

Abstract
Here we report high-resolution X-ray structures of Bacillus subtilis aspartate transcarbamoylase (ATCase), an enzyme that catalyzes one of the first reactions in pyrimidine nucleotide biosynthesis. Structures of the enzyme have been determined in the absence of ligands, in the presence of the substrate, carbamoyl phosphate, and in the presence of the bisubstrate/transition state analog N-phosphonacetyl-L-aspartate. Combining the structural data with in silico docking and electrostatic calculations, we have been able to visualize each step in the catalytic cycle of ATCase, from the ordered binding of the substrates, to the formation and decomposition of the tetrahedral intermediate, to the ordered release of the products from the active site. Analysis of the conformational changes associated with these steps provides a rationale for the lack of cooperativity in trimeric ATCases that do not possess regulatory subunits.
Keywords: pyrimidine nucleotide biosynthesis, transferase enzymes, catalytic cycle, X-ray crystal-lography
Introduction
Aspartate transcarbamoylase (E.C. 2.1.3.2, aspartate carbamoyltransferase, ATCase) plays a critical role in metabolism. It catalyzes one of the first reactions in the biosynthesis of the pyrimidine nucleotides by the transfer of the carbamoyl group from carbamoyl phosphate (CP) to L-aspartate (Asp) forming N-carbamoyl-L-aspartate (CA) and inorganic phosphate (Pi)1 (Fig. 1). The reaction mechanism is ordered with CP binding before Asp and CA departing before Pi.2 The quaternary structure of ATCases varies; however, there is one repeating theme; the core catalytic unit is composed of three chains (the catalytic chains) grouped together as a trimer (c3), exhibiting 3-fold rotational symmetry. The trimeric arrangement is required for catalysis, since the three active sites each occur at the interface between two adjacent chains with catalytic residues donated from both chains. As is the case for many other transferases, each chain of ATCase is composed of two domains, specifically CP and Asp domains, each binding their respective substrate. A closure of the domains provides assistance in lowering the activation energy via catalysis by approximation.5
Fig. 1.

The reaction of carbamoyl phosphate (CP) and L-aspartate (Asp) to form N-carbamoyl-L-aspartate (CA) and inorganic phosphate (Pi). The reaction proceeds via a tetrahedral intermediate (TET). The structure of the bisubstrate analog N-phosphonacetyl-L-aspartate (PALA) is also shown.
The structurally simplest ATCases are composed of one homotrimer of catalytic chains and exhibit no cooperativity, as observed in B. subtilis. Other organisms have more complex ATCases, as is the case in Escherichia coli, where two catalytic trimers and three regulatory dimers combine to form a dodecameric enzyme. The E. coli enzyme demonstrates both homotropic cooperativity and allosteric regulation, mechanisms that modulate the rate of pyrimidine nucleotide biosynthesis.1 Unregulated ATCases often exist as multi-enzyme complexes with other pyrimidine biosynthetic enzymes. For example, several genera of the fungi kingdom such as Saccharomyces and Neurospora display ATCase as a complex with carbamoyl phosphate synthase (CPSase).6 However, in some eukaryotes, the first three enzymes in pyrimidine nucleotide biosynthesis, CPSase, ATCase and dihydroorotase (DHOase), are fused together on a single polypeptide chain (CAD).7 These enzymes oligomerize to create a complex with at least one ATCase catalytic trimer.
The primary sequence of the catalytic chain of ATCase is highly conserved. Based upon an analysis of 250 species, 20% of the residues are conserved, including all of the residues thought to be involved in catalysis. Structural data from a variety of species have been determined but no unregulated, trimeric ATCase has had its structure determined in more that one functional form.
Numerous X-ray structures of ATCase have been reported from a variety of organisms in the presence and absence of substrates and substrate analogs,8 although most of the structures are of the E. coli enzyme either as the dodecamer or the isolated catalytic trimer. The E. coli catalytic trimer has the same quaternary structure as the B. subtilis ATCase for which structures are available in the absence of ligands9 and in the presence of PALA.10 These structures have been used to assess the allosteric mechanism based upon a comparison of the structures of the isolated catalytic trimer with the corresponding component of the holoenzyme. From these studies the details of the catalytic cycle could not be deduced since the only information available is for the enzyme in the absence of ligands or that with a bisubstrate analog bound. Although a structure of the holoenzyme in the presence and absence of CP is available,11 the conformational changes associated with ligand binding and those involved in allostery are not easy to deconvolute. By using the unregulated B. subtilis ATCase as our model system, the conformational changes involved in allostery are disconnected from those involved in substrate binding and catalysis.
Here we report a description of the entire catalytic cycle of the unregulated ATCase from B. subtilis. Using X-ray structures in the absence and presence of CP or a substrate analog, in silico docking and electrostatic calculations, we provide a molecular level glimpse into catalysis by the unregulated ATCase that helps to explain the manifestation of homotropic cooperativity in those ATCases that exhibit this property.
Results and Discussion
Overall Structure of B. subtilis ATCase
The overall topology of the B. subtilis ATCase structure is similar to that observed for c3 units of other ATCases, such as that from E. coli, and is congruous with a previously published 3Å α-carbon trace.13 All active site residues shown to be important in other ATCases14 are conserved in the B. subtilis enzyme, including Ser74 and Lys77, which are donated into the active site from an adjacent chain.
The binding of the first substrate, CP, in the ordered-binding mechanism
In the ordered-binding mechanism,2 CP must bind before Asp. A comparison of the structures of the unliganded (open form) and ATCase•CP complexes reveals extensive conformational changes induced by CP binding. Fig 2a illustrates these movements with the unliganded structure highlighted in blue and the CP-bound structure highlighted in red. These conformational changes are localized around the active site with the 40s (44–49), 70s (69–78), 100s (101–105), 120s (122–126), 160s (160–165), 180s (184–191) and 220s (212–225) loops all moving substantially toward the CP binding site in the ATCase•CP structure (CP-bound form).
