

Abstract
Background
Autophagy is essential to intracellular homeostasis and involved in the pathophysiology of a variety of diseases. Mechanisms regulating selective autophagy remain poorly understood. The COP9 signalosome (CSN) is a conserved protein complex consisting of 8 subunits (CSN1 through CSN8) and known to regulate the ubiquitin-proteasome system (UPS). However, it is unknown whether CSN plays a role in autophagy.
Methods and Results
Marked increases in LC3-II and p62 proteins were observed upon Csn8 depletion in the cardiomyocytes of mouse hearts with cardiomyocyte-restricted knockout of the gene encoding CSN subunit 8 (CR-Csn8KO). The increases in autophagosomes were confirmed by probing with GFP-LC3 and electron microscopy. Autophagic flux assessments revealed that defective autophagosome removal was the cause of autophagosome accumulation and occurred prior to a global UPS impairment in Csn8-deficient hearts. Analyzing the prevalence of different stages of autophagic vacuoles revealed defective autophagosome maturation. Down-regulation of Rab7 was found to strikingly co-localize with the autophagosome accumulation at the individual cardiomyocyte level. A significantly higher percent of cardiomyocytes with autophagosome accumulation underwent necrosis in CR-Csn8KO hearts. Chronic lysosomal inhibition with Chloroquine induced cardiomyocyte necrosis in mice. Rab7 knockdown impaired autophagosome maturation of non-selective and selective autophagy and exacerbated cell death induced by proteasome inhibition in cultured cardiomyocytes.
Conclusions: (1)
Csn8/CSN is a central regulator in not only the proteasomal proteolytic pathway but also selective autophagy; (2) likely through regulating the expression of Rab7, Csn8/CSN plays a critical role in autophagosome maturation; and (3) impaired autophagosome maturation causes cardiomyocytes to undergo necrosis.
Keywords: the COP9 signalosome, autophagy, lysosome, Rab7, necrosis
Macroautophagy (commonly known as autophagy) sequesters a portion of the cytoplasm into a double-membrane vesicle (known as an autophagosome) for fusion with, and degradation by, lysosomes. The fusion of autophagosomes with lysosomes is also known as autophagosome maturation and critical to the removal of autophagosomes. Autophagy is essential to maintaining homeostasis in the cell. Non-selective autophagy during starvation helps the cell to survive temporarily energy crisis by self-eating a portion of its cytoplasm.1 Selective autophagy, on the other hand, serves as a major executor of quality control by removing aged/damaged organelles and aggregated proteins.1–3 Alterations in autophagy were observed in many diseases and are emerging as important pathogenic factors and therapeutic targets.4–9 However, the understanding on the regulation of autophagy,10 especially the selective autophagy, remains rudimentary.
The COP9 signalosome (CSN) is an evolutionarily conserved protein complex consisting of 8 unique subunits (CSN1 through CSN8). All 8 subunits are required for CSN holo-complex assembly and functioning. The bona fide biochemical activity of CSN is cullin deneddylation, which regulates the dynamics of cullin-based RING ligases, a large family of ubiquitin E3 ligases. Hence, CSN research has so far focused on the regulation of CSN on ubiquitin-proteasome system (UPS) mediated proteolysis.11, 12
We have recently demonstrated that perinatal cardiomyocyte-restricted Csn8 knockout (CR-Csn8KO) causes dilated cardiomyopathy and premature death in mice. Impaired UPS-mediated degradation of misfolded proteins and massive cardiomyocyte necrosis were among the likely causes. Ubiquitinated proteins are accumulated in an aggregated form in CR-Csn8KO mouse hearts.12 Protein aggregation and proteasome inhibition were shown to activate autophagy in cardiomyocytes.13 Increased autophagosomes are often found to co-localize with cell death in failing hearts although the significance of this phenomenon remains mysterious.14 Hence, we sought to investigate the impact of Csn8 deficiency on cardiac autophagy and tested a link between autophagy impairment and cardiomyocyte necrosis. We have discovered that Csn8/CSN is required for autophagosome maturation and autophagosome flux in the heart and that Csn8/CSN regulates the autophagic-lysosomal pathway through supporting Rab7 expression. Impaired autophagosome maturation can cause cardiomyocyte necrosis.
METHODS
Animal models
CR-Csn8KO was achieved using the Cre-loxP system as described,12 in which transgenic Cre expression is driven by the mouse α-myosin heavy chain (Mhc6) promoter. The Csn8flox/flox::Mhc6-CreNTG littermates of CR-Csn8KO (Csn8flox/flox::Mhc6-CreTG) mice were used as control (CTL). Transgenic mice expressing the green fluorescence protein (GFP) fused LC3 (GFP-LC3) were described and generously donated by Dr. Noboru Mizushima.15 The care and use of animals in this study conform to institutional guidelines.
Western blot analyses
Protein extraction from either myocardial tissues or cultured neonatal rat ventricular cardiomyocytes (NRVMs), protein concentration determination with BCA reagents (Pierce), SDS-PAGE, immunoblotting analysis, and densitometry were performed as previously described.12
Transmission electron microscopy (TEM)
TEM was performed as we recently described.3 See Online Supplements for a detailed method.
Assessing autophagic flux in the heart
This is performed as previously described with minor modifications.3 Briefly, mice were intraperitoneally injected with bafilomycin-A1 (BFA, 3μmol/kg, Sigma). Tissues were collected 1 hour after the injection for assessing LC3-II protein levels and GFP-LC3 puncta.
Quantification of autophagic and lysosomal vesicles in myocardium
Three mouse hearts per group, 3 sections per heart, and 3 fields per section were assessed. An identical threshold was utilized to subtract the background fluorescence of both CTL and CR-Csn8KO myocardial sections. The co-localization between GFP-LC3 direct fluorescence (autophagic vacuoles) and LAMP1 immunofluorescence (lysosomal vesicles) was assessed by the MetaMorph software as described.16 The cardiomyocyte compartment area in an image was measured by the Image-Pro Plus software (LEEDS).
Cell culture and siRNA transfection
The isolation and culture of neonatal rat ventricular cardiomyocytes (NRVMs) were performed as previously described.17 For glucose deprivation (GD), myocytes were washed twice with phosphate buffered saline (PBS) and incubated with glucose-free and serum-free DMEM (Gibco). The small interference RNA (siRNA) specifically against rat Rab7 and luciferase were purchased from Qiagen.
