxR, no inhibition was observed by illudin S, and the IC50 values for AFs increase to millimolar ranges (Figure 2). Further, the relative inhibition potencies of AF and HMAF towards TrxR in the absence of pre-reduction are similar, suggesting the observed inhibition of TrxR by AFs mainly resulted from the interaction of TrxR active site with AFs, but some other interaction may also partially account for the inhibition. The inhibition of TrxR by AFs is time-dependent; within five minutes, over 60% of TrxR activity was inhibited by 50 μM AF and 7.5 μM HMAF respectively (Figure 3), suggesting a rapid reaction between TrxR and AFs.
Figure 1.
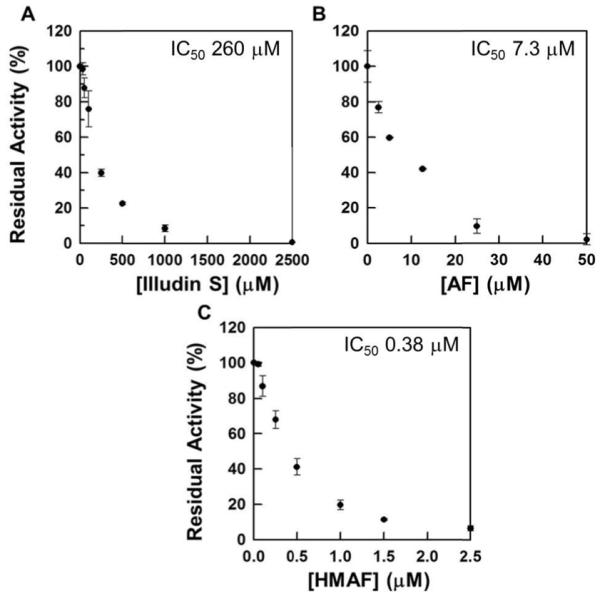
Dose-dependent inhibition of pre-reduced TrxR by illudin S and AFs. TrxR (80 nM) was first incubated with NADPH (100 μM) at 25 °C for 10 min followed by the addition of test compounds and further incubated for 2 h at 25 °C (A, Illudin S, 62.5, 125, 250, 500, 1000, 2500 μM; B, AF, 2.5, 5, 12.5, 25, 50 μM; C, HMAF, 0.125, 0.25, 0.5, 1, 1.5, 2.5 μM). The residual activity was measured with DTNB assay described in the experimental details.
Figure 2.
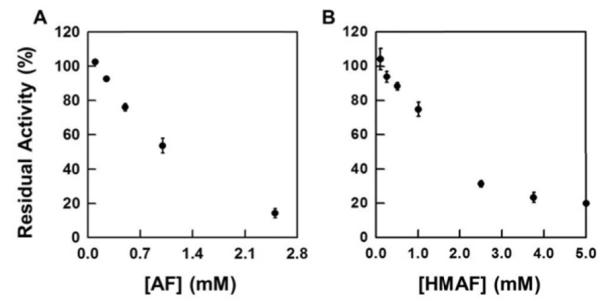
Inhibition of non-reduced TrxR by AF and HMAF. TrxR (80 nM) was incubated with the test compounds for 2 h at 25 °C (AF, 0.25, 0.5, 1.0, 2.5 mM; HMAF, 0.25, 0.5, 1.0, 2.5, 3.6, 5 mM). The residual activity was measured with DTNB assay described in the experimental details.
Figure 3.
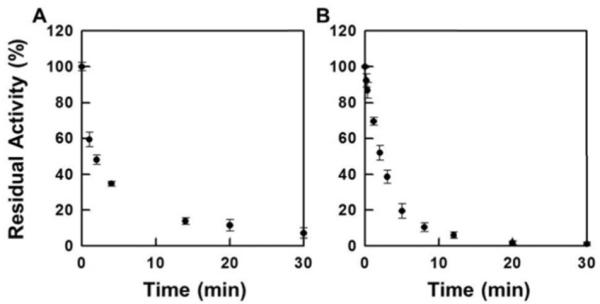
Time-dependent inhibition of TrxR by AFs. TrxR (80 nM) was first incubated with NADPH (100 μM) at 25 °C for 10 min followed bby the addition of test compounds at concentrations that would completely inhibit TrxR upon 30 min treatment (A, AF 50 μM; B, HMAF 7.5 μM) at 25 °C. To assay the enzyme activity, 100 μL of the incubation solution was taken out at different time intervals and the residual activity was measured with DTNB assay described in the experimental details. Each data point represents an average of two measurements.
TrxR inhibition by AFs is irreversible
To determine whether TrxR inhibition by AFs is reversible, gel-filtration studies were conducted. TrxR was allowed to react with AFs in the same manner as described for studies carried out to determine the concentration-dependence of inhibition. The resulting inactivated TrxR was filtered through a size exclusion column with molecular weight cut-off 6000 to remove non-covalently bound drug and the re-isolated enzyme was assayed for DTNB reduction capacity. The native TrxR activity was not recovered (Figure 4), suggesting AFs are irreversible inhibitors of TrxR.
Figure 4.
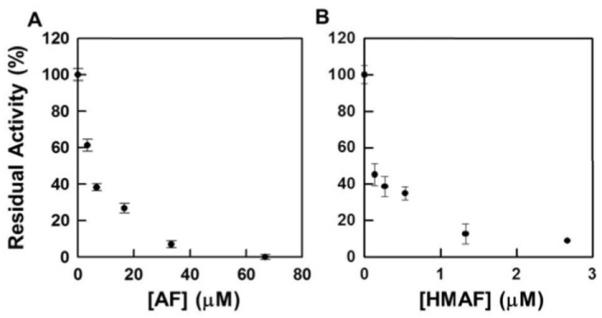
Gel-filtration analysis of AF and HMAF-inactivated TrxR. TrxR (80 nM) was first incubated with NADPH (100 μM) at 25 °C for 10 min followed by the addition of test compounds and further incubation for 2 h at 25 °C (A, AF, 3.3, 6.7, 16.7, 33.3, 66.7 μM; B, HMAF, 0.13, 0.27, 0.53, 1.33, 2.67 μM). Unbound compound was removed by Micro Bio-Spin™ P-6 pre-packed size exclusion columns. The residual activity was measured with DTNB assay described in the experimental details.
