

Abstract
Ferritin nanocages synthesize ferric oxide minerals, containing hundreds to thousands of Fe(III) diferric oxo/hydroxo complexes, by reactions of Fe(II) ions with O2 at multiple di-iron catalytic centers. Ferric–oxy multimers, tetramers, and/or larger mineral nuclei form during postcatalytic transit through the protein cage, and mineral accretion occurs in the central cavity. We determined how Fe(II) substrates can access catalytic sites using frog M ferritins, active and inactivated by ligand substitution, crystallized with 2.0 M Mg(II) ± 0.1 M Co(II) for Co(II)-selective sites. Co(II) inhibited Fe(II) oxidation. High-resolution (<1.5 Å) crystal structures show (1) a line of metal ions, 15 Å long, which penetrates the cage and defines ion channels and internal pores to the nanocavity that link external pores to the cage interior, (2) metal ions near negatively charged residues at the channel exits and along the inner cavity surface that model Fe(II) transit to active sites, and (3) alternate side-chain conformations, absent in ferritins with catalysis eliminated by amino acid substitution, which support current models of protein dynamics and explain changes in Fe–Fe distances observed during catalysis. The new structural data identify a ~27-Å path Fe(II) ions can follow through ferritin entry channels between external pores and the central cavity and along the cavity surface to the active sites where mineral synthesis begins. This “bucket brigade” for Fe(II) ion access to the ferritin catalytic sites not only increases understanding of biological nanomineral synthesis but also reveals unexpected design principles for protein cage-based catalysts and nanomaterials.
Introduction
Protein nanocages in the ferritin family have central biological functions in iron storage1–3 as well as applications in nanocatalysis and nanomaterials design.4-8 A variety of metals bind ferritins, including Cd, Tb, Ca, Mn, Mg, Zn, Cr, Pd, and Pt.6,7,9-13 The natural iron sites, which are associated with ferrous iron passing into the cage, binding, catalytic oxidation, coupling, and the release of ferric oxo products to the central nanocavity that contains the mineral, are distributed at various spatially and functionally distinct positions. In the conserved family of 24-subunit ferritin–protein nanocages (maxi-ferritins of plants animals and bacteria), the reaction at each catalytic site involves Fe(II) that binds to the di-iron sites, followed by dioxygen binding and formation of a diferric peroxo complex.14–18 The complex decays to release diferric oxo mineral precursors (eq 1).
| (1) |
Ferritin di-iron sites, which are related to di-iron cofactor sites in oxygenases,19–21 have several names: ferroxidase, Fox, FC, and oxidoreductase. To emphasize the two substrates in ferritin catalytic reactions, ferrous and dioxygen or hydrogen peroxide, the term oxidoreductase is used here as the name for the ferritin catalytic sites.
How Fe(II) reaches the active sites, deep within each of the 24 polypeptide subunits (Figure 1), remains poorly understood, although the trajectory of iron–oxy complexes after catalysis is now known.22 Knowing all the iron-binding sites in ferritin–protein cages is important not only for understanding biological function but also for designing metal sites in ferritin–protein nanocages for nanodevices and nanocatalysts.7,8,23
Figure 1.

Mg(II)- and Co(II)-binding sites in the X-ray crystal structures of frog M ferritin and special relationships of functional sites. (A) 24-subunit, self-assembling ferritin nanocages, crystallized from 100 mM BICINE, pH 9.0, with 2.0 M MgCl2. (B) 24-subunit, self-assembling ferritin nanocages, crystallized from 100 mM BICINE, pH 9.0, with 2.0 M MgCl2 and 0.1 M CoCl2. (C) A monomeric ferritin subunit viewed from the outside of the protein cage for Mg(II)–protein cocrystals. (D) A monomeric ferritin subunit viewed from the outside of the protein cage for Co(II) + Mg(II)–protein cocrystals. Mg(II), green spheres; Co(II), magenta spheres; blue squares, three-fold pores; green squares, four-fold pores; red circles, active sites; light blue, mineralization cavity.