Probing autophagosome maturation with a tandem fluorescence LC3 (tf-LC3)
To differentiate autolysosomes from autophagosomes, we infected cultured NRVMs with a recombinant adenoviral vector expressing a tf-LC3 (Ad-tf-LC3, a gift from J. Sadoshima of UMDNJ, Nework, NJ).10 The tf-LC3 is a modified LC3 (microtubule-associated protein 1 light chain 3) with N-terminal fusion of an enhanced green fluorescence protein (GFP) and a monomeric red fluorescence protein (mRFP) in tandem.18 During live cell fluorescence imaging, the tf-LC3 containing autophagosomes show as yellow puncta because both GFP and mRFP fused with LC3 fluoresce; however, the tf-LC3 labeled autolysosomes emit only red fluorescence from mRFP because the acidic environment of autolysosomes quenches GFP but not mRFP.18 In our experiments, direct fluorescence images of live cultured NRVMs were captured with an epi-fluorescent microscope (Zeiss Axiovert 200M). The number of green, red, and yellow fluorescent puncta in each cell was manually counted from at least 3 different myocyte preparations. At least 50 cells per group were scored in each experiment.
LDH activity assay
This was performed with a Cytotoxicity Detection Kit (LDH) (Roche) as previously described.3
Propidium iodide (PI) uptake assay
Cultured NRVMs were incubated with PI-containing medium (5μg/mL) for 10 min. The cells were then washed with cold PBS for 3 times, fixed with 4% paraformaldehyde, counterstained with DAPI (Sigma-Aldrich), and imaged by an epi-fluorescence microscope (Zeiss Axiovert 200M). The numbers of PI positive cells were normalized by the nuclei numbers revealed by DAPI. A total of over 3000 cells from10 random fields in each group were scored.
Statistical Analysis
All continuous variables are presented as mean ± SD unless otherwise indicated. Differences between two groups were evaluated for statistical significance using Student’s t-test when sample size was appropriate and the population distributed normally; otherwise, the Mann-Whitney U test was used and the data were summarized using standard box plots. When difference among 3 or more groups was evaluated, one-way analysis of variance (ANOVA) or when appropriate, 2-way ANOVA, followed by the Holm-Sidak test for pair-wise comparisons were performed. The P value <0.05 were considered statistically significant but a Bonferroni correction was applied to the comparisons of the same parameter at multiple ages.
RESULTS
Csn8 deficiency increases autophagosomes in the heart
Conversion of LC3-I to LC3-II and the incorporation of LC3-II into autophagic membranes mark important events in autophagic activation and autophagosome formation. Therefore, the protein abundance of LC3-II is commonly used as a biochemical parameter of the steady level of autophagosomes.1 As previously reported,12 the depletion of Csn8 protein in CR-Csn8KO hearts occurred between postnatal day 1 and day 7. Concurrently, significant increases in LC3-II proteins were observed in CR-Csn8KO hearts by 1 week of age and the increase maintained thereafter (Figure 1A, B). Interestingly, western blot analyses revealed no significant change in beclin1, autophagy related protein 5 (Atg5), and Atg7 in CR-Csn8KO hearts, compared with their littermate CTL (Figure 1C). To track autophagosomes, transgenic GFP-LC3 whose punctate distribution marks autophagosomes,15 was cross-bred into the CR-Csn8KO background. GFP-positive puncta (Figure 1D; see Supplementary Figure I for puncta quantification) and GFP-LC3-II protein levels (Figure 1E, 1F) were significantly increased in CR-Csn8KO hearts. These data indicate that autophagosomes were significantly increased in CR-Csn8KO hearts. We then performed TEM to detect autophagic vacuoles in the cardiomyocytes of CR-Csn8KO and CTL hearts. We found massive increases in autophagosomes in CR-Csn8KO hearts. These autophagic vacuoles frequently enclose mitochondria and most do not show signs of lysis of their contents, suggesting that they are early stage autophagosomes (Figure 1G).
Figure 1. Increased abundance of autophagosomes in CR-Csn8KO hearts.

In all figures, either Csn8KO or KO is used to indicate CR-Csn8KO. A, Representative western blot images for the indicated proteins in myocardium from mice of the indicated age. GAPDH was probed as loading control. B, A summary of the densitometric data of LC3-II from the experiments as illustrated in panel A. *p<0.05, #p<0.01 vs. CTL, Student’s t-test. C, Western blot analyses for the indicated proteins in the heart at 3 weeks (wk). D, Confocal micrographs of myocardial GFP-LC3 direct fluorescence from CTL/GFP-LC3 and CR-Csn8KO/GFP-LC3 mice at 3 weeks. Bar=10 μm. E and F, Western blot analyses of myocardial GFP-LC3 in mice as described in D. Representative images (E) and a summary of the densitometric quantification (F) are shown. n = 4 for each group; #p<0.01 vs. CTL. G, Electron micrographs of ventricular myocardium from 3-week-old mice. Representative images from CTL (a) and CR-Csn8KO (b~e) are shown. Panels c~e are magnified images respectively from the indicated areas of panel b, illustrating examples of autophagosomes (arrows). Bar=1 μm.
Autophagosome removal is impaired prior to UPS proteolytic function impairment in Csn8 deficient hearts
p62/SQSTM1 is a substrate of selective autophagy and accumulates when the autophagic-lysosomal pathway is disrupted.19 Our western blot analyses revealed that p62 protein levels in Csn8-deficient hearts were significantly increased at 1 week of age and the increase became more pronounced at 3 weeks (Figure 2A, 2B). Marked increases of p62-positive foci were also observed by immunofluorescence (186 ± 22 vs. 8 ± 3 dots/104 μm2 for CTLs, p=0.0001). The p62-positive dots were largely co-localized with GFP-LC3 puncta (Supplementary Figure S1), suggesting that the accumulated p62 proteins are mostly associated with autophagosomes. These findings are consistent with a decrease in autophagosome removal in CR-Csn8KO hearts. This is indeed confirmed by directly comparing autophagic flux in CR-Csn8KO and CTL hearts. CR-Csn8KO and CTL mice at 2 and 3 weeks of age were intraperitoneally injected with saline or bafilomycin-A1 (BFA, 3μmol/kg), a widely used lysosomal inhibitor.1 At 2 weeks of age, BFA treatment increased LC3-II significantly in both the CTL and the CR-Csn8KO hearts, compared with their corresponding saline treated controls; but the increase is much less in the CR-Csn8KO mice than in the CTL mice (25% vs. 270%). At 3 weeks of age, the BFA treatment caused a 2.8-fold increase of LC3-II over the saline treatment in the CTL mouse hearts. However in the CR-Csn8KO hearts where LC3-II was already high at the baseline, BFA failed to further elevate the LC3-II level (Figure 2C, 2D). As a control, the BFA treatment induced similar degrees of LC3-II accumulation in the livers of CR-Csn8KO and CTL mice at both 2 and 3 weeks of age (Supplementary Figure S2). Consistently, BFA treatment significantly accumulated GFP-LC3 puncta in CTL hearts, but failed to do so in the hearts of CR-Csn8KO mice at 3 weeks (Supplementary Figure S3). These findings indicate that Csn8 deficiency impairs autophagosome removal, leading to accumulation of autophagosomes in cardiomyocytes. To delineate the temporal relationship between defective autophagy and the impaired UPS-mediated protein degradation in CR-Csn8KO hearts, we further determined the time course of UPS malfunction by introducing GFPdgn, a validated reverse reporter of UPS function,20 into CR-Csn8KO and CTL littermate mice by cross-breeding as previously described.12 GFPdgn proteins were significantly increased in the hearts of CR-Csn8KO mice at 3 weeks but not at 2 weeks of age, compared with their littermate controls (Figure 2E, 2F). These data indicate that impaired degradation of UPS substrates in the CR-Csn8KO heart is not discernible until after 2 weeks of age, which is after impaired autophagosome removal has occurred.