Fig. 2.

Structural changes of the α-carbon backbone associated with the binding of CP and PALA. (a,b) Structure of one catalytic chain of B. subtilis ATCase along with the 70s loop from the adjacent chain (70s loop*), and (c,d) one catalytic chain of E. coli ATCase along with the 80s loop from the adjacent chain (80s loop*) (a,c). The structure of the unliganded enzyme is shown with blue highlights, while the structure of the ATCase•CP complex is shown with red highlights, CP represented as spheres, and active site residues as sticks. (b,d) The structure of the ATCase•CP complex is shown with blue highlights, while the structure of the ATCase•PALA complex is shown with red highlights, PALA represented as spheres, and active site residues as sticks. The color spectral change from blue to red corresponds to 30 structures calculated linearly between the two determined X-ray structures. PDB entries 1ZA1,11 1ZA2,11 and 1D0925 were used for (c) and (d).
The movement of the 40s loop, specifically Arg49, dramatically reconfigures the active site pocket in the CP-bound form of the enzyme. Using Castp,15 the volume of the active site cavity was calculated and is visualized in Fig 3a–d. The binding of CP reduces the volume of the active site cavity by one-half, from 1848 Å3 (Fig. 3a) to 918 Å3 (Fig. 3b). In order to help understand how the substrates bind to the enzyme and induce the conformational changes (Fig. 2a,b), DELPHI16 was used to calculate the electrostatic potentials of each of the structures and the potentials were mapped onto the corresponding surface (Fig. 4a–d). In addition GLIDE, in extra precision (XP) mode,19 was used to dock substrates to the unliganded and ATCase•CP structures as a tool to identify potential binding sites. Shown in Fig. 4a is a sliced image of the surface of the unliganded enzyme. An area of significant positive electrostatic potential is observed where CP docks. The position of CP in Fig. 4a corresponds to the CP pose with highest affinity to the unliganded enzyme. The docked position of CP is not the site observed in the ATCase•CP structure (compare the positions of CP in Fig. 4a and c).
Fig. 3.
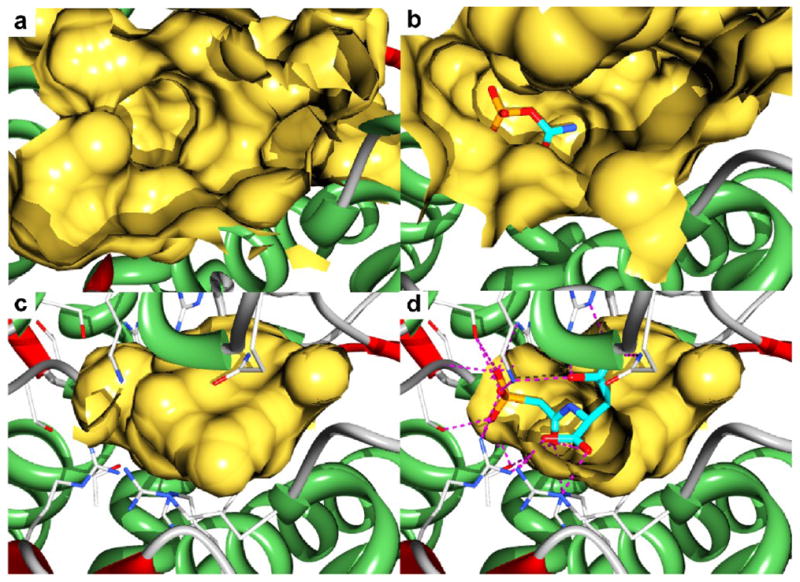
X-ray structures of the B. subtilis ATCase showing the active site cavity as calculated by Castp15 (a-d). (a) The active site of the enzyme in the absence of ligands. (b) The active site of the ATCase•CP complex with CP shown as sticks. (c) The active site of the ATCase•PALA complex. (d) A cut away view into the active site cavity of the ATCase•PALA complex showing the interactions (dotted lines) between the active site residues and PALA (sticks).
Fig. 4.

X-ray structures of the B. subtilis ATCase showing the active site electrostatic potential mapped onto the surface as calculated by DELPHI16 (a-d). Also shown, represented as sticks, are (a) CP, (b) Asp, (c) Asp, and (d) CA docked into the structures. (a,b) Solvent accessible surface representation of the structure of the B. subtilis ATCase in the absence of ligands, (c) in the presence of CP, and (d) in the presence of PALA. The surface was sliced to reveal details of the active site. The electrostatic potential of the structure was mapped onto the surface (−10 kT/e (red) to +10 kT/e (blue)). The CP in (c) is that determined in the ATCase•CP complex, and in (d) the Pi corresponds to the position of the phosphonate of PALA.
Analysis of the structural data, electrostatics, and docking results suggests that the initial binding of CP, particularly the phosphate portion, induces conformational changes of the 40s, 70s, 100s and 120s loops. The conformational changes of the 40s loop, in particular Arg49, reorient CP into a higher-affinity CP binding site in the ATCase•CP structure (Fig. 3b). The GLIDE XP docking scores for CP binding to the unliganded structure and the ATCase•CP structure with CP removed are −6.8 and −9.6 kcal/mole, respectively. These in silico docking calculations reveal that CP interacts mainly with residues Arg49, Thr50, Gln130 and Arg281 of the unliganded enzyme. All of these interactions are maintained after the conformational changes induced by CP binding except for Arg281. In addition, new interactions are observed between CP and Ser47, Thr48, Arg99, His127 and Ser74 from the adjacent chain. All of the interactions to CP in the ATCase•CP structure (Fig. 5a) explain the enhanced CP affinity to the active site conformation of the ATCase•CP structure compared to the unliganded active site as predicted by the GLIDE XP calculations.