Alkylation of cysteines at TrxR active site
An affinity assay involving pH-selective BIAM-alkylation of free Cys vs Sec residues was carried out for the inactivated protein resulting from treatment with AFs. The purpose of the experiment was to determine whether active site Sec and Cys in TrxR were chemically modified by AFs. Despite multiple potential TrxR alkylation sites, the pH- and time-dependent selective alkylation of active site residues by BIAM has been optimized and established previously, and applied to other TrxR-alkylating compounds (40-42). Per the published procedures, the enzyme was allowed to react with varying concentrations of AFs, then labeled with BIAM. Due to the pKa difference of active site residues Cys 497 (pKa 8.3) and Sec 498 (pKa 5.2) (16), only the free selenol of Sec is modified at pH 6.5 and both active site residues are modified at pH 8.5. The results of the experiment were that at pH 6.5, the intensity of the BIAM label was diminished in intensity for low micromolar AF and HMAF (25 μM and 5 μM, respectively). The same trend was observed with increasing concentrations of AFs, suggesting selective modification of the free selenol of Sec by AFs (Figure 5). Likewise, at pH 8.5 and for the highest levels of AFs (125 μM AF and 25 μM HMAF), BIAM labeling was almost abolished as compared to that at pH 6.5. These data suggest that both active site Cys and Sec residues were modified by both AF and HMAF.
Figure 5.
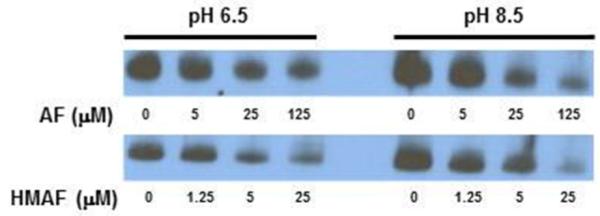
Free C-terminus redox-active site Sec and Cys of TrxR were detected by biotin-conjugated iodoacetamide. Different concentrations of AFs were added to NADPH (200 μM) pre-reduced TrxR (0.9 μM) and incubated at 25 °C for 2 h. BIAM was added to label active site free Sec at pH 6.5 and free Cys and Sec at pH 8.5. Results are representative of three independent experiments.
Identification of the covalent adduct between AF and TrxR
Since AF irreversibly inhibited the DTNB reduction activity of TrxR, and the modification appears to occur at the C-terminus active site, we analyzed the trypsin digest of the covalently modified TrxR by LC/MS to evaluate the chemical pathway involved. A singly charged ion corresponding to the active site peptide product of the tryptic digest was observed by ion trap MS analysis (m/z 1142.4, Figure 6A). Due to the presence of selenium in the peptide sequence (SGGDILQSGCysSecG), the isotope envelope is uniquely diagnostic. A singly charged ion corresponding to AF-mediated monoalkylation of the active site tryptic peptide was observed (m/z 1358.6, Figure 6B), and the associated mass change (m/z 216) is consistent with formation of an adduct via direct conjugate addition at the α,β-unsaturated carbonyl. Under similar experimental conditions, the HMAF-modified peptide was not identified. However, unlike controls (i.e. DMSO-treated TrxR tryptic digest), for the HMAF-treated sample, a 60% reduction of the extracted ion intensity of the active site peptide product was observed (Figure S1). Extensive experimentation with different digestion conditions, including other proteases such as V8 E and Asp-N, still did not provide diagnostic information. Likewise, MALDI ionization (QSTAR XL and Biflex III) and nanospray ionization (4000 QTRAP), and extensive optimization of ionization parameters, did not result in a corresponding signal for a modified active-site peptide. These results suggest that modification by HMAF drastically suppresses the ionization efficiency of the peptide.
Figure 6.

LC/MS analysis of AF-modified TrxR active-site peptide (SGGDILQSGCysSecG). A, Average mass for the unmodified peptide (m/z 1142.4) from an untreated sample of enzyme as a blank control; B, average mass for the AF-modified peptide (m/z 1358.6) from a treated sample; C, proposed mechanism of reaction of AF with Sec at TrxR active site.
AFs do not impact Gpx activity or result in covalent modification
The test compounds were evaluated as potential Gpx inhibitors. Thus, Gpx was treated with varying concentrations of AFs and illudin S and residual enzyme activity was measured by a GR-coupled assay, however none of the tested drugs caused a decrease in enzyme activity (Table S1). By contrast, and as a positive control, Gpx was treated with iodoacetamide (IAA), and under the conditions of the general assay IAA inhibited Gpx activity with an IC50 of 2.4 mM (Figure S2). While IAA has not been characterized explicitly as a Gpx inhibitor previously, it is a common alkylating agent known to efficiently modify free cysteine and selenocysteine residues, and it was found to be an effective positive control for monitoring loss of Gpx activity. Furthermore, after Gpx was incubated with AFs for 3 h and unbound compound removed with a Microcon® Y-10 centrifuge filter (NMWL 10,000), whole protein mass spectrometry analysis indicated only the intact Gpx peak (Figure S3). These data suggest that neither AFs nor illudin S impact Gpx activity, and do not modify or interact with the enzyme active site.
Evidence for TrxR inhibition by AFs in whole cells
Human cervical cancer cells (HeLa) are highly sensitive to AFs, with a reported IC50 of 20 ng/mL HMAF three day continuous treatment (43). Illudin S was reported to be around 30-fold more toxic in vitro than AF (44). To test whether TrxR might be involved in the observed HeLa cell sensitivity toward AFs, we first compared the cytotoxicity of the three compounds to HeLa cells (Figure S4). At the concentration range tested for the three compounds, illudin S is at least 100-fold more toxic than AF. Cells were treated with illudin S, AF, or HMAF at concentrations that maintain 80% cell viability upon 12 h treatment. After washing and cell lysis, residual TrxR activity was determined. A dose-dependent loss of TrxR activity resulted from each of the compounds tested (Figure 7). At equitoxic concentrations, AF (0.2 μM) and HMAF (0.2 μM) inhibited TrxR activity by 20% (Figure 7), and increasing concentrations of each drug inhibited cellular TrxR activity more. Thus, TrxR activity was inhibited by 35% and 60% with 1 μM and 4 μM AF treatment, respectively. A more drastic inhibition of activity was seen with HMAF treatments, as 1 μM HMAF led to 60% TrxR reduction and 4 μM eliminated cellular TrxR activity. Cellular TrxR was inhibited 20% and 50% by 0.2 and 1 μM illudin S, respectively. Higher concentrations of illudin S inhibited cell viability more than 40%. Overall, the relative potencies of the three compounds for reducing cellular TrxR activity (at a given toxicity level) were in the same order as inhibition potencies in a cell-free system.
Figure 7.

Inhibition of TrxR activity in HeLa cells by illudin S and AFs. HeLa cells were exposed to individual compound. A, illudin S: 0.04, 0.20, 1.00 μM; B, AF 0.2, 1.0, 4.0 μM; C, HMAF: 0.2, 1.0, 4.0 μM) for 12 h and then cellular TrxR activity was measured. Asterisks represent a significant difference relative to controls: * p < 0.05, ** p < 0.01.