Figure 2. Autophagic flux assessments.

A, and B, Time course of changes in p62 protein expression in CR-Csn8KO mouse hearts. Representative images (A) and a summary of densitometry data (B) of western blot analyses of p62 are shown. *p<0.05, #p<0.01 vs. CTL; Student’s t-test. C and D, Autophagic flux assays based on bafilomycin-A1 (BFA) induced changes in the endogenous LC3-II protein level. Three-week-old CTL and CR-Csn8KO mice were treated with BFA (3 μmol/kg, i.p.) or vehicles and sacrificed 1 hour after the BFA injection. LC3 protein levels in ventricular myocardium and the liver tissue were quantified using western blot analysis. Representative western blot images (C) and densitometric quantification (D) of LC3 proteins are presented. n=4 mice for each group. *p<0.05, #p<0.01, 2-way ANOVA followed by Holm-Sidak test; N.S., not significant. E and F, Probing myocardial UPS proteolytic function in CR-Csn8KO mice using a surrogate UPS substrate (GFPdgn). Through cross-breeding, transgenic GFPdgn was introduced into CR-Csn8KO and CTL mice, The resultant CTL/GFPdgn and CR-Csn8KO/GFPdgn mice at 2 and 3 weeks of age were examined for myocardial protein levels of GFPdgn. Representative images of western blot analyses for GFPdgn (E) and a summary of changes in GFPdgn protein levels (F) are shown. L.C., a non-specific band used as loading control. *p<0.05 vs. CTL.
Changes in lysosomal genesis in Csn8 deficient hearts
The decreased removal of autophagosomes can be caused by a decrease in lysosomes. Hence we investigated the impact of Csn8 deficiency on lysosomal genesis in the heart. Compared with CTL, the levels of two major lysosomal membrane sialoglycoproteins (LAMP-1 and LAMP-2) were markedly increased in CR-Csn8KO hearts, so were the intermediate and mature forms of cathepsin-D, but not the cathepsin-D precursor (Figure 3A, Supplementary Figure S4). Cathepsin-D activity was also increased (Figure 3B). Consistently, double immunostaining of LAMP1 and cathepsin-D showed that LAMP1-positive vesicles and cathepsin D positive vesicles were both significantly increased (Figure 3C, 3D); however, the density of vesicles positive for both LAMP1 and cathepsin-D, which is indicative of lysosomes, remained unchanged in CR-Csn8KO cardiomyocytes (Figure 3D). Interestingly, the relative abundance of CathD+/LAMP1+ dots, to either CathD+ or LAMP1+ dots, were all significantly reduced in CR-csn8KO hearts (Figure 3E, 3F). A plausible interpretation of these results is that Csn8 deficient cardiomyocytes attempt to increase their lysosomal synthesis by synthesizing more cathepsin-D and LAMP’s but these lysosomal components fail to productively assemble a greater number of lysosomes. Nonetheless, it appears that a normal number of lysosomes are available for fusion with autophagosomes because the total number of lysosomes was not decreased in the CR-Csn8KO cardiomyocytes.
Figure 3. Analyses of lysosomal genesis in mouse hearts.

A, , Western blot analyses of indicated lysosomal proteins. B, Changes in cathepsin D activities. C~F, Confocal microscopic analyses of LAMP1 and cathepsin D (Cath D) positive vesicles. Perfusion fixed ventricular myocardial sections from the CTL and CR-Csn8KO mice were double-immunofluorescence stained for LAMP1 (red), a lysosomal membrane protein, and cathepsin-D (green), a lysosomal protease. Representative confocal micrographs are shown in panel C. The number densities of cathepsin D positive (Cath D+), LAMP1 positive (LAMP1+), and Cath D/LAMP1 double positive (Cath D+/LAMP1+) vesicles are summarized in panel D. The percentage of the Cath D/LAMPdouble positive vesicles (i.e., lysosomes) over the total number of Cath D+ vesicles or over the total number of LAMP1+ vesicles are respectively presented in panels E and F. *p<0.05 vs. CTL.
Fusion between autophagosomes and lysosomes is impaired in Csn8 deficient hearts
The removal of autophagosomes by lysosomes occurs after the autophagosome fuses with the lysosome to form autolysosomes. Therefore, the accumulation of autophagic vacuoles could result from a blockage in the fusion between autophagosomes with lysosomes or from a defect in lysosomal proteolysis in the autolysosomes.1 A defect in the fusion is characterized by a reduced amount of autolysosomes relative to autophagosomes or lysosomes; whereas a defect in lysosomal proteolysis after fusion would increase the relative prevalence of autolysosomes.1 We measured the density of autophagic vacuoles, lysosomes, and autolysosomes respectively through quantification of GFP-LC3 puncta (autophagosomes and autolysosomes), LAMP1 positive dots (lysosomes and autolysosomes), and GFP-LC3::LAMP1 double positive dots (autolysosomes only). Confocal microscopy was performed with myocardial cryosections from the CR-Csn8KO and CTL mice (Figure 4A). To better understand the changes in membrane dynamics caused by Csn8 deficiency, we sought to compare the changes in the relative abundance of the different stages of autophagic vesicles in CR-Csn8KO hearts with those observed in the heart of GFP-LC3 tg mice that had undergone 12 hours of starvation (the Starved group), or had received an intravenous injection of proteasomal inhibitor MG262 (the MG262 group) (see Supplementary Figure S5 for representative confocal micrographs). Both starvation and proteasome inhibition are known to activate autophagy and increase autophagic flux.
Figure 4. Analyses of the changes in autophagic vacuole dynamics caused by Csn8 deficiency, starvation, and proteasome inhibition.

A, , Confocal microscopic analyses of the abundance of autophagic vacuoles, lysosomes, and autolysosomes in mouse hearts. The GFP-LC3 transgene was introduced to the CR-Csn8KO and CTL background through cross-breeding and the resultant CTL/GFP-LC3 and CR-Csn8KO/GFP-LC3 mice at 3-weeks were examined at the baseline. Cryosections of ventricular myocardium were immunostained for LAMP1 (red) to identify lysosomes. Representative duo-color images of GFP-LC3 direct fluorescence (green) and LAMP1 immunofluorescence (red) are shown. The insets are enlarged images of asterisk-marked areas. Arrowheads point to GFP- and LAMP1- co-localized dots. Bar=10 μm. B~D, Box plots summarizing changes in the density of autophagic vacuoles (B), lysosomes (C), and autolysosomes (D). E and F, Box plots summarizing changes in the relative density of autolysosomes among total number of lysosomal vacuoles (E) or among total number of autophagic vacuoles (F) in the heart of CR-Csn8KO, Starved, or MG262-treated mice. Mouse grouping was the same as described in Figure 4A. The numbers of LAMP1 positive (LAMP1 dots, all lysosomes), GFP-LC3 positive (GFP dots, all autophagic vacuoles), and GFP::LAMP1 double positive (GFP::LAMP1 dots, autolysosomes) puncta were analyzed using the images collected as described in panel A (see Methods for details). *#p<0.01; N.S., not significant; the Mann-Whitney U test.