Fig. 5.

Substrates, products and analogs bound to ATCase. The active site orientation is the same as in Fig. 3. An asterisk indicates that the residue was donated into the active site from the adjacent chain. The label on Thr48 has been omitted for clarity. (a) Structure of the ATCase•CP complex with Asp bound, as determined by docking. A red arrow with the head at the carbonyl carbon of CP from the α-amino nitrogen of Asp demonstrates the first chemical event in the reaction to form TET. (b) Structure of the ATCase•PALA complex with the tetrahedral intermediate (TET) docked in place of PALA. (c) Structure of the ATCase•PALA complex with CA docked into the active site in place of PALA. In addition, a Pi was substituted for the phosphonate of PALA. (d) Electron density maps of the ATCase•PALA structure overlaid on the final refined coordinates. A Fo − Fc electron density map with PALA omitted from the calculation is shown in green contoured at 5σ, and a 2Fo − Fc electron density map shown in blue contoured at 2σ. (e) The active site of the ATCase•PALA complex with a Pi at the position occupied by the phosphonate of PALA (Pi1). Pi2 corresponds to one of three binding sites for Pi determined in the GLIDE docking calculations. (f) A possible path of Pi release from the active site. In addition to the Pi1 and Pi2 positions shown in (e), two additional possible Pi binding sites are shown, Pi3 and Pi4. Pi4 is closest to an exit from the active site. For clarity docked substrates are shown with pink carbon atoms, while substrates or analogs determined by crystallography are shown with cyan carbons.
The binding of the second substrate, Asp, in the ordered-binding mechanism
A feature of the kinetics of many ATCases including the B. subtilis enzyme is very strong substrate inhibition by Asp.20 A possible explanation for the observed substrate inhibition is that as-partate binds in a nonproductive mode with its two carboxylates reversed. However, GLIDE XP docking calculations on the unliganded structure provide another explanation. When Asp is docked to the unliganded enzyme, it docks to approximately the same positively charged pocket as does CP with a GLIDE XP score of −6.5 kcal/mole (compare Fig. 4a and b). The ability of Asp to bind to the same site as CP in the unliganded enzyme explains the strong substrate inhibition exhibited by Asp.
The conformational changes induced by the binding of CP dramatically alter the active site pocket (compare Fig. 3a and b) and electrostatics of the active site (compare Fig. 4a and c). In the unliganded structure, the portion of the active site where Asp would be expected to bind in order to react with CP is, essentially, neutral in charge. However, in the ATCase•CP structure, there exists a positively charged pocket adjacent to CP. In order to visualize how Asp might bind to the ATCase•CP structure, Asp was docked to the ATCase•CP structure using the XP mode of GLIDE. Shown in Fig. 5a is the highest affinity Asp docking pose with a GLIDE XP score of −8.4 kcal/mole. PROPKA was used to calculate the pKa of the amino group of Asp in the active site of the ATCase•CP complex: the calculation gave a pKa value less than 2, such that at physiological pH Asp would bind to the ATCase•CP complex with a neutral amino group, poised for nucleophilic attack.
The reaction between CP and Asp to form the tetrahedral intermediate
Stopped-flow small-angle X-ray scattering (SAXS) has been used to establish that when Asp is mixed with the E. coli ATCase•CP complex, there is an alteration in the structure of the enzyme on the millisecond time scale.23 Although these experiments were performed on a type of ATCase with both catalytic and regulatory chains, the structure of the E. coli ATCase•PALA complex after the Asp-induced conformational change has the same overall structure as the B. subtilis ATCase•PALA complex. Therefore, it would be expected that the structure of the B. subtilis AT-Case•PALA complex corresponds to the overall structure of the enzyme immediately upon the addition of Asp to the ATCase•CP complex. For the B. subtilis enzyme, these conformational changes are shown in Fig. 2b, with the blue highlighting the ATCase•CP structure and the red highlighting the ATCase•PALA structure (closed form). The conformational changes associated with Asp binding involve substantial motions of the 180s and 220s loops, which undergo smaller motions upon CP binding to the unliganded enzyme. However, the portions of the structure involved in the creation of the CP binding site are less altered upon the conversion to the closed form of the active site (compare Fig. 2a and b).
As indicated above, the conformational changes associated with the binding of CP not only create a physical binding pocket for Asp but substantially change the electrostatic environment of the active site (Fig. 4c). Shown in Fig. 5a is the highest affinity pose of Asp docked into the ATCase•CP structure. The Asp amino group nitrogen is 3.4Å from the carbonyl of CP and perpendicular to its plane, at an optimal distance and orientation for the nucleophilic attack. Furthermore, the interactions to the carbamoyl group of CP observed in the ATCase•CP structure would further promote nucleophilic attack by stabilizing the negative charge on the carbonyl oxygen of the transition state.
Asp docks to the ATCase•CP complex (Fig. 5a) such that its α-carboxylate interacts with Lys77 of the adjacent chain. Conformational changes that occur as Asp binds to the ATCase•CP complex include the closure of the 220s loop towards the active site, allowing interactions to form between the β-carboxylate of Asp and Arg211 and Gln213. The structural data suggest that Lys77 first positions Asp for nucleophilic attack, then acts as a bridge between the two substrate molecules, holding them together as the transition state forms (compare Fig. 5a and b). The binding of Asp causes the closing of the two domains of the catalytic chain creating the environment necessary for the formation of the tetrahedral intermediate (TET). As shown in Fig. 1, the structural similarity between PALA and TET suggests that PALA can be used to model the structure of the AT-Case•TET complex.