Although the ordering of potency for HMAF > AF > illudin is consistent between the enzyme inhibition data and the cell-based activity data, there is a significant dampening effect observed for cellular reductase activity inhibition vs. cell-free enzyme inhibition for illudin. Namely, the IC50 for illudin-mediated inhibition of enzyme activity is 260 μM, or almost two orders of magnitude less potent than HMAF, while at an equitoxic concentration, illudin already elicits a 50% reduction in cell-based reductase activity. Thus, compared to AF and HMAF, illudin S is disproportionately active in cells. These data underscore that illudin S and AFs share core structural features, however there appear to be fundamental differences in their toxicity profiles. Potential cross-talk between unknown pathways, which may be efficiently modulated by illudin S and related to its overall high toxicity, and TrxR may account for the significant impact on cellular reductase activity while the molecule has a comparatively low impact on free enzyme activity.
Selenite-enhanced sensitivity of cells towards AFs
Studies carried out here to test the potential influence of TrxR induction on cytotoxicity demonstrated that cells could be favorably sensitized by selenite preconditioning. It is known that culture medium supplemented with sodium selenite (Na2SeO3) will induce TrxR expression in breast cancer MCF-7 cells and liver cancer HepG2 cells (45, 46). In the present study, HeLa cells were cultured in media containing 1 μM sodium selenite for three consecutive days. 1 μM of selenite is also close to physiological Se concentration, and higher concentrations of added selenite (2 and 3 μM) caused cytotoxicity (observed as cells significantly detached from culture dish). A saturated level of TrxR was observed after three day induction by selenite. By Western blotting analysis and measurement of cellular TrxR activity after selenite supplementation, cellular TrxR protein levels and TrxR activity increased four-fold (Figure S5). We then investigated whether changes in sensitivity toward illudin S and AFs were associated with the increased protein levels and activities. Results indicated that increasing the cellular level of TrxR makes cells more sensitive toward AFs, but not towards illudin S (Figure 8), which is consistent with the inhibition potency of these compounds with purified TrxR. After induction by 1 μM selenite for three continuous days, HeLa cell sensitivity towards 100 μM AFs was increased about 50% compared to the cell sensitivity without selenite induction.
Figure 8.

Differential sensitivity of HeLa cells towards illudin S and AFs. HeLa cells were seeded on 96-well plates (1000 cells/well) and cultured in medium with (red/right) or without (blue/left) addition of sodium selenite (1 μM) for three continuous days and then changed with medium containing test compounds (illudin S, 0.2, 0.5, 1, 2 μM; AFs, 10, 20, 50, 100 μM) for 12 h. Cytotoxicity was measured with MTS assay (Promega). Asterisks represent a significant difference between cytotoxicities resulting from no selenite and 1 μM selenite medium: * p < 0.05, ** p < 0.01.
Discussion
The cellular function of Se has been attributed mainly to its presence in selenoproteins and the correlation of selenium administration and level of selenoenzymes has been extensively studied (2, 10, 47-52). Se is incorporated into selenoproteins in the form of selenocysteine either during protein synthesis or through post-translational processes (52-54). Selenoproteins such as TrxR and Gpx are involved in cellular antioxidant defense and redox signaling. The active site Sec residue of these proteins has a higher propensity to react with electrophiles compared to its Cys counterpart (16) and therefore, the potential to be more easily inactivated by alkylating agents. Key electrophilic features of illudin S and AFs include a cyclopropane ring and an α,β-unsaturated ketone, which may react by direct addition or coupled with reductive bioactivation (28-32). Increased reductase-mediated bioactivation by NADPH-dependent enzyme or enzymes, and subsequent nucleophilic attack at the cyclopropane ring by DNA is thought to be an important process contributing to AF toxicity, but does not fully account for differences in cytotoxicity profiles (27, 55-57), and their potential for inactivating key cellular proteins as a contributing factor is a hypothesis under investigation (18, 27). In correlation with their reactivity with small thiols, illudin S and AFs were expected to have the same relative reactivities toward thiol-containing enzymes (35, 58), but in a previous study a surprising finding was that AFs are much more reactive than illudin S toward the cellular redox-regulating enzyme GR (18).
The thioredoxin system comprising TrxR, its protein substrate thioredoxin and NADPH, together with its homologous glutathione system comprising GR, NADPH and GSH, is the major regulator of intracellular redox balance, exerting a wide range of activities in oxidant defense, cell viability, and proliferation. Patterns of GR inhibition by illudin S and AF have been defined, but the potential influence of the more reactive Sec residue within TrxR, and similarly Gpx, is unknown. Elevated levels of TrxR were found in many types of cancer cell lines, especially ovarian and prostate cancer which are both very sensitive to HMAF (9). TrxR is very similar in both structure and mechanism to GR (59). They both use NADPH as a cofactor to reduce the conserved internal disulfide center. TrxR is distinct from GR by the presence of a C-terminus redox center (Gly-Cys-Sec-Cys) which is functionally equal to GR’s substrate GSSG (60, 61). The flexibility of the C-terminus redox center not only allows TrxR to have a broad range of substrates, but also makes it extraordinarily reactive towards electrophiles due to the reactive and solvent accessible Sec. The Sec was reported to be targeted by many anti-cancer compounds, such as curcumin, motexafin gadolinium, flavonoids, and quinines (40, 62). TrxR regulates cell proliferation by controlling DNA systhesis and antioxidant defense with reduced Trx. In normal cells, TrxR is critical for maintaining intracellular proteins in their reduced states and defending against oxidative stress. In malignant cells, however, TrxR supports tumor growth and progression, and inactivation may induce or promote cell death. Here we find that NADPH-reduced TrxR is inhibited by AFs with low micromolar IC50s, which has similar potency as TrxR inhibitors characterized to date. Examples include curcumin (63), carmustine (64), gold complexes (65), platinum compounds (64, 66), and 2-chloroethyl ethyl sulfide (CEES) (42). The IC50 of illudin S toward TrxR determined in this study was 100-fold higher than that of the AFs. This trend is reverse in order relative to cytotoxicity, but is consistent with its diminished ability to inhibit GR (18).
The related homotetrametic enzyme Gpx is comprised of two asymmetric units containing two dimers, each with two selenocysteines at the active sites in the form of Se− in the resting state and that directly participate in the process of hydroperoxide reduction of Gpx. There was no data obtained in this study to suggest any chemical reactivity of the drugs toward the active site of the protein resulting in inhibition of activity. Unlike the flexible C-terminus active site of TrxR, the active site of Gpx locates at the N-terminal ends of long α-helices, surrounded by aromatic side-chains. This structural context may therefore protect Sec from AFs (67). For relatively high concentrations of drug (≥ 1000 μM illudin S, ≥ 20 μM AF, ≥ 2 μM HMAF) we observed an increase in Gpx activity (Table S1) that could not be explained on the basis of our experiments. To our knowledge there is no precedent or good rationale for this unusual observation, yet the data for lower concentrations show no change in activity and offer no suggestion that these compounds are Gpx inhibitors.