Compared with the corresponding control groups, the density of autophagic vacuoles (i.e., GFP-LC3 puncta) was significantly increased in the cardiomyocytes of all three groups (CR-Csn8KO, the Starved, and the MG262 treated) (Figure 4B). However, the density of lysosomes (i.e., LAMP1 positive puncta) was increased only in the CR-Csn8KO group but not in the Starved or the MG262 groups (Figure 4C). Notably, the density of autolysosomes (i.e., GFP-LC3/LAMP1 double positive puncta) was significantly increased in the Starved and the MG262 groups but not in CR-Csn8KO (Figure 4D). Consequently, the abundance of autolysosomes in CR-Csn8KO cardiomyocytes is remarkably decreased relative to either autophagic vacuoles or lysosomal vacuoles (Figure 4E and 4F). By contrast, the Starved and the MG262 groups contain a higher relative abundance of autolysosomes to all lysosomal vacuoles and unchanged relative to autophagic vacuoles (Figure 4E and 4F), compared with their respective control groups. These analyses reveal that the nature of the increased autophagic vacuoles in CR-Csn8KO hearts differs sharply from that caused by starvation or proteasome inhibition in that Csn8 deficiency impairs autophagosome maturation whereas starvation and proteasome inhibition enhances autophagosome maturation. Furthermore, these data strongly suggest that a reduced ability of autophagosomes to fuse with lysosomes contributes, at least in part, to the defective removal of autophagosomes in the CR-Csn8KO mouse hearts.
Severe impairment of autophagosome maturation may be a cause of cardiomyocyte necrosis in CR-Csn8KO mice
As we previously reported,12 a marked increase in cardiomyocyte necrosis, as detected by Evan’s blue dye (EBD) uptake and increased leukocyte infiltration, is present in CR-Csn8KO mouse hearts before elevated apoptosis becomes discernible. The necrotic nature of the EBD-positive cardiomyocytes is confirmed by intracellular positive staining for mouse endogenous Ig (Supplementary Figure S6) and the increased uptake of intravenously injected rabbit anti-desmin antibodies (data not shown). Based on the findings described so far, we reasoned that a greater autophagosome accumulation in a cardiomyocyte of the CR-Csn8KO heart is indicative of more severe impairment in autophagosome maturation in the cell. The heterogeneity of GFP-LC3 puncta accumulation among cardiomyocytes in CR-Csn8KO hearts (Figure 1D, 4A) allows us to determine whether impaired autophagosome maturation correlates to the increased necrosis at the individual cardiomyocyte level. Quantification of GFP-LC3 puncta in the EBD-negative cardiomyocytes of CR-Csn8KO hearts showed that the 95% confidence range of the GFP-LC3 puncta number density is from 1 to 16 per 100 μm2. Accordingly, all cardiomyocytes (EBD+ and EBD−) in CR-Csn8KO hearts were divided into two groups: Group A containing 16 or fewer GFP-LC3 puncta per 100μm2 and Group B containing >16 puncta per 100 μm2. The incidence of increased EBD uptake cells was analyzed between the two groups. Group B showed a significantly higher percent of cells that are EBD positive than Group A (52% vs. 16%; p<0.0001; Figure 5A, 5B and Supplementary Table 1). These data suggest that impairment of autophagic maturation might be an underlying cause for increased cardiomyocyte necrosis in Csn8 deficient hearts. The sufficiency of blocking autophagic flux to promote cardiomyocyte necrosis in intact mice was then demonstrated by treating adult GFP-LC3 tg mice respectively with Chloroquine (CQ; 10 mg/kg, i.p., daily) or saline for 3 weeks. Increased EBD positive cardiomyocytes and myocardial CD45 cell infiltration were detected in the CQ-treated mice, compared with the vehicle treated mice (Figure 5C, 5D).
Figure 5. Impaired autophagosome maturation causes cardiomyocyte necrosis in mice.
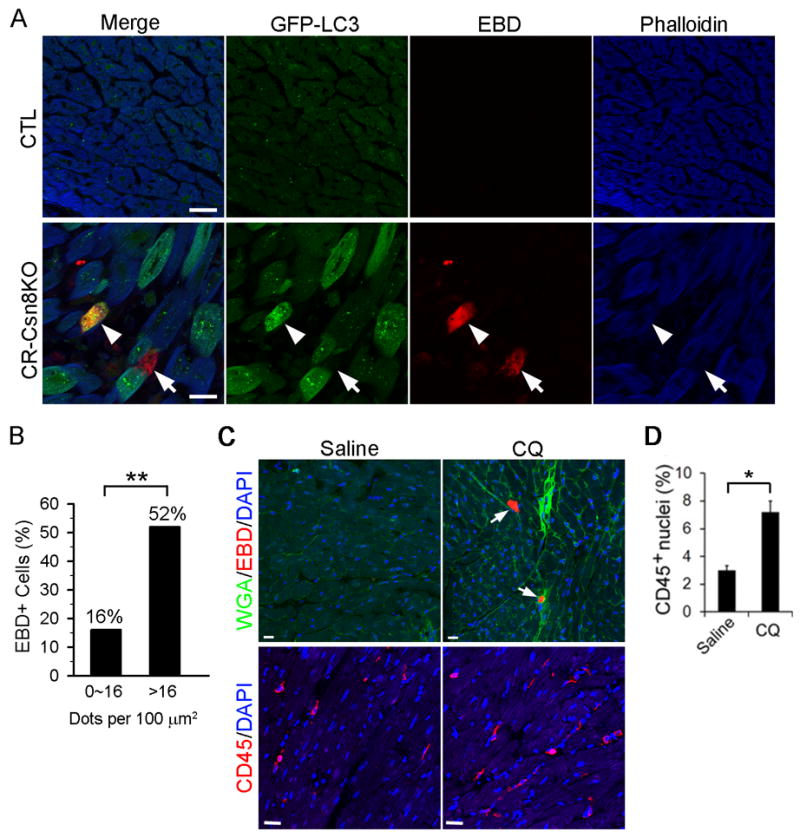
A, and B, Cardiomyocytes with severe accumulation of autophagosomes more likely undergo necrosis (arrowhead). GFP-LC3::CR-Csn8KO and GFP-LC3::CTL mice at 3 weeks were administered with Evan’s blue dye (EBD, 100 mg/kg, i.p.) 18 hours before the heart was flushed with saline and fixed with 4% paraformaldehyde via retrograde perfusion through the abdominal aorta. Cryosections of ventricular myocardium were stained with Alexa-568 conjugated Phalloidin to identify cardiomyocytes. The sections were imaged with fluorescence confocal microscopy for GFP-LC3 (green), EBD (red), and F-actin (blue). Representative confocal micrographs (A) and a comparison of the prevalence of EBD positive cells in CR-Csn8KO hearts between the cardiomyocytes containing 16 or fewer GFP-LC3 puncta per 100 μm2 and those showing >16 puncta per 100 μm2 (B) are presented. Scale bar=20 μm; **p<0.0001, Fisher’s exact test. C and D, chronic blockade of autophagic flux causes cardiomyocytes necrosis (arrows) in wild type mice. Adult mice were injected with Chloroquine (CQ, 10 mg/kg, daily) or saline for 3 weeks. At the end of the experiment, mice were injected with EBD and ventricular myocardium samples were processed to detect cardiomyocyte EBD uptake (C) as described in panel A. The sections were counterstained with wheat germ agglutinin (WGA, green) and DAPI (blue) to reveal cell membrane and the nuclei. An adjacent set of sections were immunostained for CD45 (red) and the nuclei were stained with DAPI (blue) for quantification of the percentage of CD45 positive nuclei among all nuclei in the field (D). Bar=20μm; n=3 mice/group; *p<0.05.