Shown in Fig. 5d is a closeup of the PALA binding site in the ATCase•PALA complex. A total of 26 interactions position PALA in the active site, of which three are hydrophobic in nature, and are not shown in Fig. 5d. The plethora of interactions to PALA explains the nanomolar affinity of this bisubstrate analog.24
In order to validate the in silico dockings, PALA was docked into the ATCase•PALA complex with PALA removed. PALA docked within the cavity shown in Fig. 3c. All of the highest affinity poses determined from the docking calculations have the phosphonate in exactly the same position as observed in the X-ray structure. Albeit, there were variations in the position of the β-carboxylate within this set of poses. The GLIDE XP docking score of the highest affinity pose was −11.7 kcal/mole. For comparison, the structure of the proposed tetrahedral intermediate (TET) was also docked to the ATCase•PALA complex with PALA removed using exactly the same protocol. As seen in Fig. 5b, the tetrahedral intermediate interacts with ATCase in essentially the same manner as does PALA. However, the number of interactions increases from 26 to 28, two of which are hydrophobic in nature. Arg49, Lys77 and Gln130 form additional interactions to TET that are not observed in the ATCase•PALA complex. Three of these interactions are to atoms that are not available in the PALA structure. Two interactions are formed to the amino group on the tetrahedral carbon: one from the side-chain of Gln130 and the other from the backbone carbonyl of Pro249. The side chains of Arg49 and Lys77 also interact with the bridging phosphate oxygen. The GLIDE XP docking score of the highest affinity pose of TET was −13.6 kcal/mole, almost 2 kcal/mole more negative than that calculated for PALA.
The closure of the domains upon Asp binding further reduces the volume of the active site by an additional one-half, from 918 Å3 in the ATCase•CP complex (Fig. 3b) to 411 Å3 in the ATCase•PALA complex (Fig. 3c), essentially forcing Asp toward CP. The closure of the domains also dramatically alters the electrostatic environment of the active site (Fig. 4d). Castp calculations indicate that the active site cavity in the ATCase•PALA complex has no exit. This closed, positively charged cavity provides the perfect environment to promote the formation of the transition state and to stabilize its negative charge.
The ATCase•PALA complex and the docked TET to the ATCase•PALA structure with PALA removed each show an interaction between the backbone carbonyl of Ala250 and the Asp amino group (Fig. 5b,d). Ala250 acts as a hydrogen-bond acceptor as Asp is brought into proximity of the CP-binding domain, increasing the nucleophilicity of the Asp amine. Ala250 is not conserved in all ATCases, however, since the interaction with Asp involves the backbone carbonyl of Ala250, the nature of the side chain is unimportant. Interestingly, this particular alanine is flanked by two pro-line residues, which are conserved. Moreover, this primary-sequence feature results in Ala250 having phi/psi angles in a forbidden region of the Ramachandran plot. The unusual phi/psi angles at this position are observed in other ATCase structures even when alternate residues are present.
The breakdown of the tetrahedral intermediate to form CA and Pi
The breakdown of the tetrahedral intermediate involves the collapse of the oxyanion and the release of Pi as shown in Fig. 1. For the E. coli enzyme, Gouaux and Lipscomb26 proposed that the conformation of the transition state for the breakdown of TET involves an intramolecular proton transfer where a phosphate oxygen acts as a general base, accepting a proton from the ionized secondary amine. The interaction between the phosphate oxygen and the amine proton completes a six-membered ring in a chair conformation, supporting their modeling and mechanism for the decomposition of the transition state.
Analysis of the structure of TET docked into the B. subtilis enzyme (Fig. 5b) indicates the conformation of the transition state would, most likely, be in a distorted chair conformation. The structural data of the ATCase•PALA complex and the conformation of TET docked into that structure with PALA removed suggest an alternate mechanism, which utilizes an enzyme-mediated proton transfer with Arg99 acting as a general acid. Using PROPKA, the pKa values of all ionizable amino acid side chains in the structure were calculated. The pKa of Arg99 was calculated to be 6.2 as compared to the average pKa of 12.0 for all the other Arg residues in the structure. This decrease in the pKa can be explained by the highly electropositive pocket in which Arg99 lies (Fig. 4d). This extremely low pKa value allows Arg99 to act as an acid at physiological pH. The phosphate oxygen acceptor was 2.1Å from the labile Arg99 proton. Protonation of the phosphate group makes it a better leaving group and promotes product formation.
The conformational changes needed for the release of the first product, CA
There is sufficient room in the closed form of the active site pocket (Fig. 3c,d) to not only accommodate PALA or TET but also the products CA and Pi. However, the combination of CA and Pi takes up sufficiently more space than PALA or TET, due to the required separation between the two products as opposed to the unimolecular nature of PALA or TET. We propose that the closed form of the enzyme, as exemplified by the ATCase•PALA complex (Fig. 3c) is transient in nature. As the transition state decomposes, the products separate and destabilize the closed form. Based upon modeling, the 220s loop that closed in toward the active site during Asp binding now opens at least partially. We estimate that the conformation of the catalytic chain must relax back between 30–40% towards the CP-bound conformation before an exit route forms that is large enough for CA to leave the active site and be released into solution.
The exit of the second product, Pi, from the active site
Once CA leaves the active site, the remaining product Pi can leave the active site via the same opening. In silico docking calculations were performed on the ATCase•PALA complex with PALA removed and Pi positioned in place of the PALA phosphonate. Three different Pi locations were identified with GLIDE XP docking scores of −7.2, −7.1 and −6.3 kcal/mole (Fig. 5e,f). As the XP docking scores become more positive, the relative position of the Pi becomes farther from the position occupied by the phosphonate portion of PALA and closer to the exit from the active site. These three positions suggest an exit route of Pi from the active site (Fig. 5e,f).