Trapping Fe(II) substrate in ferritin–protein crystals has been difficult due to the speed (millisecconds) of catalytic oxidation to the diferric peroxo intermediate and decay to the diferric oxo product (seconds, eq 1). However, structural information on Fe(II) in the ferritin di-iron site has been obtained by variable-temperature, variable-field magnetic circular dichroism/circular dichroism (VTVH MCD/CD)20 and in crystals of a heme-containing bacterioferritin from Escherichia coli, where the first iron added is retained at the catalytic site and appears to behave as a cofactor.2,24
Here we characterize the path of metal ions from the solution to the catalytic center using X-ray diffraction of active and inactive ferritin–protein cages cocrystallized with Co(II) + Mg(II). These crystal structures reveal the following:
A line of metal ions in the channels connects pores in the external cage to the inner cavity surface. Three metal ions occur around the pore entrance to the cavity, as if directed toward the three active sites linked to each pore.
Metal sites also occur along the inner surface of the protein cage between the pores and the active sites.
Metal ligands at the active sites adopt alternate orientations that are lost when amino acid substitution eliminates catalysis. These results identify the protein flexibility associated with activity that was suggested previously by the changes in Fe–Fe distances during the catalytic cycle.16
Experimental Procedures
Protein Preparation
The Fe(II)-site mutants (Gln137Glu, Asp140His, and Gln137Glu/Asp140His) of frog M ferritin were generated using the QuikChange method (Stratagene) in the pET-3a vector encoding wild-type frog M ferritin.21 Ferritin variants were expressed in E. coli BL21(DE3) pLysS21,25 and purified as described previously.25-27 The buffer of purified samples was exchanged with 100 mM 3-(N-morpholino)propanesulfonic acid (pH 7.0) and 100 mM NaCl for storage at 4 °C, during which time catalytic activity was constant. The Bradford method (Biorad) was used with bovine serum albumin as a standard to determine protein concentrations.
Protein Crystallization
Crystals were grown by the hanging-drop vapor-diffusion method in drops containing 2 μL of 5–20 mg/mL protein solution mixed with an equal volume of the precipitant solution, 2.0 M MgCl2 and 100 mM N,N-bis(2-hydroxyethyl)glycine (BICINE) (pH 9.0), with and without 100 mM CoCl2. Cubic crystals formed within 1–2 days at 4 °C. The crystals were transferred to precipitant solution containing 20% (v/v) ethylene glycol as a cryoprotectant and kept at least 5 min before freezing in liquid nitrogen.
Data Collection
X-ray diffraction data were collected at 100 K at 11 111 eV on Beamline 8.3.1 at the Advanced Light Source, Lawrence Berkeley National Laboratory. The data were processed using the programs ELVES, MOSFLM, and SCALA.28-30 Data collection statistics are summarized in Table S1 (Supporting Information). Co(II) was distinguished from Mg(II) by anomalous scattering.
Refinement
Initial models were obtained by molecular replacement using EPMR31 with a single subunit of the structure of wild-type frog M ferritin (PDB code 1MFR),27 excluding Mg(II) and water molecules, as the search model. Structures were refined using the program REFMAC532 and modified on the basis of 2mFo – DFc and mFo – DFc electron-density maps using COOT.33 Water molecules were positioned initially using ARP/wARP.33,34 Co(II) ions were placed on the basis of the anomalous Fourier difference maps and geometry. Metal-ion occupancy was manually adjusted to minimize difference density. Positions of Mg(II) ions were determined on the basis of the coordination geometry. The atomic temperature factors were refined anisotropically, and the quality of the model was evaluated using the WHAT IF Web server.35 The refinement parameters are summarized in Table S1. Figures showing the structures were prepared using PyMOL (www.pymol.org).36
Protein Data Bank Codes
The atomic coordinates and structural factors have been deposited in the Protein Data Bank with accession codes 3KA3, 3KA4, 3KA6, 3KA8, and 3KA9 for Mg(II)-wild type, Co(II)-wild type, Co(II)-Gln137Glu, Co(II)-Asp140His, and Co(II)-Gln137Glu/Asp140His, respectively.