Rab7 down-regulation co-localizes with, and correlates to, autophagosome accumulation in the cardiomyocytes of CR-Csn8KO mouse hearts
The mechanisms underlying autophagosome maturation remain poorly understood. Nevertheless, a small G protein Rab7 has been shown to play an indispensible role in autophagosome maturation and lysosomal genesis in cultured cells.21 It was recently shown that increased Rab7 expression is required for starvation-induced autophagy in cardiomyocytes.10 To explore the molecular mechanisms underlying the defective selective autophagy in Csn8-deficient cardiomyocytes, we measured Rab5, Rab7, and Rab11 protein expression and found a significant up-regulation of both Rab5 and Rab11 but a remarkable down-regulation of Rab7 in CR-Csn8KO mouse hearts (Figure 6A, 6B). Furthermore, a potential causal relationship between Rab7 down-regulation and the impaired autophagosome removal is suggested by our examination of Rab7 cellular distribution in CTL/GFP-LC3 and CR-Csn8KO/GFP-LC3 mouse hearts. In the CTL, Rab7 is readily detectable using immunofluorescence and is, as expected, located in the cytoplasm with a comparable expression level among different cardiomyocytes. In CR-Csn8KO hearts, the overall immunofluorescence of Rab7 was decreased, which is consistent with results of the western blot analysis. Rab7 expression appeared uneven among different cardiomyocytes on the same section. More interestingly, the accumulation of GFP-LC3 puncta (i.e. autophagosomes) was most intense in the cardiomyocytes with the lowest Rab7 protein expression (Figure 6C). This remarkable co-localization between the down-regulation of Rab7 and autophagosome accumulation in individual cells strongly suggests that Rab7 is likely a nexus through which Csn8/CSN regulates autophagosome maturation.
Figure 6. Down-regulation of Rab7 in Csn8 deficient mouse hearts.

A, and B, Representative images (A) and a summary of densitometry data (B) of western blot analysis of Rab5, Rab7, and Rab11 in mouse hearts at 3 weeks. n=4 mice per group; *p<0.05 vs. CTL. C, Representative confocal micrographs to illustrate the co-localization between Rab7 down-regulation and autophagosome accumulation at the individual cardiomyocyte level in CR-Csn8KO hearts. Scale bar=10 μm.
Down-regulation of Rab7 impairs autophagosome maturation and exacerbates the induction of cell death by proteasome inhibition in cultured cardiomyocytes
To begin testing a causal relationship between the Rab7 down-regulation and the impaired autophagosome maturation in Csn8 deficient cardiomyocytes, we next investigated the impact of siRNA-mediated Rab7 knockdown (Rab7KD) on autophagosome maturation under the baseline condition and during starvation or proteasome inhibition in cultured NRVMs. We measured autophagic flux by assessing the changes in biochemical markers (LC3-II, p62) in presence and absence of BFA-mediated lysosomal inhibition. Under basal condition in the absence of BFA treatment, Rab7KD significantly increased both LC3-II and p62, compared with the control siRNA (siLuci) group (Figure 7A, 7B). BFA-induced lysosome inhibition led to a marked increase in LC3-II and p62 levels in the control siRNA group; but this increase was significantly less in the Rab7KD group (Figure 7C, 7D). These findings indicate that Rab7 down-regulation is sufficient to impair basal autophagic flux.
Figure 7. Rab7 knockdown (Rab7KD) impairs autophagic flux in cultured cardiomyocytes.

NRVMs were transfected with siRNAs against either luciferase (siLuci) or Rab7 (siRab7) for 48 hours before subsequent treatments. A~D, Rab7KD impaired baseline autophagic flux. Representative images (A, C) and the summary of densitometry data (B, D) from western blot analyses for the indicated proteins are shown. E~H, Rab7KD attenuated glucose deprivation (GD) or proteasome inhibition induced increases in autophagic flux. To induce non-selective autophagy cells were subjected to GD for 4 hours. To activate selective autophagy, cells were treated proteasome inhibitor bortezomib (BZM, 20nM) for 12 hours. To assess autophagic flux cells were treated with BFA (100nM) or vehicle 3 hours before harvesting the cells. Representative images (E, G) and summaries of densitometry data (F, H) from western blot analyses are shown. *p<0.05 vs. siLuci.
Nutrition starvation by glucose deprivation (GD) activates non-selective autophagy and increases autophagic flux.1 GD-induced increase of autophagic flux was measured by the difference of LC3-II protein levels between the BFA-treated and vehicle-treated cells. GD activated autophagic flux was significantly less in Rab7KD cells than in the siLuci transfected cells (Figure 7E, 7F). Furthermore, we tested the effect of Rab7KD on proteasome inhibition activated selective autophagy. As expected, proteasome inhibition by bortezomib (BZM) increased autophagic flux. The BZM-induced autophagic flux was also significantly attenuated by Rab7KD (Figure 7E, 7H). These data indicate that Rab7 is required for autophagic flux activated by either GD or proteasome inhibition in cardiomyocytes.