Lack of cooperativity in the B. subtilis unregulated ATCase
One of the properties of the B. subtilis ATCase that distinguishes it from some other ATCases is its lack of homotropic cooperativity. The structure of the B. subtilis ATCase•CP complex, reported here, helps clarify the lack of cooperativity in c3 ATCases. Although the wild-type B. subtilis ATCase is not cooperative, amino acid substitutions can result in the manifestation of cooperativity in the B. subtilis ATCase. Specifically, the R99A mutation results in a cooperative enzyme with a Hill coefficient of 1.5.27 As seen in Fig. 5a, Arg99 is critically involved in the binding of CP. Although the R99A mutation allows the B. subtilis ATCase to manifest cooperativity, it has a detrimental influence on both the binding of CP and Asp. In order for the R99A B. subtilis ATCase to exhibit cooperativity, the enzyme shifts between two states that differ in activity and/or affinity for substrate The altered binding of the substrates to the R99A enzyme essentially creates a two-state system that did not previously exist, providing an explanation for the observed cooperativity of this mutant.27
How is it then that the E. coli ATCase holoenzyme (c6r6) exhibits cooperativity while the E. coli c3 ATCase and the wild-type B. subtilis ATCase do not? A comparison of the structural changes induced by the binding of CP to the B. subtilis ATCase and the E. coli ATCase holoenzyme provides an explanation. As seen in Fig. 2a, CP binding induces dramatic alterations to the B. subtilis enzyme structure, with major conformational changes involving the 40s, 70s, 100s, 120s, 160s, 180s and 220s loops. The binding of CP to the E. coli holoenzyme induces much less dramatic conformational changes, with only the equivalent of the 40s and 70s loops undergoing major structural alterations26 (Fig. 2c). The reduced magnitude of the conformational changes is partially due to stabilizing interactions between the catalytic and regulatory chains that restrict some of the loop motions observed in the B. subtilis enzyme.
The conformational changes induced by the saturation of the unliganded B. subtilis ATCase with CP results in the creation of active sites that have high-activity and high-affinity for Asp. Thus, Asp binding does not induce cooperativity. In contrast, the R99A B. subtilis ATCase does not bind CP in the correct position to induce the necessary conformational changes. Asp binding to one of the active sites in the R99A ATCase•CP complex can transmit conformational changes to the other active sites, enhancing their affinity for Asp. In the case of the E. coli holoenzyme, the binding of CP alone does not shift the active site conformation sufficiently to create the high-affinity binding site; rather, it is only the binding of Asp to the E. coli ATCase•CP complex that causes the necessary loop movements inducing cooperativity. This explanation is supported by SAXS measurements that showed CP has little or no influence on the quaternary structure of the E. coli holoenzyme,30 while the binding of Asp to the E. coli ATCase•CP complex induces the allosteric structural transition which manifests itself in the observed kinetic cooperativity.23 Furthermore, CP binding induces much larger quaternary conformational changes in versions of E. coli ATCase that have weakened interactions either between the regulatory and catalytic chains31 or between the two catalytic trimers in the holoenzyme.32
The unique set of three X-ray crystallographic structures of B. subtilis ATCase reported here, in the absence and presence of a substrate and a bisubstrate analog in combination with electrostatic calculations and ligand docking has provided sufficient data to visualize each step in the catalytic cycle of ATCase. The steps being the ordered binding of the substrates, the formation of the tetrahedral intermediate followed by its decomposition to the products, the conformational changes associated with these steps, and the ordered release of the products from the active site. In addition, this work has helped us understand the manifestation of homotropic cooperativity in some AT-Cases.
Materials and Methods
Materials
Chemicals were purchased from commercial sources and used as received with the exception of CP, which was purified before use by precipitation from 50% (v/v) ethanol and was stored desiccated at −20° C prior to use.1 PALA was obtained from the National Cancer Institute.
Protein expression and purification
B. subtilis ATCase was over-expressed in M9 minimal media using the E. coli strain EK1104 [F− ara, thi, Δ(pro-lac), ΔpyrB, pyrF±, rpsL] transformed with plasmid pEK17120 containing the B. subtilis pyrB gene under the control of the E. coli pyrBI promoter33 The purification procedure is a variation on that performed previously.20 First, ion-exchange chromatography was employed using a Q-Sepharose Fast Flow (GE Healthcare) column (11 cm x 2.5 cm). The fractions containing AT-Case were pooled and dialyzed against 0.05 M Tris-acetate, 2 mM 2-mercaptoethanol pH 8.3 (Low Q buffer). The protein solution was then brought to 20% ammonium sulfate saturation and purified by hydrophobic-interaction chromatography employing a Phenyl Sepharose (GE Healthcare) column (8.5 cm x 2 cm). The protein was eluted using a linear gradient from Low Q buffer + 20% ammonium sulfate to Low Q buffer. The fractions containing pure ATCase, as confirmed by SDS-PAGE,34 were pooled and dialyzed against storage buffer (40 mM KH2PO4, 2 mM 2-mercaptoeth-anol, and 2 mM EDTA, pH 7.0). Protein concentration was determined by A280 measurement using the extinction coefficient of B. subtilis ATCase, 0.57 cm2 • mg−1.35
Crystallization, X-ray data collection and processing
B. subtilis ATCase was crystallized by the hanging drop vapor diffusion method as previously reported.13 The enzyme, at a final concentration of 12 mg/ml, was mixed with an equal volume of crystallization buffer (1.7 M (NH4)2SO4, 0.1 M Tris-HCl, pH 8.5, 2.0% PEG 200) and equilibrated over a reservoir of 0.5 ml of crystallization buffer at 20 ºC. Bar-shaped crystals grew to average dimensions of 0.5 x 0.3 x 0.1 mm within one week. These crystals were used directly to determine the structure of the unliganded enzyme. To determine the structure of the ATCase•CP complex, these crystals were equilibrated in crystallization buffer containing 13.3 mM CP for one hour prior to freezing. For the ATCase•PALA structure, the enzyme was co-crystallized with PALA (2 mM) by the hanging drop vapor diffusion method, based upon conditions identified by a crystal screen at the Hauptman-Woodward Institute (Buffalo, NY). The drop consisted of equal volumes of 10 mg/ml enzyme solution and crystallization buffer (0.1 M potassium bromide, 0.1 M CAPS buffer, pH 10.0, 18% PEG 8000) over a reservoir of 0.5 ml of crystallization buffer. Hexagonal crystals grew to average dimensions of 0.2 x 0.2 x 0.05 mm within one week.