AF and HMAF are both irreversible TrxR inhibitors, however, only HMAF is an irreversible GR inhibitor. TrxR and GR are homodimeric flavoproteins of the same family and are similar in primary sequence and structure, but GR lacks the Sec residue in the C-terminal active site. A possible rationale for the difference in AFs reactivity is that Sec that may undergo a hindered conjugate addition reaction (Scheme 6C) with the α,β-unsaturated ketone that is common to AFs. Therefore, TrxR is capable of reacting with both AF and HMAF. In the case of GR inhibition by HMAF (18), however, evidence suggests that a substitution reaction occurs with the methylenehydroxy substituent that is specific to HMAF. Thus, the differences in the chemical reaction mediating the modification may account for the combined observations.
On the basis of the BIAM labeling experiment performed here (Figure 5), both active site residues are alkylated at low micromolar concentrations of AF and HMAF. This affinity assay provides mechanistic information regarding how AFs compare to other TrxR inhibitors. AFs irreversibly inhibit TrxR to a similar degree as previously characterized inhibitors, but the manner in which they modify important active site residues differs. Our results demonstrate that like curcumin (polyphenol), carmustine (nitrosourea), and 1-chloro-2,4-dinitrobenzene (DNCB, nitroaromatic), AFs modify Sec and Cys. In contrast, CEES (half-mustard) (42) and 5-iodoacetamidofluorescein (iodoacetamide conjugate) (68) inhibit TrxR by only modifying the Sec residue. Whether there is medicinal relevance in selectively targeting Sec vs. both active site residues is not understood, however existing data suggests possible differences in triggering molecular pathways. For example, alkylation of Sec and Cys by curcumin (40) and DNCB (69) results in irreversible inhibition and high induction of NADPH oxidase, which produces reactive oxygen species (ROS). Cisplatin-derivatization of TrxR inhibits enzyme activity but does not induce NADPH oxidase activity (70), and Sec-targeting CEES is capable of producing (ROS) (42), but it has not been demonstrated whether the generation of ROS also occurs via induction of NADPH oxidase. Interestingly, it has been shown that alkylated TrxR (with DNCB or cisplatin) promotes apoptosis to a similar extent as truncated TrxR lacking its two C-terminal residues (71), but ROS can be generated as long as a functional N-terminal redox active motif remains (72).
Mass spectrometry analysis suggests the adduct was formed following a direct alkylation pathway with a mass change of m/z 216, which is also consistent with our proposed HMAF-GR adduct formed by direct attack at the α,β-unsaturated ketone followed by opening the cyclopropane ring by water molecule. Although blotting results suggest that the neighboring cysteine was also modified by higher concentration of AFs, no ions corresponding to the bis-adducts were observed, possibly due to the poor ionization efficiency of the alkylated peptides as previously discussed for GR-HMAF (18). Because of the unique Se isotope envelope, it is easy to confirm the identity of corresponding peptide modifications. We only observed singly charged ions corresponding to the unmodified active peptide (SGGDILQSCysSecG, m/z 1142.4) and a mono-adduct of AF. Significant reduction (> 60%) on the extracted ion intensity corresponding to the active site tryptic peptide (m/z 1142.4) was observed for the AFs-treated TrxR compared to the control (Figure S1), suggesting that the reaction between TrxR and AFs occurred at the active site, which is consistent to the BIAM-labeling experiment. Similar results have been reported for 4-hydroxynonenal modification of the TrxR active site, in which also only a singly charged modified peptide mass peak was identified with the Se-specific isotopic mass distribution (73).
In cancer cells, TrxR expression is up to 10-fold higher compared with normal cells (6, 74). Addition of Se to cell media has been reported to induce TrxR expression 10–30 fold by itself or in combination with sulforaphane (45, 46). An approximately 3.6-fold induction of TrxR protein levels in HeLa cells supplemented with 1 μM selenite is associated with a 4.2-fold increase in activity (Figure S5). Preliminary tests for potential synergies of Se plus sulforaphane co-induction suggested that no significant additional increases resulted, and that sulforaphane displayed potentially confounding direct inhibitory properties (Data not shown). Therefore, we used Se alone to induce TrxR and test the impact on drug sensitivity (75).
After selenite-mediated TrxR induction, HeLa cells become more sensitive to AFs, but not to illudin S. By analogy, high TrxR has been observed in various cancers. AFs may be selectively toxic in part because TrxR inhibition contributes to toxicity more significantly in the case of high TrxR cancer cells than low TrxR normal cell. Enhanced antitumor activity has been observed for the combination therapy of Se compounds and anticancer drugs such as taxol, doxorubincin, irinotecan, platinum agents, 5-fluorouracil and campthotecin (76-78). Further, cisplatin and doxorubincin (17) have been characterized as TrxR inhibitors, suggesting increasing TrxR levels with Se may enhance cell sensitivity towards anticancer drugs.
Conclusion
In this study we evaluated potential interactions between the natural product illudin S and its AF analogues toward the Se-dependent cellular redox-regulating enzymes Gpx and TrxR as isolated enzymes, and in cell-based models. The inhibition potencies of illudin S and AFs are in the same order for TrxR as what has been previously observed for the thiol-based redox-regulating enzyme GR, i.e., HMAF > AF > illudin S. However, AF and HMAF irreversibly target the TrxR active site Sec and Cys residues more efficiently than the GR active site Cys residues, which is consistent with the existence of the more reactive Sec in the TrxR active site. In contrast, Gpx is not inhibited by AFs, suggesting that the TrxR active site is more accessible and reactive (18). For both TrxR and GR, illudin S was a weaker inhibitor than AFs, which is opposite to their reactivity toward small molecule thiols, suggesting that the polar substituents that decorate the five membered ring may interfere with enzyme binding, and that the flat fulvene structure binds efficiently. Furthermore, AFs abrogate cellular TrxR activity and, for HeLa cells, are more potent cytotoxins after selenite-induced TrxR overexpression. The data obtained in this study contributes to a broader understanding of chemical and biochemical factors involved in regulating cancer cell susceptibility toward alkylating agents, specifically the potential for selectively targeting Se-dependent TrxR expression to enhance cytotoxicity.
Supplementary Material
Acknowledgements
We thank Dr. Jiachang Gong for providing synthetic AF and HMAF and Brock Matter for assistance regarding mass spectrometry data. We thank Dr. Arne Holmgren and Dr. Jun Lu (Karolinska Institute, Stockholm, Sweden) for generously providing recombinant TrxR used for a preliminary screen of potential TrxR inhibition by AFs. Mass spectrometry was carried out in the Analytical Biochemistry Facility of the Masonic Cancer Center.