To further demonstrate that Rab7 down-regulation impairs autophagosome maturation during not only non-selective autophagy but also proteasome inhibition activated selective autophagy, we employed a tf-LC3 (GFP-mRFP-LC3). After incorporation into autophagosomes, the tf-LC3 can effectively differentiate autophagosomes from autolysosomes because the acidic environment in autolysosomes quenches GFP but not mRFP fluorescence. The tf-LC3 was expressed in cultured NRVMs via Ad-tf-LC3 infection. We found a modest but statistically significant increase of the yellow puncta but not red puncta in Rab7KD cardiomyocytes at the baseline (Supplementary Figure S7, Figure 8B), suggesting that Rab7 is required for basal autophagy in the cultured NRVMs. Importantly, GD or BZM treatment markedly increased both yellow and red-only puncta. However, Rab7KD exacerbated the GD- or BZM- induced increases in the yellow puncta but significantly attenuated the GD- or BZM- induced increase in the red-only puncta, compared with the control siRNA treated cells (Figure 8A, 8B; Supplementary Figure S8). These results demonstrate compellingly that Rab7 down-regulation is sufficient to impair autophagosome maturation in both non-selective and selective autophagy in cardiomyocytes.
Figure 8. Rab7 down-regulation disrupts autophagosome maturation and increases proteasome inhibition-induced cell death in cultured neonatal rat ventricular myocytes (NRVMs).

A, and B, Changes in autolysosome formation as probed with tf-LC3. Cultured NRVMs were infected with Ad-tf-LC3 24hrs after transfection of siRab7 or siLuci (as control); 24hrs later, the cells were treated with bortezomib (BZM, 20nM) for 12hrs or subjected to glucose deprivation (GD) for 4hrs before the cells were imaged live for the GFP (green) and mRFP (red) signals from tf-LC3. Representative epi-fluorescent images (A) and a summary (B) of the changes of autophagic vacuole abundance with indicated treatments. Insets are the enlarged images of the indicated area. Arrowheads, red-only puncta (autolysosome); arrows, green and red superimposed puncta (autophagosomes); scale bar=50μm; *p<0.05; #p<0.01. C~E, Rab7 knockdown exacerbates BZM-induced cell death. BZM (10nM) treatment was initiated 48hrs after the siRNA transfection. At the indicated time points of BZM treatment, culture media were sampled for measuring lactate dehydrogenase (LDH) activities (C). At 24hrs of BZM treatment, propidium iodide (PI) was applied to the cell culture. Ten minutes later, the unbound PI was thoroughly removed and the cells were then fixed in 4% paraformaldehyde, counter-stained with DAPI, and imaged for PI and DAPI staining. Representative epi-fluorescence micrographs are shown in panel D. The percent of PI-positive (red) nuclei among all DAPI-stained nuclei (blue) was determined from 3 independent repeats (E). Scale bar=100 μm; *p<0.01 vs. siLuci; #p< 0.01 vs. siLuci+BZM.
Lastly, we determined the effect of Rab7 down-regulation on proteasome inhibition induced cell death. Cardiomyocytes undergoing necrosis or late apoptosis lose their cell membrane integrity, which can be assessed by measuring the leakage of lactate dehydrogenase (LDH) into the culture medium and the uptake of a cell membrane-impermeable dye, propidium iodide (PI), which will then bind to double stranded DNA and fluoresce.14 A low dose of BZM (10nM) significantly increased LDH activities in the medium and PI-uptake in a time-dependent manner, which was significantly augmented by Rab7KD (Fig. 8C~8E). These data indicate that Rab7 down-regulation disrupts autophagosome maturation and sensitizes cardiomyocytes to proteasome inhibition-induced cell death.
DISCUSSION
The deletion of the Csn8 gene in cardiomyocytes destabilizes other CSN subunits;12 therefore, the observed phenotypes in CR-Csn8KO mice may not be attributed exclusively to the Csn8 deficiency but to the CSN holo-complex as well. CSN is known to regulate UPS proteolytic function by modulating the functioning of cullin-based ubiquitin E3 ligases.11, 12 In the present study, we have expanded for the first time the regulatory role of CSN to macroautophagy, another critical catabolic and quality control pathway in the cell. We have demonstrated here that (1) Csn8/CSN is indispensable for macroautophagy in cardiomyocytes of intact mice; (2) Csn8/CSN is required for autophagosome maturation or the fusion between autophagosomes and lysosomes; (3) Csn8/CSN regulates autophagosome maturation likely by regulating Rab7 expression; and (4) impaired autophagosome removal in mouse hearts appears to promote cardiomyocytes necrosis.
Csn8/CSN is required for autophagosome maturation in heart muscle cells
We have collected compelling evidence that autophagic flux is impaired in the cardiomyocytes of CR-Csn8KO mice. Marked increases in LC3-II, indicative of increased autophagosomes, were observed upon Csn8 depletion in cardiomyocytes. The increases in autophagosomes were confirmed by probing with a transgenic GFP- LC3 and by TEM examination of autophagic vacuoles. Autophagic flux assessment by blocking lysosome-mediated autophagosome removal revealed that defective autophagosome removal rather than an increase in autophagosome formation causes the autophagosome accumulation in Csn8 deficient cardiomyocytes.
Defective autophagosome removal can be caused by either inadequate lysosomal proteolytic activities or defective fusion between autophagosomes and lysosomes. Our biochemical and morphological analyses show that the total number of lysosomes (i.e., vesicles positive for both LAMP1 and cathepsin-D) remained unchanged in Csn8 deficient cardiomyocytes, inferring that a defect in autophagosome-lysosome fusion may be the cause. Indeed, compared with the cardiomyocytes in the littermate controls, autolysosomes as revealed by co-localization between LAMP1 and GFP-LC3 puncta failed to increase, albeit a significant increase in autophagic vacuoles, in the cardiomyocytes of CR-Csn8KO mouse hearts. These data show that autophagosome-lysosome fusion is impaired in the cardiomyocytes of CR-Csn8KO mice. Therefore, Csn8/CSN is required for autophagosome maturation in at least heart muscle cells. Corroborating these findings, Pearce et al. recently reported that CSN2 knockdown accumulated autophagosomes in cultured K562 cells;22 however, they did not investigate whether the increase in autophagosome was caused by increased formation or decreased removal.