In each case, crystals were transferred into cryoprotectant consisting of 20% glycerol in crystallization buffer before freezing in liquid nitrogen and data collection on Beamline X29 at the National Synchrotron Light Source at Brookhaven National Laboratory (Upton, NY). The diffraction data were integrated, scaled, and averaged using HKL2000.36 Data collection and refinement statistics are shown in Table I.
Table 1.
Data collection and refinement statistics
| Unliganded | CP-Bound | PALA-Bound | |
|---|---|---|---|
| Data collection | |||
| Space Group | C2 | C2 | C2 |
| Cell Dimensions: | |||
| a, b, c (Å) | 253.5, 153.0, 51.1 | 251.9, 153.0, 51.2 | 189.7, 141.4, 114.8 |
| α, β, γ (°) | 90.0, 96.9, 90.0 | 90.0, 96.1, 90.0 | 90.0, 110.0, 90.0 |
| Resolution (Å)a | 50–2.20 (2.28– 2.20) | 50.0–2.10 (2.18–2.10) | 50–2.60 (2.69– 2.60) |
| Rsym(%)a | 0.088 (0.52) | 0.085 (0.61) | 0.126 (0.61) |
| Average (I/σ)a | 20.02 (3.21) | 22.55 (3.31) | 17.38 (2.98) |
| Completeness (%)a | 99.9 (99.8) | 100.0 (100.0) | 100.0 (100.0) |
| Redundancya | 7.2 (6.8) | 7.5 (7.5) | 7.2 (6.8) |
| Refinement | |||
| Resolution (Å) | 49.17–2.20 | 34.12–2.10 | 47.28–2.58 |
| Reflections | 98,205 | 111,454 | 88,161 |
| Rwork/Rfree | 0.17/0.20 | 0.17/0.20 | 0.18/0.22 |
| Number of atoms | |||
| Protein | 6,974 | 6,922 | 13,752 |
| Waters | 583 | 624 | 720 |
| R.m.s. deviations | |||
| Bond lengths (Å) | 0.007 | 0.008 | 0.007 |
| Angles (°) | 1.01 | 1.06 | 0.74 |
| Mean B value (Å2) | 45.4 | 47.8 | 36.6 |
Values in parentheses are for the highest resolution shell
Structure solution and data refinement
The structure of the unliganded enzyme was solved by molecular replacement using as the initial model an E. coli c3 ATCase extracted from a holoenzyme structure in the T-state (PDB entry 1ZA1)11 after removal of water and ligand molecules. Molecular replacement was performed using the AutoMR module of PHENIX.37 The structure of the ATCase•CP complex was refined directly using the unliganded structure as the initial model. Poor fitting loops in the ATCase•CP complex were rebuilt using the AutoBuild module of PHENIX. The structure of the ATCase•PALA complex was solved by molecular replacement using BALBES38 using as the initial model an E. coli c3 ATCase extracted from a holoenzyme structure in the R-state (PDB entry 1D09).25 The final AT-Case•PALA structure contained two c3 units in the asymmetric unit as suggested by the Matthews coefficient. The AutoBuild module of PHENIX was used for initial model building, then waters were removed from the active site and the LigandFit module of PHENIX was used to place the six PALA molecules into the asymmetric unit. Manual rebuilding, addition of waters and CP and Pi modeling were performed using COOT.39 Waters were accepted if they were within hydrogen bonding distance of either main chain or side chain atoms. Automated refinement using non-crystallographic symmetry restraints were initially used but were turned off during final refinements performed by PHENIX. The final structures were validated using MolProbity and PROCHECK.42 Residues 292–304 (C-terminus) were omitted in all chains of each structure due to weak electron density.
Docking with glide
Docking of substrates to the B. subtilis structure in the absence and presence of ligands was performed using GLIDE (Schrödinger, Inc.) in extra-precision mode (XP). The Protein Preparation Wizard of MAESTRO (Schrödinger, Inc.) was used to assign bond orders, add hydrogen atoms, optimize the hydrogen-bonding network and to refine the hydrogen atom positions, using the OPLS2005 force field, while restraining the protein to a maximum displacement of 0.3 Å. Ligands were prepared for docking using the LigPrep module and all possible ionization states were generated. The Glide XP docking score was used to evaluate the results. In order to validate the GLIDE results CP was docked into the ATCase•CP structure with CP removed, and PALA was docked in the ATCase•PALA structure with PALA removed. The average difference in atomic positions between the docked CP and the CP position determined in the structure was 0.63Å, while the root mean square deviation of the two conformations was 0.22Å. The average difference in atomic position between the docked PALA and the PALA position determined in the structure was 0.31Å, while the root mean square deviation of the two conformations was 0.27Å.
Accession codes
Coordinates and structure factors for the unliganded, CP and PALA bound B. subtilis ATCases have been deposited in the Protein Data Bank under accession codes 3R7D, 3R7F, 3R7L respectively.
Three X-ray structures were determined of B. subtilis aspartate transcarbamoylase.