Results
High-Resolution Crystal Structures of Mg(II)– and Co(II)– Ferritin–Protein Nanocages Reveal Pathways for Metal-Ion Entry and Active-Site Access
To obtain crystals with a resolution high enough to see side-chain conformations in a wild-type sequence that avoided the amino acid substitutions used to facilitate crystallization,10 we used the M ferritin of the bullfrog, Rana catesbeiana. Frog ferritin, which assembles into a classic cage containing 24 identical subunits, has been used frequently as a model because of the favorable phylogenetic position of the amphibian in comparison to other vertebrates (see ref 1 and the Supporting Information). Moreover, in kinetic and spectroscopic studies, different iron species in the early stages of the catalytic cycle are conveniently trapped.16,17,20,21 In a previous study of frog M ferritin, however, trigonal crystals (space group P3121) obtained from 0.1 M sodium acetate, pH 4.6, 0.1 M MgCl2, and 25% (NH4)2SO4 diffracted to only 2.8 Å resolution.37 In those crystals, which are stabilized by contacts between α4–α5 loops in different nanocages, significant regions of the protein were unresolved.27 We found that crystals grown from 100 mM BICINE, pH 9.0, 2.0 M MgCl2 ± 0.1 M CoCl2 diffracted to higher resolution (<1.5 Å). Using these new cubic crystals (space group F432), which resemble many other ferritin crystals,10,12,26,38–41 we could model all residues in frog M ferritin except the C-terminal Ser175.
With a Cα root-mean-square-deviation (rmsd) of 0.19 Å, the overall frog M ferritin structure is essentially identical to that observed previously (PDB 1MFR).27 Residues located for the first time include Arg59, Glu60, Lys82, Glu88, Gln101, Lys115, Asp119, Glu136, Lys139, and Glu158. In addition, the electron density of the C-terminal residues 172–174, missing in the previous structure, was present in the new structure. Numerous ordered water molecules were observed at the 24 oxidoreductase sites, in the eight ion channels, and on the shell surfaces. Ten Mg(II) and eight Co(II) metal ions with occupancies ≥25% were observed bound to each subunit. Of the 10 Mg(II) per subunit, three bind to residues in three subunits, making the total number of Mg(II) sites per cage 192. In addition, of the eight Co(II) ions per subunit, one is shared by residues from three subunits (Glu 130) and one is shared by four subunits, making the total number of Co(II) sites per cage 152. The conformational flexibility and low (25%) occupancy of some of the metal sites suggest that the total number of metal ions bound at any one time may be lower.
The positions of the Mg(II) in each subunit were assigned on the basis of the geometry and the relatively short distance between Mg(II) ion and ligand (amino acid or water molecules) of ~2.1 Å. The coordination geometry of almost all Mg(II) ions in the current structure is octahedral. Two of the 10 Mg(II) ions were at the di-iron oxidoreductase centers,27 although the ligands for Mg(II) are different from those previously reported. Five Mg(II) ions are observed as [Mg(H2O)6]2+. Remarkably, multiple Mg(II) ions formed a line inside the three-fold-symmetric pores that connect the external surface to the central cavity (Figures 1 and 2).
Figure 2.
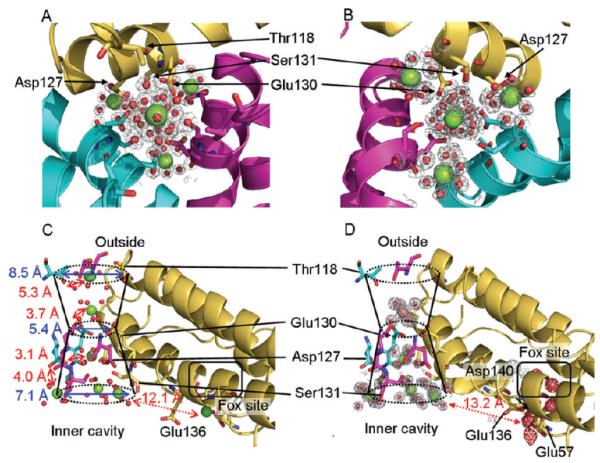
Hydrated Mg(II) and Co(II) ions form a line through the three-fold-symmetric ion channels of ferritin–protein cages. A line of four metal ions is observed in the channel from the outer to the inner edge of the protein cage, plus two more at the inner edge of the protein; metal residue distances range from 3.1 to 5.3 Å. The three metals ions accumulated at the channel exits into the cavity suggest metal ions being distributed from one pore to the three active sites ringing each pore. Top: Views through the channels from the pores. Each of the three subunits is displayed in a different color. (A) From outside of the cage. (B) From inside of the cage at the channel exit into the central cavity. Bottom: Views of the ion channel with outer and inner pores formed by three subunits. (C) One of the three-fold-symmetric pores in ferritin cocrystallized with Mg(II); six Mg(II) ions link the ion channel to the two at the active site. (D) One of the pores in ferritin–protein cages, cocrystallized with Mg(II) and Co(II), showing one Co(II) ion and four Mg(II) ions in the ion channel and four Co(II) ions at the catalytic centers. Mg(II), green spheres; Co(II), magenta spheres; blue, dimensions of the funnel-shaped ion channel; red, Mg residue distances and the distance between the metal ion bound to Asp127 and a metal ion at the inner cavity surface near the catalytic sites. The gray mesh represents the 2Fo – Fc electron density map at 1.5σ level.