Csn8/CSN regulates autophagosome maturation likely through modulating Rab7 expression
The regulation on autophagosome maturation is poorly understood. In cell culture studies, Rab7 was shown to be indispensable for autophagosome-lysosome fusion and lysosomal genesis;21 but this remains to be tested in intact animals. Hariharan et al. recently demonstrated that upregulation of Rab7 plays an important role in mediating starvation-induced increases in autophagic flux in cardiomyocytes.10 The present study suggests that down-regulation of Rab7 likely mediates the Csn8-deficiency induced impairment of autophagosome maturation. This contention is supported by multiple lines of evidence. First, the protein expression of Rab7 but not Rab5 or Rab11 was significantly decreased in CR-Csn8KO mouse hearts. Second, perhaps due to differential onset of Csn8KO in various cardiomyocytes of a heart, the degree of Rab7 protein down-regulation varied among cardiomyocytes in the heart; and more importantly, the down-regulation of Rab7 strikingly co-localized with, and correlated to, GFP-LC3 accumulation. The highest GFP-LC3 puncta increase is found in the cardiomyocytes with the lowest Rab7 protein expression. Third, we failed to decrease Rab7 protein levels in cultured NRVMs by overexpressing GFP-LC3 (data not shown). Finally, Rab7KD in cultured NRVMs recapitulates the autophagosome maturation impairment phenotype observed in CR-Csn8KO mouse hearts. Rab7KD impairs the autophagosome maturation not only at baseline but also during GD and proteasome inhibition, both known to increase autophagic flux. GD activates non-selective autophagy while, likely through upregulation of p62, proteasome inhibition activates selective autophagy. The critical role of Rab7 in starvation-induced autophagy in cardiomyocytes was recently delineated by Sadoshima and his colleague.10 Our data demonstrate for the first time in cardiomyocytes that Rab7 is essential to autophagosome maturation in selective autophagy. To fully prove that Rab7 downregulation mediates the autophagic defect in CR-Csn8KO hearts, it will be important to test whether cardiomyocyte-restricted Rab7 overexpression rescues the defect in CR-Csn8KO mice. At this point, we could not rule out that a Rab7-independent mechanism may also contribute to impaired autophagosome maturation in Csn8 deficient hearts.
The mechanism by which Csn8/CSN regulates Rab7 expression is currently unknown but a potential link has been implicated in the existing literature. First, we have shown that the F-Box protein Atrogin1 is down-regulated in Csn8CKO hearts,12 which is consistent with the prevalent theory that CSN-mediate deneddylation stabilize F-Box proteins;11 second, Atrogin1, which is a target gene of FoxO1/FoxO3, promotes FoxO1/FoxO3 transactivation via a feed forward mechanism that depends on K63-linked polyubiquitination of the FoxO proteins by Atrogin1;23 and finally Rab7 is a target gene of the FoxO transcription factors, at least during autophagy activation.10 Indeed, we found that Rab7 mRNA levels are significantly decreased in CR-Csn8KO mouse hearts (data not shown). These evidences are consistent with a proposition that CSN may regulate the transcription of Rab7 via Atrogin1 and FoxO proteins.
Failure of autophagosome maturation promotes cardiomyocyte necrosis
As we described previously, massive cardiomyocyte necrosis was observed in CR-Csn8KO mouse hearts as early as 3 weeks of age when increased apoptosis and congestive heart failure are not discernible. Here we found impaired autophagosome removal in CR-Csn8KO hearts as early as 1 week of age, or two weeks before a global impairment of UPS proteolytic function was evident or increases in cardiomyocyte necrosis were detected.12 This temporal relationship represents the first line of evidence for a causal relationship between autophagy impairment and cardiomyocytes necrosis. Second, a significantly higher percent of cardiomyocytes with severe accumulation of autophagosomes showed increased EBD uptake compared with those with a relatively lower autophagosome density in CR-Csn8KO hearts. Third, blocking autophagosome maturation by Rab7KD exacerbates proteasome inhibition induced LDH leakage and PI uptake in cultured NRVMs. And finally, chronic inhibition of autophagic flux by CQ significantly increased EBD uptake and CD45 infiltration in mouse hearts with wild type Csn8.
Notably, mice with similarly achieved cardiomyocyte-restricted Atg5 knockout show impaired autophagosome formation;19 but they do not display the severe abnormal phenotypes as we observed in CR-Csn8KO mice which have impaired autophagosome removal. This discrepancy suggests that impaired autophagosome removal may be more detrimental than decreased autophagosome formation, as recently hypothesized by Gottlieb and Mentzer.9 One potential mechanism by which impaired autophagosome maturation causes cardiomyocyte necrosis is that when cardiomyocytes cannot self-digest the defective organelles or protein aggregates sequestered by autophagosomes, they may alternatively dump these highly inflammatory autophagosome contents to the extracellular space.9 This can conceivably have at least two consequences: first, the cell loses the opportunity to recycle the ingredients; second, the dumped content may act as autocrinal and/or paracrinal factors to trigger inflammation and even activate the programmed necrosis pathway.
Taken together, we have uncovered a novel function of Csn8 as an important player in the autophagosome removal stage of selective autophagy. The autophagy defect can be detected in CR-Csn8KO hearts when Csn8 protein is just depleted (1-week), or two weeks before the onset of UPS functional deficiency and discernible cardiomyopathy.12 Chronic inhibition of autophagy was shown to compromise the degradation of UPS substrates in non-cardiac cells.24 Hence, it is very possible that the accumulation of a surrogate UPS substrate (GFPdgn) observed in the later age of CR-Csn8KO mouse hearts may also be attributable to the impairment of the autophagic pathway. A working model for the role of Csn8/CSN in the cardiomyocytes of postnatal hearts can be summarized as follow: Csn8/CSN suppresses the necrotic pathway by enhancing autophagosome maturation via supporting Rab7 expression. Given that autophagy dysfunction and cardiomyocyte necrosis have been observed in congestive heart failure of various etiologies,14 it will be important to decipher how Csn8/CSN regulates Rab7 expression and how impaired autophagosome maturation causes necrosis. Considering that CSN has already been established as an important player in the UPS, our study, which places CSN in the autophagy-mediated degradation pathway, further underscores the prominent role of CSN in catabolic processes in the cell.
Supplementary Material
Clinical Summary.
(Macro)autophagy sequesters portions of the cytoplasm into double-membrane vesicles (known as autophagosomes) for fusion with, and degradation by, lysosomes. Autophagy is essential to intracellular homeostasis and involved in the pathophysiology of a variety of diseases. Targeting autophagy can potentially become a therapeutic strategy to treat heart disease and this is being intensively explored. Thus, it is critical to understand better the mechanisms that regulate autophagy, especially the selective autophagy that removes defective organelles and protein aggregates. The COP9 signalosome (CSN) is a conserved protein complex consisting of 8 subunits (CSN1 through CSN8) and known to regulate the ubiquitin-proteasome system (UPS), another major intracellular proteolytic pathway. We report that mice with perinatal cardiomyocyte-restricted ablation of the Csn8 gene (CR-Csn8KO) display massive cardiomyocyte necrosis, develop dilated cardiomyopathy, and die prematurely. However, it is unknown whether CSN plays a role in autophagy. Here we report that the fusion between autophagosomes and lysosomes is impaired in the cardiomyocytes of CR-Csn8KO mice; as a result, the removal of autophagosomes is impaired. This appears to be caused by a down-regulation of Rab7, a small G-protein known to be indispensable to the fusion process. Impaired autophagosome removal is likely responsible for the massive cardiomyocyte necrosis in CR-Csn8KO mice because cardiomyocytes with more severe autophagosome accumulation undergo necrosis more often in CR-Csn8KO mice and chronic lysosomal inhibition with Chloroquine is sufficient to cause cardiomyocyte necrosis in mice. Therefore, we conclude that Csn8/CSN regulates both the UPS and autophagy in the heart.