Funding Support We acknowledge the National Cancer Institute (R01 CA123007) for support of this research. The Analytical Biochemistry Facility of the Masonic Cancer Center, University of Minnesota, is supported in part by the Cancer Center Support Grant (CA 77598).
Footnotes
Supporting Information Available: Data from Gpx activity studies, extracted ion chromatograms from TrxR tryptic digests, LC/MS spectra derived from AFs-treated Gpx, cytotoxicity data for HeLa cells, data regarding induction of cellular TrxR by selenite. This material is available free of charge via the Internet at http://pubs.acs.org.
Abbreviations: Se, selenium; Sec, selenocysteine; TrxR, thioredoxin reductase; Gpx, glutathione peroxidase; DTNB, 5,5′-dithiobis(2-nitrobenzoic acid); AF, acylfulvene, HMAF, hydroxymethylacylfulvene; GR, glutathione reductase; BIAM, biotin-conjugated iodoacetamide; DMEM, Dulbecco’s modified Eagle’s medium; DMSO, dimethyl sulfoxide; Trx, thioredoxin; BCA, bicinchoninic acid; TBS, Tris-buffered saline; CEES, 2-chloroethyl ethyl sulfide; DNCB; 1-chloro-2,4-dinitrobenzene; ROS, reactive oxygen species; MTS, 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium; NMWL, nominal molecular weight limit; PVDF, polyvinylidene fluoride; TFA, trifluoroacetic acid, H2O2, hydrogen peroxide; IAA, iodoacetamide.
References
- 1.Selenius M, Rundlöf A-K, Olm E, Fernandes AP, Björnstedt M. Selenium and the selenoprotein thioredoxin reductase in the prevention, treatment and diagnostics of cancer. Antioxid. Redox Signaling. 2010;12:867–880. doi: 10.1089/ars.2009.2884. [DOI] [PubMed] [Google Scholar]
- 2.Rayman MP. Selenium in cancer prevention: a review of the evidence and mechanism of action. Proc. Nutr. Biochem. 2005;64:527–542. doi: 10.1079/pns2005467. [DOI] [PubMed] [Google Scholar]
- 3.Zeng H, Combs GF., Jr. Selenium as an anticancer nutrient: roles in cell proliferation and tumor cell invasion. J. Nutr. Biochem. 2008;19:1–7. doi: 10.1016/j.jnutbio.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 4.Sechi S, Chait BT. Modification of cysteine residues by alkylation. A tool in peptide mapping and protein identification. Anal. Chem. 1998;70:5150–5158. doi: 10.1021/ac9806005. [DOI] [PubMed] [Google Scholar]
- 5.Forstrom JW, Zakowski JJ, Tappel AL. Identification of the catalytic site of rat liver glutathione peroxidase as selenocysteine. Biochemistry. 1978;17:2639–2644. doi: 10.1021/bi00606a028. [DOI] [PubMed] [Google Scholar]
- 6.Gladyshev VN, Jeang KT, Stadtman TC. Selenocysteine, identified as the penultimate C-terminal residue in human T-cell thioredoxin reductase, corresponds to TGA in the human placental gene. Proc. Natl. Acad. Sci. U.S.A. 1996;93:6146–6151. doi: 10.1073/pnas.93.12.6146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bellinger FP, Raman AV, Reeves MA, Berry MJ. Regulation and function of selenoproteins in human disease. Biochem. J. 2009;422:11–22. doi: 10.1042/BJ20090219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu J, Jiang C. Selenium and cancer chemoprevention: hypotheses integrating the actions of selenoproteins and selenium metabolites in epithelial and non-epithelial target cells. Antioxid. Redox Signaling. 2005;7:1715–1727. doi: 10.1089/ars.2005.7.1715. [DOI] [PubMed] [Google Scholar]
- 9.Berggren M, Gallegos A, Gasdaska JR, Gasdaska PY, Warneke J, Powis G. Thioredoxin and thioredoxin reductase gene expression in human tumors and cell lines, and the effects of serum stimulation and hypoxia. Anticancer Res. 1996;16:3459–3466. [PubMed] [Google Scholar]
- 10.Esworthy RS, Baker MA, Chu FF. Expression of selenium-dependent glutathione peroxidase in human breast tumor cell lines. Cancer Res. 1995;55:957–962. [PubMed] [Google Scholar]
- 11.Nuray NN, Tandoğan B, Türkoğlu MA, Demirer S. Is it useful to determine glutathione peroxidase and thioredoxin reductase activities for comparisons of malign and benign breast diseases? Turk. J. Biochem. 2009;34:187–194. [Google Scholar]
- 12.Holmgren A. Bovine thioredoxin system. Purification of thioredoxin reductase from calf liver and thymus and studies of its fuction in disulfide reduction. J. Biol. Chem. 1977;252:4600–4606. [PubMed] [Google Scholar]
- 13.May JM, Mendiratta S, Hill KE, Burk RF. Reduction of dehydroascorbate to ascorbate by the selenoenzyme thioredoxin reductase. J Biol. Chem. 1997;272:22607–22610. doi: 10.1074/jbc.272.36.22607. [DOI] [PubMed] [Google Scholar]
- 14.Prabhakar R, Vreven T, Morokuma K, Musaev DG. Elucidation of the mechanism of selenoprotein glutathione peroxidase (GPx)-catalyzed hydrogen peroxide reduction by two glutathione molecules: a density functional study. Biochemistry. 2005;44:11864–11871. doi: 10.1021/bi050815q. [DOI] [PubMed] [Google Scholar]
- 15.Asahi M, Fujii J, Takao T, Kuzuya T, Hori M, Shimonishi Y, Taniguchi N. The oxidation of selenocysteine is involved in the inactivation of glutathione peroxidase by nitric oxide donor. J Biol. Chem. 1997;272:19152–19157. doi: 10.1074/jbc.272.31.19152. [DOI] [PubMed] [Google Scholar]
- 16.Huber RE, Criddle RS. Comparison of the chemical properties of selenocysteine and selenocystine with their sulfur analogs. Arch. Biochem. Biophys. 1967;122:164–173. doi: 10.1016/0003-9861(67)90136-1. [DOI] [PubMed] [Google Scholar]