Structures were determined in the absence and presence of substrates and analogs
In silico docking was used to discern how the reactive substrates bind.
Electrostatic calculations were used to determine the alterations of pKa values.
Data were combined to visualize each step in the catalytic mechanism.
Acknowledgments
This work was supported in part by Grant GM26237 from the National Institutes of Heath. Data for the crystals were measured at Beamline X29 of the National Synchrotron Light Source. We thank Howard Robinson of Brookhaven National Laboratory for data collection and assistance with data processing. Use of the National Synchrotron Light Source, Brookhaven National Laboratory, was supported by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-98CH10886.
Abbreviations used
- ATCase
aspartate transcarbamoylase
- CP
carbamoyl phosphate
- Asp
L-as-partate
- CA
N-carbamoyl-L-aspartate
- Pi
inorganic phosphate
- CPSase
carbamoyl phosphate synthetase
- DHOase
dihydroorotase
- SAXS
stopped-flow small-angle X-ray scattering
- PALA
N-phosphonacetyl-L-aspartate
- TET
tetrahedral intermediate
- PEG
polyethylene glycol
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Gerhart JC, Pardee AB. Enzymology of control by feedback inhibition. J Biol Chem. 1962;237:891–896. [PubMed] [Google Scholar]
- 2.Wedler FC, Gasser FJ. Ordered substrate binding and evidence for a thermally induced change in mechanism for E. coli aspartate transcarbamylase. Arch Biochem Biophys. 1974;163:57–68. doi: 10.1016/0003-9861(74)90454-8. [DOI] [PubMed] [Google Scholar]
- 3.Krause KL, Voltz KW, Lipscomb WN. Structure at 2.9-Å resolution of aspartate carbamoyltransferase complexed with the bisubstrate analogue N-(phosphonacetyl)-L-aspartate. Proc Natl Acad Sci USA. 1985;82:1643–1647. doi: 10.1073/pnas.82.6.1643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robey EA, Schachman HK. Regeneration of active enzyme by formation of hybrids from inactive derivatives: implications for active sites shared between polypeptide chains of aspartate transcarbamoylase. Proc Natl Acad Sci USA. 1985;82:361–365. doi: 10.1073/pnas.82.2.361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jencks WP. Catalysis in Chemistry and Enzymology. McGraw-Hill; New York: 1969. [Google Scholar]
- 6.Hervé G, Nagy M, Le Gouar M, Penverne B, Ladjimi M. The carbamoyl phosphate synthetase-aspartate transcarbamoylase complex of Saccharomyces cerevisiae: molecular and cellular aspects. Biochem Soc Trans. 1993;21:195–8. doi: 10.1042/bst0210195. [DOI] [PubMed] [Google Scholar]
- 7.Grayson DR, Evans DR. The isolation and characterization of the aspartate transcarbamylase domain of the multifunctional protein, CAD. J Biol Chem. 1983;258:4123–4129. [PubMed] [Google Scholar]
- 8.Lipscomb WN. Aspartate Transcarbamoylase from Escherichia coli: Activity and Regulation. Adv Enzymol. 1994;68:67–151. doi: 10.1002/9780470123140.ch3. [DOI] [PubMed] [Google Scholar]
- 9.Beernink PT, Endrizzi JA, Alber T, Schachman HK. Assessment of the allosteric mechanism of aspartate transcarbamoylase based on the crystalline structure of the unregulated catalytic subunit. Proc Natl Acad Sci U S A. 1999;96:5388–5393. doi: 10.1073/pnas.96.10.5388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Endrizzi JA, Beernink PT, Alber T, Schachman HK. Binding of bisubstrate analog promotes large structural changes in the unregulated catalytic trimer of aspartate transcarbamoylase: implications for allosteric regulation. Proc Natl Acad Sci U S A. 2000;97:5077–5082. doi: 10.1073/pnas.090087197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wang J, Stieglitz KA, Cardia JP, Kantrowitz ER. Structural basis for ordered substrate binding and cooperativity in aspartate transcarbamoylase. Proc Natl Acad Sci U S A. 2005;102:8881–8886. doi: 10.1073/pnas.0503742102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ke H-M, Honzatko RB, Lipscomb WN. Structure of unligated aspartate carbamoyltransferase of Escherichia coli at 2.6-Å resolution. Proc Natl Acad Sci USA. 1984;81:4027–4040. doi: 10.1073/pnas.81.13.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Stevens RC, Reinisch KM, Lipscomb WN. Molecular Structure of Bacillus subtilis Aspartate Transcarbamoylase at 3.0 Å Resolution. Proc Natl Acad Sci U S A. 1991;88:6087–6091. doi: 10.1073/pnas.88.14.6087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stevens RC, Chook YM, Cho CY, Lipscomb WN, Kantrowitz ER. Escherichia coli aspartate carbamoyltransferase: The probing of crystal structure analysis via site-specific mutagenesis. Protein Eng. 1991;4:391–408. doi: 10.1093/protein/4.4.391. [DOI] [PubMed] [Google Scholar]
- 15.Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, Liang J. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res. 2006;34:W116–8. doi: 10.1093/nar/gkl282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gilson M, Honig B. Calculations of Electrostatic Potentials in an Enzyme Active Site. Nature. 1987;330:84–86. doi: 10.1038/330084a0. [DOI] [PubMed] [Google Scholar]
- 17.Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, Shaw DE, Francis P, Shenkin PS. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47:1739–49. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 18.Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, Banks JL. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J Med Chem. 2004;47:1750–9. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- 19.Friesner RA, Murphy RB, Repasky MP, Frye LL, Greenwood JR, Halgren TA, Sanschagrin PC, Mainz DT. Extra precision glide: docking and scoring incorporating a model of hydrophobic enclosure for protein-ligand complexes. J Med Chem. 2006;49:6177–96. doi: 10.1021/jm051256o. [DOI] [PubMed] [Google Scholar]