Acknowledgments
We thank the Imaging Core of the Division of Basic Biomedical Sciences for assistance in confocal microscopy, Ms. Andrea Jahn for outstanding technical assistance in maintaining mouse colonies and genotype determination, Mr. Suleman Said for technical support on TEM sample preparation, and Dr. Evelyn Schlenker for statistical assistance. Dr. X. Wang is a recipient of the Established Investigator Award of the American Heart Association.
Sources of Funding
This work was in part supported by NIH grants R01HL085629 and R01HL072166, American Heart Association grants 0740025N (to X. W.) 0625738Z (to H.S.), and 11PRE5730009 (to M.J.R.). The Imaging Core is supported an NIH grant (5P20RR015567).
Footnotes
Conflict of Interest Disclosures: None.
References
- 1.Mizushima N, Yoshimori T, Levine B. Methods in mammalian autophagy research. Cell. 2010;140:313–326. doi: 10.1016/j.cell.2010.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tannous P, Zhu H, Johnstone JL, Shelton JM, Rajasekaran NS, Benjamin IJ, Nguyen L, Gerard RD, Levine B, Rothermel BA, Hill JA. Autophagy is an adaptive response in desmin-related cardiomyopathy. Proc Natl Acad Sci U S A. 2008;105:9745–9750. doi: 10.1073/pnas.0706802105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zheng Q, Su H, Ranek MJ, Wang X. Autophagy and p62 in cardiac proteinopathy. Circ Res. 2011;109:296–308. doi: 10.1161/CIRCRESAHA.111.244707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kassiotis C, Ballal K, Wellnitz K, Vela D, Gong M, Salazar R, Frazier OH, Taegtmeyer H. Markers of autophagy are downregulated in failing human heart after mechanical unloading. Circulation. 2009;120:S191–197. doi: 10.1161/CIRCULATIONAHA.108.842252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sciarretta S, Hariharan N, Monden Y, Zablocki D, Sadoshima J. Is autophagy in response to ischemia and reperfusion protective or detrimental for the heart? Pediatr Cardiol. 2011;32:275–281. doi: 10.1007/s00246-010-9855-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xie M, Morales CR, Lavandero S, Hill JA. Tuning flux: Autophagy as a target of heart disease therapy. Curr Opin Cardiol. 2011;26:216–222. doi: 10.1097/HCO.0b013e328345980a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fleming A, Noda T, Yoshimori T, Rubinsztein DC. Chemical modulators of autophagy as biological probes and potential therapeutics. Nat Chem Biol. 2011;7:9–17. doi: 10.1038/nchembio.500. [DOI] [PubMed] [Google Scholar]
- 8.Gustafsson AB, Gottlieb RA. Autophagy in ischemic heart disease. Circ Res. 2009;104:150–158. doi: 10.1161/CIRCRESAHA.108.187427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gottlieb RA, Mentzer RM. Autophagy during cardiac stress: Joys and frustrations of autophagy. Annu Rev Physiol. 2010;72:45–59. doi: 10.1146/annurev-physiol-021909-135757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of foxo by sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res. 2010;107:1470–1482. doi: 10.1161/CIRCRESAHA.110.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wei N, Serino G, Deng XW. The cop9 signalosome: More than a protease. Trends Biochem Sci. 2008;33:592–600. doi: 10.1016/j.tibs.2008.09.004. [DOI] [PubMed] [Google Scholar]
- 12.Su H, Li J, Menon S, Liu J, Kumarapeli AR, Wei N, Wang X. Perturbation of cullin deneddylation via conditional csn8 ablation impairs the ubiquitin-proteasome system and causes cardiomyocyte necrosis and dilated cardiomyopathy in mice. Circ Res. 2011;108:40–50. doi: 10.1161/CIRCRESAHA.110.230607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tannous P, Zhu H, Nemchenko A, Berry JM, Johnstone JL, Shelton JM, Miller FJ, Jr, Rothermel BA, Hill JA. Intracellular protein aggregation is a proximal trigger of cardiomyocyte autophagy. Circulation. 2008;117:3070–3078. doi: 10.1161/CIRCULATIONAHA.107.763870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Palumbo A, Gay F, Bringhen S, Falcone A, Pescosta N, Callea V, Caravita T, Morabito F, Magarotto V, Ruggeri M, Avonto I, Musto P, Cascavilla N, Bruno B, Boccadoro M. Bortezomib, doxorubicin and dexamethasone in advanced multiple myeloma. Ann Oncol. 2008;19:1160–1165. doi: 10.1093/annonc/mdn018. [DOI] [PubMed] [Google Scholar]
- 15.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y. In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell. 2004;15:1101–1111. doi: 10.1091/mbc.E03-09-0704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mokin M, Keifer J. Quantitative analysis of immunofluorescent punctate staining of synaptically localized proteins using confocal microscopy and stereology. J Neurosci Methods. 2006;157:218–224. doi: 10.1016/j.jneumeth.2006.04.016. [DOI] [PubMed] [Google Scholar]
- 17.Kumarapeli AR, Su H, Huang W, Tang M, Zheng H, Horak KM, Li M, Wang X. Alpha b-crystallin suppresses pressure overload cardiac hypertrophy. Circ Res. 2008;103:1473–1482. doi: 10.1161/CIRCRESAHA.108.180117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kimura S, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged lc3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- 19.Nakai A, Yamaguchi O, Takeda T, Higuchi Y, Hikoso S, Taniike M, Omiya S, Mizote I, Matsumura Y, Asahi M, Nishida K, Hori M, Mizushima N, Otsu K. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13:619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 20.Kumarapeli AR, Horak KM, Glasford JW, Li J, Chen Q, Liu J, Zheng H, Wang X. A novel transgenic mouse model reveals deregulation of the ubiquitin-proteasome system in the heart by doxorubicin. FASEB J. 2005;19:2051–2053. doi: 10.1096/fj.05-3973fje. [DOI] [PubMed] [Google Scholar]
- 21.Gutierrez MG, Munafo DB, Beron W, Colombo MI. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117:2687–2697. doi: 10.1242/jcs.01114. [DOI] [PubMed] [Google Scholar]
- 22.Pearce C, Hayden RE, Bunce CM, Khanim FL. Analysis of the role of cop9 signalosome (csn) subunits in k562; the first link between csn and autophagy. BMC Cell Biol. 2009;10:31. doi: 10.1186/1471-2121-10-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li HH, Willis MS, Lockyer P, Miller N, McDonough H, Glass DJ, Patterson C. Atrogin-1 inhibits akt-dependent cardiac hypertrophy in mice via ubiquitin-dependent coactivation of forkhead proteins. J Clin Invest. 2007;117:3211–3223. doi: 10.1172/JCI31757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Korolchuk VI, Mansilla A, Menzies FM, Rubinsztein DC. Autophagy inhibition compromises degradation of ubiquitin-proteasome pathway substrates. Mol Cell. 2009;33:517–527. doi: 10.1016/j.molcel.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.