- 17.Witte AB, Anestal K, Jerremalm E, Ehrsson H, Arner ES. Inhibition of thioredoxin reductase but not of glutathione reductase by the major classes of alkylating and platinum-containing anticancer compounds. Free Radical Biol. Med. 2005;39:696–703. doi: 10.1016/j.freeradbiomed.2005.04.025. [DOI] [PubMed] [Google Scholar]
- 18.Liu X, Sturla SJ. Profiling patterns of glutathione reductase inhibition by the natural product illudin S and its acylfulvene analogues. Mol. BioSyst. 2009;5:1013–1024. doi: 10.1039/b904720d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Anchel M, Hervey A, Robbins WJ. Antibiotic substances from Basidiomycetes+. VII. Clitocybe illudens. Proc. Natl. Acad. Sci. U.S.A. 1950;36:300–305. doi: 10.1073/pnas.36.5.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McMorris TC, Anchel M. Fungal metabolites. The structures of the novel sesquiterpenoids illudin-S and -M. J. Am. Chem. Soc. 1965;87:1594–1600. doi: 10.1021/ja01085a031. [DOI] [PubMed] [Google Scholar]
- 21.McMorris TC, Kelner MJ, Wang W, Diaz MA, Estes LA, Taetle R. Acylfulvenes, a new class of potent antitumor agents. Experientia. 1996;52:75–80. doi: 10.1007/BF01922420. [DOI] [PubMed] [Google Scholar]
- 22.McMorris TC, Kelner MJ, Wang W, Yu J, Estes LA, Taetle R. Hydroxymethyl)acylfulvene: an illudin derivative with superior antitumor properties. J. Nat. Prod. 1996;59:896–899. doi: 10.1021/np960450y. [DOI] [PubMed] [Google Scholar]
- 23.Kelner MJ, McMorris TC, Taetle R. Preclinical evaluation of illudins as anticancer agents: basis for selective cytotoxicity. J. Natl. Cancer. Inst. 1990;82:1562–1565. doi: 10.1093/jnci/82.19.1562. [DOI] [PubMed] [Google Scholar]
- 24.Kelner MJ, McMorris TC, Estes L, Wang W, Samson KM, Taetle R. Efficacy of HMAF (MGI-114) in the MV522 metastatic lung carcinoma xenograft model nonresponsive to traditional anticancer agents. Invest. New Drugs. 1996;14:161–167. doi: 10.1007/BF00210787. [DOI] [PubMed] [Google Scholar]
- 25.MacDonald JR, Muscoplat CC, Dexter DL, Mangold GL, Chen SF, Kelner MJ, McMorris TC, Von Hoff DD. Preclinical antitumor activity of 6-hydroxymethylacylfulvene, a semisynthetic derivative of the mushroom toxin illudin S. Cancer Res. 1997;57:279–283. [PubMed] [Google Scholar]
- 26.Kelner MJ, McMorris TC, Montoya MA, Estes L, Uglik SF, Rutherford M, Samson KM, Bagnell RD, Taetle R. Characterization of MGI 114 (HMAF) histiospecific toxicity in human tumor cell lines. Cancer Chemother. Pharmacol. 1999;44:235–240. doi: 10.1007/s002800050972. [DOI] [PubMed] [Google Scholar]
- 27.Herzig MC, Arnett B, MacDonald JR, Woynarowski JM. Drug uptake and cellular targets of hydroxymethylacylfulvene (HMAF) Biochem. Pharmacol. 1999;58:217–225. doi: 10.1016/s0006-2952(99)00085-4. [DOI] [PubMed] [Google Scholar]
- 28.Dick RA, Yu X, Kensler TW. NADPH alkenal/one oxidoreductase activity determines sensitivity of cancer cells to the chemotherapeutic alkylating agent irofulven. Clin. Cancer Res. 2004;10:1492–1499. doi: 10.1158/1078-0432.ccr-03-0162. [DOI] [PubMed] [Google Scholar]
- 29.Gong J, Vaidyanathan VG, Yu X, Kensler TW, Peterson LA, Sturla SJ. Depurinating acylfulvene-DNA adducts: characterizing cellular chemical reactions of a selective antitumor agent. J. Am. Chem. Soc. 2007;129:2101–2111. doi: 10.1021/ja0665951. [DOI] [PubMed] [Google Scholar]
- 30.Jaspers NG, Raams A, Kelner MJ, Ng JM, Yamashita YM, Takeda S, McMorris TC, Hoeijmakers JH. Anti-tumour compounds illudin S and Irofulven induce DNA lesions ignored by global repair and exclusively processed by transcription- and replication-coupled repair pathways. DNA Repair. 2002;1:1027–1038. doi: 10.1016/s1568-7864(02)00166-0. [DOI] [PubMed] [Google Scholar]
- 31.Koeppel F, Poindessous V, Lazar V, Raymond E, Sarasin A, Larsen AK. Irofulven cytotoxicity depends on transcription-coupled nucleotide excision repair and is correlated with XPG expression in solid tumor cells. Clin. Cancer Res. 2004;10:5604–5613. doi: 10.1158/1078-0432.CCR-04-0442. [DOI] [PubMed] [Google Scholar]
- 32.Amato RJ, Perez C, Pagliaro L. Irofulven, a novel inhibitor of DNA synthesis, in metastatic renal cell cancer. Invest. New Drugs. 2002;20:413–417. doi: 10.1023/a:1020649827173. [DOI] [PubMed] [Google Scholar]
- 33.Neels JF, Gong J, Yu X, Sturla SJ. Quantitative correlation of drug bioactivation and deoxyadenosine alkylation by acylfulvene. Chem. Res. Toxicol. 2007;20:1513–1519. doi: 10.1021/tx7001756. [DOI] [PubMed] [Google Scholar]
- 34.Tanaka K, Inoue T, Tezuka Y, Kikuchi T. Michael-type addition of illudin S, a toxic substance from Lampteromyces japonicus, with cysteine and cysteine-containing peptides in vitro. Chem. Pharm. Bull. 1996;44:273–279. doi: 10.1248/cpb.44.273. [DOI] [PubMed] [Google Scholar]
- 35.McMorris TC, Yu J. Reaction of antitumor hydroxymethylacylfulvene (HMAF) with thiols. Tetrahedron. 1997;53:14579–14590. [Google Scholar]
- 36.McMorris TC, Yu J, Ngo HT, Wang H, Kelner MJ. Preparation and biological activity of amino acid and peptide conjugates of antitumor hydroxymethylacylfulvene. J. Med. Chem. 2000;43:3577–3580. doi: 10.1021/jm0000315. [DOI] [PubMed] [Google Scholar]
- 37.Tada M, Yamada Y, Bhacca NS, Nakanishi K, Ohashi M. Structure and reactions of illudin-S (Lampterol) Chem. Pharm. Bull. 1964;12:853–855. doi: 10.1248/cpb.12.853. [DOI] [PubMed] [Google Scholar]
- 38.Promega, editor. CellTiter 96® AQueous One Solution Cell Proliferation Assay. 2009. pp. a–13. [Google Scholar]