The eight Co(II) ions observed in the ferritin cocrystals (Figures 1B,D and 2D) were assigned unambiguously on the basis of the strong scattering and anomalous scattering effects of cobalt; Co(II) binding in the presence of the 20-fold excess of Mg(II) required for crystallization shows sites’ selectivity for Co(II). Selective binding of Co(II) to ferritins near the active sites is also indicated by the 70% inhibition of Fe(II) oxidation in solution by Co(II), while Mg(II) has no significant effect (<5%). In the presence of the eight bound Co(II) ions, five Mg(II) ions were displaced in the pores or into new sites near the active sites. One Co(II)-specific site was observed at the end of the Fe(III) channel in each subunit, where the premineral products of oxidoreductase catalysis exit the protein cage and enter the nanocavity.22 This Co(II) site is at the end of the long axis of each subunit, near the four-fold axes. Other Co(II)-specific sites occur at the di-iron active sites where alternate conformations of paired Co(II) ions are observed (Figure 3; Tables S2 and S3, Supporting Information).
Figure 3.

Alternate Co(II) site conformations at the catalytic centers in ferritin–protein nanocages coincide with activity and suggest intermediate Fe(II)-binding sites between the pores and the active centers. Residues that bind Co(II) in protein crystals are shown as stick models; residues with alternate conformations are shown in green and cyan. (A–D) Co(II) binding at active sites in ferritin Mg(II) + Co(II) protein cages with or without altered Fe(II) ligands; Co(II) is an Fe(II) inhibitor. All His substitutions resulted in inactivation, and the very conservative change, Gln137Glu, eliminated the normal reaction pathway and resulted in intermediates and mineral products with altered spectroscopic properties.21 (A) Wild-type ferritin; (B) Asp140His ferritin; (C) ferritin with two substitutions, Asp140His and Gln137Glu; and (D) Gln137Glu ferritin. The appearance of four Co(II) ions indicates multiple conformations of Co(II)-binding sites that are coincident with activity; the inactive proteins in panels B and C have only one set of Co(II) ligands. (E) “Snapshots” of possible intermediates for Fe(II) moving from the pores to the active centers, based on Co(II) binding and plausible geometry of the coordination structure for each Co(II) (see Tables S2 and S3, Supporting Information). Red mesh, Co(II) anomalous scattering with 3.0σ level; gray mesh, 2Fo – Fc electron density map with 1.5σ level; green, Mg(II); magenta, Co(II).
A Line of Metal Ions Reveals the Connection between the External Pores at Three-Fold Axes and the Internal Mineralization Cavity
The paths of ions through the apparently solid protein shell have not been elucidated, despite the observation of bound metal ions, e.g., Mg, Ca, Zn, Cd, and Pd, in ferritin–protein cages around the exterior surfaces near the three-fold axes and pores.6,12,13,38,42,43 In contrast to mitochondrial ferritin, which binds [Mg(H2O)6]2+ at the three-fold pore,12 in frog M ferritin, four hydrated Mg(II) ions form a 15.5 Å line that passes through channels connecting the pores on the external surface to the inner pores that exit into the cavity of the protein cage (Figure 2). One of the Mg(II) ions in the center of the channel is flanked by two other Mg(II) ions at the inner and outer pore exits (Figure 2). The three metal ions at the inner end of the channels may indicate how Fe(II) substrate is distributed by each of eight pores to each cluster of three active sites in the protein cage.