- 20.Baker DP, Aucoin JM, Williams MK, DeMello LA, Kantrowitz ER. Overexpression and Purification of the Trimeric Aspartate Transcarbamoylase from Bacillus subtilis. Prot Exp Purif. 1995;6:679–684. doi: 10.1006/prep.1995.1089. [DOI] [PubMed] [Google Scholar]
- 21.Li H, Robertson AD, Jensen JH. Very fast empirical prediction and rationalization of protein pKa values. Proteins. 2005;61:704–21. doi: 10.1002/prot.20660. [DOI] [PubMed] [Google Scholar]
- 22.Bas DC, Rogers DM, Jensen JH. Very fast prediction and rationalization of pKa values for protein-ligand complexes. Proteins. 2008;73:765–83. doi: 10.1002/prot.22102. [DOI] [PubMed] [Google Scholar]
- 23.Tsuruta H, Sano T, Vachette P, Tauc P, Moody MF, Wakabayashi K, Amemiya Y, Kimura K, Kihara H. Structural kinetics of the allosteric transition of aspartate transcarbamoylase produced by physiological substrates. FEBS Lett. 1990;263:66–68. doi: 10.1016/0014-5793(90)80706-o. [DOI] [PubMed] [Google Scholar]
- 24.Collins KD, Stark GR. Aspartate transcarbamylase: Interaction with the transition state analogue N-(phosphonacetyl)-L-aspartate. J Biol Chem. 1971;246:6599–6605. [PubMed] [Google Scholar]
- 25.Jin L, Stec B, Lipscomb WN, Kantrowitz ER. Insights into the mechanism of catalysis and heterotropic regulation of E. coli aspartate transcarbamoylase based upon a structure of enzyme complexed with the bisubstrate analog N-phosphonacetyl-L-aspartate at 2.1 Å. Proteins: Struct Funct Genet. 1999;37:729–742. [PubMed] [Google Scholar]
- 26.Gouaux JE, Krause KL, Lipscomb WN. The catalytic mechanism of Escherichia coli aspartate carbamoyltransferase: A molecular modeling study. Biochem Biophys Res Commun. 1987;142:893–897. doi: 10.1016/0006-291x(87)91497-5. [DOI] [PubMed] [Google Scholar]
- 27.Stebbins JW, Kantrowitz ER. Conversion of the Non-Cooperative Bacillus subtilis Aspartate Transcarbamoylase into a Cooperative Enzyme by a Single Amino Acid Substitution. Biochemistry. 1992;31:2328–2332. doi: 10.1021/bi00123a017. [DOI] [PubMed] [Google Scholar]
- 28.Koshland DE, Nemethy G, Filmer D. Comparison of Experimental Binding Data and Theoretical Models in Proteins Containing Subunits. Biochemistry. 1966;5:365–385. doi: 10.1021/bi00865a047. [DOI] [PubMed] [Google Scholar]
- 29.Monod J, Wyman J, Changeux JP. On the Nature of Allosteric Transitions: A Plausible Model. J Mol Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- 30.Hervé G, Moody MF, Tauc P, Vachette P, Jones PT. Quaternary structure changes in aspartate transcarbamylase studied by X-ray solution scattering; signal transmission following effector binding. J Mol Biol. 1985;185:189–199. doi: 10.1016/0022-2836(85)90190-1. [DOI] [PubMed] [Google Scholar]
- 31.Chan RS, Sakash JB, Macol CP, West JM, Tsuruta H, Kantrowitz ER. The role of intersubunit interactions for the stabilization of the T State of Escherichia coli aspartate transcarbamoylase. J Biol Chem. 2002;277:49755–49760. doi: 10.1074/jbc.M208919200. [DOI] [PubMed] [Google Scholar]
- 32.Sakash JB, Chan RS, Tsuruta H, Kantrowitz ER. Three of the six possible intersubunit stabilizing interactions involving Glu239 are sufficient for restoration of the homotropic and heterotropic properties of Escherichia coli aspartate transcarbamoylase. J Biol Chem. 2000;275:752–758. doi: 10.1074/jbc.275.2.752. [DOI] [PubMed] [Google Scholar]
- 33.Nowlan SF, Kantrowitz ER. Superproduction and rapid purification of E. coli aspartate transcarbamoylase and its catalytic subunit under extreme derepression of the pyrimidine pathway. J Biol Chem. 1985;260:14712–14716. [PubMed] [Google Scholar]
- 34.Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- 35.Brabson JS, Switzer RL. Purification and properties of Bacillus subtilis aspartate transcarbamoylase. J Biol Chem. 1975;250:8664–8669. [PubMed] [Google Scholar]
- 36.Otwinowski Z, Minor W. Processing of X-ray diffraction data collected in oscillation mode. In: Carter CW Jr, Sweet RM, editors. Methods Enzymol. Vol. 276. Academic Press; NY: 1997. pp. 307–326. [DOI] [PubMed] [Google Scholar]
- 37.Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC, Zwart PH. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. 2010;D66:213–21. doi: 10.1107/S0907444909052925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Long F, Vagin AA, Young P, Murshudov GN. BALBES: a molecular-replacement pipeline. Acta Crystallogr D Biol Crystallogr. 2008;64:125–32. doi: 10.1107/S0907444907050172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and Development of Coot. Acta Cryst. 2010;D66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen VB, Arendall WB, 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Davis IW, Leaver-Fay A, Chen VB, Block JN, Kapral GJ, Wang X, Murray LW, Arendall WB, 3rd, Snoeyink J, Richardson JS, Richardson DC. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–83. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Laskowski RA, MacArthur MW, Moss DS, Thornton JM. PROCHECK: A program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26:283–291. [Google Scholar]