- 39.Flohé L, Günzler WA. Assays of glutathione peroxidase. Methods Enzymol. 1984;105:114–121. doi: 10.1016/s0076-6879(84)05015-1. [DOI] [PubMed] [Google Scholar]
- 40.Fang J, Lu J, Holmgren A. Thioredoxin reductase is irreversibly modified by curcumin: a novel molecular mechanism for its anticancer activity. J Biol. Chem. 2005;280:25284–25290. doi: 10.1074/jbc.M414645200. [DOI] [PubMed] [Google Scholar]
- 41.Kim J-R, Yoon HW, Kwon K-S, Lee S-R, Rhee SG. Identification of proteins containing cysteine residues that are sensitive to oxidation by hydrogen peroxide at neutral pH. Anal. Biochem. 2000;283:214–221. doi: 10.1006/abio.2000.4623. [DOI] [PubMed] [Google Scholar]
- 42.Jan Y-H, Heck DE, Gray JP, Zheng H, Casillas RP, Laskin DL, Laskin JD. Selective targeting of selenocysteine in thioredoxin reductase by the half mustard 2-chloroethyl ethyl sulfide in lung epithelial cells. Chem. Res. Toxicol. 2010;23:1045–1053. doi: 10.1021/tx100040k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Poindessous V, Koeppel F, Raymond E, Cvitkovic E, Waters SJ, Larsen AK. Enhanced antitumor activity of irofulven in combination with 5-fluorouracil and cisplatin in human colon and ovarian carcinoma cells. Int. J. Oncol. 2003;23:1347–1355. [PubMed] [Google Scholar]
- 44.Kelner MJ, McMorris TC, Montoya MA, Estes L, Uglik SF, Rutherford M, Samson KM, Bagnell RD, Taetle R. Characterization of acylfulvene histiospecific toxicity in human tumor cell lines. Cancer Chemother. Pharmacol. 1997;41:237–242. doi: 10.1007/s002800050734. [DOI] [PubMed] [Google Scholar]
- 45.Gallegos A, Berggren M, Gasdaska JR, Powis G. Mechanisms of the regulation of thioredoxin reductase activity in cancer cells by the chemopreventive agent selenium. Cancer Res. 1997;57:4965–4970. [PubMed] [Google Scholar]
- 46.Zhang J, Svehlikova V, Bao Y, Howie AF, Beckett GJ, Williamson G. Synergy between sulforaphane and selenium in the induction of thioredoxin reductase 1 requires both transcriptional and translational modulation. Carcinogenesis. 2003;24:497–503. doi: 10.1093/carcin/24.3.497. [DOI] [PubMed] [Google Scholar]
- 47.Yu SY, Ao P, Wang LM, Huang SL, Chen HC, Lu XP, Liu QY. Biochemical and cellular aspects of the anticancer activity of selenium. Biol. Trace Elem. Res. 1988;15:243–255. doi: 10.1007/BF02990141. [DOI] [PubMed] [Google Scholar]
- 48.Novoselov SV, Calvisi DF, Labunskyy VM, Factor VM, Carlson BA, Fomenko DE, Moustafa ME, Hatfield DL, Gladyshev VN. Selenoprotein deficiency and high levels of selenium compounds can effectively inhibit hepatocarcinogenesis in transgenic mice. Oncogene. 2005;24:8003–8011. doi: 10.1038/sj.onc.1208940. [DOI] [PubMed] [Google Scholar]
- 49.Gundimeda U, Schiffman JE, Gottlieb SN, Roth B, Gopalakrishna R. Negation of the cancer-preventive actions of selenium by overexpression of protein kinase Cε and selenoprotein thioredoxin reductase. Carcinogenesis. 2009;30:1553–1561. doi: 10.1093/carcin/bgp164. [DOI] [PubMed] [Google Scholar]
- 50.Renko K, Hofmann PJ, Stoedter M, Hollenbach B, Behrends T, Kohrle J, Schweizer U, Schomburg L. Down-regulation of the hepatic selenoprotein biosynthesis machinery impairs selenium metabolism during the acute phase response in mice. FASEB J. 2009;23:1758–1765. doi: 10.1096/fj.08-119370. [DOI] [PubMed] [Google Scholar]
- 51.Karunasinghe N, Ferguson LR, Tuckey J, Masters J. Hemolysate thioredoxin reductase and glutathione peroxidase activities correlate with serum selenium in a group of New Zealand men at high prostate cancer risk. J. Nutr. 2006;136:2232–2235. doi: 10.1093/jn/136.8.2232. [DOI] [PubMed] [Google Scholar]
- 52.Allmang C, Wurth L, Krol A. The selenium to selenoprotein pathway in eukaryotes: more molecular partners than anticipated. Biochim. Biophys. Acta. 2009;1790:1415–1423. doi: 10.1016/j.bbagen.2009.03.003. [DOI] [PubMed] [Google Scholar]
- 53.Lu J, Holmgren A. Selenoproteins. J. Biol. Chem. 2009;284:723–727. doi: 10.1074/jbc.R800045200. [DOI] [PubMed] [Google Scholar]
- 54.Donovan J, Copeland PR. Threading the needle: getting selenocysteine into proteins. Antioxid. Redox Signaling. 2010;12:881–892. doi: 10.1089/ars.2009.2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gong J, Neels JF, Yu X, Kensler TW, Peterson LA, Sturla SJ. Investigating the role of stereochemistry in the activity of anticancer acylfulvenes: synthesis, reductase-mediated bioactivation, and cellular toxicity. J. Med. Chem. 2006;49:2593–2599. doi: 10.1021/jm051104t. [DOI] [PubMed] [Google Scholar]
- 56.Woynarowska BA, Woynarowski JM, Herzig MC, Roberts K, Higdon AL, MacDonald JR. Differential cytotoxicity and induction of apoptosis in tumor and normal cells by hydroxymethylacylfulvene (HMAF) Biochem. Pharmacol. 2000;59:1217–1226. doi: 10.1016/s0006-2952(00)00254-9. [DOI] [PubMed] [Google Scholar]
- 57.Woynarowski JM, Napier C, Koester SK, Chen SF, Troyer D, Chapman W, MacDonald JR. Effects on DNA integrity and apoptosis induction by a novel antitumor sesquiterpene drug, 6-hydroxymethylacylfulvene (HMAF, MGI 114) Biochem. Pharmacol. 1997;54:1181–1193. doi: 10.1016/s0006-2952(97)00321-3. [DOI] [PubMed] [Google Scholar]
- 58.McMorris TC, Kelner MJ, Wang W, Moon S, Taetle R. On the mechanism of toxicity of illudins: the role of glutathione. Chem. Res. Toxicol. 1990;3:574–579. doi: 10.1021/tx00018a013. [DOI] [PubMed] [Google Scholar]