The Mg(II) ions nearest the external pore exit at the three-fold axes of the ferritin–protein cage are coordinated to at least four ordered water molecules. The amino acid closest to the Mg(II) at the external pore opening, Thr118, is too far away to interact directly with the metal (hydroxyl oxygen–Mg(II) = 5.3 Å). The next Mg(II) ion in the channels, also hydrated, is bound at a constriction in the channels to 5.4 Å, from 8.5 Å. The Mg(II) ion is in the center of the channel and is hydrogen-bonded through coordinated waters to carboxylates of conserved residues Asp127 and Glu130 and to the hydroxyl group of Ser131 contributed by the three subunits that make up each pore (Figure 2C). The next Mg(II) ion also is coordinated through water to Asp127 from each subunit. A fourth hydrated Mg(II) ion binds at the end of the channel where the pore connects the channel to the internal cavity of the cage, and the channel is widened to 7.1 Å. The ions at the end of the channel are a short distance from residues in the active sites (Figure 2C,D).
When frog M ferritin is cocrystallized with 0.1 M Co(II) in the presence of 2 M Mg(II), [Co(H2O)6]2+ is hydrogen-bonded to Glu130 at the channel constriction, displacing [Mg(H2O)6]2+ and preventing Mg(II) binding at the external channel entrance (Figure 2D). The presence of four Co(II) ions in the region of each active site (Figures 2D and 3A) contrasts with Mg(II) (Figure 2C). While Co(II) hydrates occupy metal-binding sites between the three-fold pore and the active site, the difference in the redox/hydrolytic chemistry of Co(II) prevents formation of cobalt oxide minerals inside ferritin–protein cages. The smaller number of Co(II) ions in the entry channels likely reflects a higher affinity of the active sites for Co(II) than Mg(II) and the inhibitory effects we observed of Co(II) on Fe(II) oxidation.
Multiple Metal-Binding Sites and Alternate Conformations at the Oxidoreductase Site
Around the oxidoreductase center, we observe four Co(II) ions (Figure 3B), designated as Co1, Co2, Co3, and Co4, with 0.70, 0.50, 0.30, and 0.50 occupancy, respectively. The refined occupancies of these ions suggest that the binding sites are metastable, and the conformational heterogeneity may reflect the superposition of bound and free sites. The amino acids that display two conformations in the Co(II)–ferritin cocrystals are conserved active-site residues Glu57, Glu103 (in the Fe2 site), Gln137, Asp140, conserved Tyr30, and frog M ferritin specific His54 (Figure 3B). More of the flexible residues are in or near the Fe2 site, the ferritin-specific site of the di-iron protein catalyst family, with the weaker Fe ligand constellation that is required for rapid iron oxidation21 and likely for product (diferric oxo) release.
The location of the Co1–Co2 pair is quite similar to the oxidoreductase center sites, Fe1 and Fe2, required for catalysis and to the Zn sites in the crystal structure of human H ferritin (PDB 2CIH13). However, the ligands coordinating the Co1–Co2 pair (Tables S2 and S3, Supporting Information) do not match those defined by mutagenesis and spectroscopy with the natural substrate Fe(II).20,21,25 For example, Co2 is 4.1 Å from the carbonyl oxygen of the Fe ligand Gln137 and 4.5 Å from the carboxylate of Asp140. Moreover, the Co(II) ligands include His54, which is not conserved in ferritin sequences. The Co(II) ligand environment is completed by a water molecule (W2) bridging Co1 and Co2. A similar bridging water has been observed in a di-Co(II) model complex.44 The Co1–Co2 distance of 4.2 Å is longer than the Fe–Fe distances in ferritin, measured using EXAFS at 3.4, 2.5, and 3.0 Å for the di-Fe(II), diferric peroxo, or diferric oxo/hydroxo forms, respectively.16 The different coordination environment for Co1–Co2 and Fe1–Fe2 will contribute to the resistance of Co(II) oxidation in ferritin and the Co(II) inhibition of Fe(II) oxidation.