- 59.Arscott LD, Gromer S, Schirmer RH, Becker K, Williams CH., Jr. The mechanism of thioredoxin reductase from human placenta is similar to the mechanisms of lipoamide dehydrogenase and glutathione reductase and is distinct from the mechanism of thioredoxin reductase from Escherichia coli. Proc. Natl. Acad. Sci. U.S.A. 1997;94:3621–3626. doi: 10.1073/pnas.94.8.3621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Williams CH, Arscott LD, Muller S, Lennon BW, Ludwig ML, Wang PF, Veine DM, Becker K, Schirmer RH. Thioredoxin reductase two modes of catalysis have evolved. Eur. J. Biochem. 2000;267:6110–6117. doi: 10.1046/j.1432-1327.2000.01702.x. [DOI] [PubMed] [Google Scholar]
- 61.Zhong L, Holmgren A. Essential role of selenium in the catalytic activities of mammalian thioredoxin reductase revealed by characterization of recombinant enzymes with selenocysteine mutations. J Biol. Chem. 2000;275:18121–18128. doi: 10.1074/jbc.M000690200. [DOI] [PubMed] [Google Scholar]
- 62.Hashemy SI, Ungerstedt JS, Avval F. Zahedi, Holmgren A. Motexafin gadolinium, a tumor-selective drug targeting thioredoxin reductase and ribonucleotide reductase. J. Biol. Chem. 2006;281:10691–10697. doi: 10.1074/jbc.M511373200. [DOI] [PubMed] [Google Scholar]
- 63.Dal Piaz F, Braca A, Belisario MA, De Tommasi N. Thioredoxin system modulation by plant and fungal secondary metabolites. Curr. Med. Chem. 2010;17:479–494. doi: 10.2174/092986710790226165. [DOI] [PubMed] [Google Scholar]
- 64.Urig S, Becker K. On the potential of thioredoxin reductase inhibitors for cancer therapy. Semin. Cancer Biol. 2006;16:452–465. doi: 10.1016/j.semcancer.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 65.Bindoli A, Rigobello MP, Scutari G, Gabbiani C, Casini A, Messori L. Thioredoxin reductase: a target for gold compounds acting as potential anticancer drugs. Coord. Chem. Rev. 2009;253:1692–1707. [Google Scholar]
- 66.Tonissen KF, Di Trapani G. Thioredoxin system inhibitors as mediators of apoptosis for cancer therapy. Mol. Nutr. Food Res. 2009;53:87–103. doi: 10.1002/mnfr.200700492. [DOI] [PubMed] [Google Scholar]
- 67.Ladenstein R, Epp O, Bartels K, Jones A, Huber R, Wendel A. Structure analysis and molecular model of the selenoenzyme glutathione peroxidase at 2.8 A resolution. J. Mol. Biol. 1979;134:199–218. doi: 10.1016/0022-2836(79)90032-9. [DOI] [PubMed] [Google Scholar]
- 68.Sun QA, Wu Y, Zappacosta F, Jeang KT, Lee BJ, Hatfield DL, Gladyshev VN. Redox regulation of cell signaling by selenocysteine in mammalian thioredoxin reductases. J. Biol. Chem. 1999;274:24522–24530. doi: 10.1074/jbc.274.35.24522. [DOI] [PubMed] [Google Scholar]
- 69.Nordberg J, Zhong L, Holmgren A, Arner ES. Mammalian thioredoxin reductase is irreversibly inhibited by dinitrohalobenzenes by alkylation of both the redox active selenocysteine and its neighboring cysteine residue. J. Biol. Chem. 1998;273:10835–10842. doi: 10.1074/jbc.273.18.10835. [DOI] [PubMed] [Google Scholar]
- 70.Arnér ES, Nakamura H, Sasada T, Yodoi J, Holmgren A, Spyrou G. Analysis of the inhibition of mammalian thioredoxin, thioredoxin reductase, and glutaredoxin by cis-diamminedichloroplatinum (II) and its major metabolite, the glutathione-platinum complex. Free Radical Biol. Med. 2001;31:1170–1178. doi: 10.1016/s0891-5849(01)00698-0. [DOI] [PubMed] [Google Scholar]
- 71.Anestål K, Arnér ESJ. Rapid induction of cell death by selenium-compromised thioredoxin reductase 1 but not by the fully active enzyme containing selenocysteine. J. Biol. Chem. 2003;278:15966–15972. doi: 10.1074/jbc.M210733200. [DOI] [PubMed] [Google Scholar]
- 72.Anestål K, Prast-Nielsen S, Cenas N, Arnér ESJ. Cell death by SecTRAPs: thioredoxin reductase as a prooxidant killer of cells. PLoS One. 2008;3:e1846. doi: 10.1371/journal.pone.0001846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cassidy PB, Edes K, Nelson CC, Parsawar K, Fitzpatrick FA, Moos PJ. Thioredoxin reductase is required for the inactivation of tumor suppressor p53 and for apoptosis induced by endogenous electrophiles. Carcinogenesis. 2006;27:2538–2549. doi: 10.1093/carcin/bgl111. [DOI] [PubMed] [Google Scholar]
- 74.Tamura T, Stadtman TC. A new selenoprotein from human lung adenocarcinoma cells: purification, properties, and thioredoxin reductase activity. Proc. Natl. Acad. Sci. U.S.A. 1996;93:1006–1011. doi: 10.1073/pnas.93.3.1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hu Y, Urig S, Koncarevic S, Wu X, Fischer M, Rahlfs S, Mersch-Sundermann V, Becker K. Glutathione- and thioredoxin-related enzymes are modulated by sulfur-containing chemopreventive agents. Biol. Chem. 2007;388:1069–1081. doi: 10.1515/BC.2007.135. [DOI] [PubMed] [Google Scholar]
- 76.Vadgama JV, Wu Y, Shen D, Hsia S, Block J. Effect of selenium in combination with Adriamycin or Taxol on several different cancer cells. Anticancer Res. 2000;20:1391–1414. [PubMed] [Google Scholar]
- 77.Fakih M, Cao S, Durrani FA, Rustum YM. Selenium protects against toxicity induced by anticancer drugs and augments antitumor activity: a highly selective, new, and novel approach for the treatment of solid tumors. Clin. Colorectal Cancer. 2005;5:132–135. doi: 10.3816/ccc.2005.n.026. [DOI] [PubMed] [Google Scholar]
- 78.Rudolf E, Radocha J, Cervinka M, Cerman J. Combined effect of sodium selenite and campthotecin on cervical carcinoma cells. Neoplasma. 2004;51:127–135. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.