The ligand environments of Co1, Co2, Co3, and Co4 (Figure 3; Table S3) suggest different sites where Fe(II) binds on the way to the active site. Co2,Co3 and Co3,Co4 are likely alternate binding sites until two Fe(II) ions, which bind independently,20 bind at the Fe1 and Fe2 sites. The Co–Co distances for the possible di-Co sites are 3.1–4.2 Å with a bridging carboxylate ligand (Glu58 for Co1–Co2, Glu103 for Co2–Co3, and Asp140 for Co3–Co4), like those observed in other Co–Co sites.44 A reasonable path for Fe(II) from three-fold pores to the catalytic centers is indicated by Co3–Co4/Co2–Co3 binding (Figure 3E). The Co4 site connects the inner exit of the three-fold channels from the outside of the shell to the active site because, in addition to Glu57 and Glu136, it contains Asp140, which is part of the active-site Fe2 ligand set. Given the higher occupancy of the Co4 site, it may be that Glu57 and Glu136 are gates for Fe(II) binding at the active sites (Figure 3C). The multiple Co(II)-binding sites may explain the presence of a so-called C or Fe3 site.11,24 Flexibility of the metal ligands around the ferritin oxidoreductase centers is needed for the catalytic cycle.16
High-Resolution Structures of Ferritin–Co(II) Cocrystals with Catalytic Sites Modified by Ligand Substitutions Support a Relationship between Ligand Structural Flexibility and Activity
To explore the role of alternate structures in turnover, we determined the structures of inactive ferritin mutants21 with the Asp140His/Gln137Glu or Asp140His substitution in the catalytic site. Histidine and glutamate occur at the Fe2 site of di-iron oxygenases centers.45,46 Both mutations alter Co(II) binding (Figure 3; Table S2). Co(II) bound to His140 and Glu137 at the Fe2 site in the variant with two amino acid substitutions, and conformational heterogeneity was eliminated (Figure 3B,C). Moreover, the B-factor values around the Fe2 sites in His140 ferritins were significantly lower than in wild-type (Asp140) ferritin (Figure S2, Supporting Information). Only two Co(II) environments were observed in the active sites in the Asp140His mutant, whether Gln or Glu was at position 137, in contrast to four arrangements in the wild-type active site. In each of these proteins, loss of ligand flexibility coincided with loss of catalytic activity.
Minor changes in the active-site ligands affect ferritin catalysis. For example, in the Gln137Glu variant, the diferric peroxo intermediate (eq 1) is replaced with an unstable, as yet unidentified intermediate.21 The mutation also alters Co(II) binding, although side-chain orientation is preserved, and two alternate sets of two Co(II) ions were observed in the Co(II)–ferritin crystal structures (Figure 3D). When a carboxylate replaces the amide, coordination of Glu103 to Co2 is monodentate, and a water molecule (W3), coordinated to Co2 when Gln is present, is absent. Based on solution properties, such subtle structural differences in ferritins affect not only the properties of catalytic intermediates and side reactions but also the spectroscopic properties of the mineral itself.21
Additional Co(II) and Mg(II) Binding Sites
We observed several additional metal ion sites in the ferritin crystals distributed throughout the protein cage: (1) the outer shell surface near two-fold axes of each cage and between two protein cages; (2) the center of the four-fold cage axes; and (3) the inner cage surface at the end of each subunit helix bundle, where mineral nuclei enter the cavity,22 and near the four-fold axes, at junctions of pairs of subunits.
On the outer surface near the two-fold axes, three hydrated Mg(II) ions bind in the presence or absence of Co(II) (Figure S1, Supporting Information). The ions bind near residues Lys82 and Lys83 in a long loop and bridged to Glu80 in a neighboring ferritin cage in the crystals. Glu80 also forms a Cd(II) bridge in crystals of horse spleen9 and human H ferritin.9,10,12,13
On the four-fold axes lined with helix 5 from each of four subunits, a Co(II) is coordinated to each His169, Cl−, and a water molecule (Figure S1). Cl− was identified by weak anomalous scattering, comparable to S, and the presence of Br, identified by anomalous scattering in ferritin crystallized from 1 M MgBr2. Metal ions have been observed in the same place in other eukaryotic ferritins12,13 and in the prokaryotic Helico-bactor pylori ferritin. In the divergent H. pylori ferritin, the four-fold opening is proposed as an iron entry channel.47 By contrast, in frog M ferritin, only a single ion is found in these sites. A common functional role for the four-fold openings remains uncertain.
On the inner surface of the ferritin cage, metal ions bind to sites on the two-fold axes and at the four-fold axes. Co(II) is coordinated by conserved residues Glu60 and Glu63, three waters, and His56 from an adjacent subunit (Figure S1). Other metal ions have been observed in the same place.6,13,42 Glu60 and Glu63 are required for mineral formation in recombinant L-subunit ferritins (animal-specific subunits without catalytic sites)48 but not in recombinant H-subunit (catalytically active) ferritins.49 These different requirements for Glu60 and Glu63 suggest a role for these residues independent of catalysis, such as moving Fe(II) dissolved from the mineral surface out of the protein cage. At another metal ion site on the inner surface near the four-fold axes (Figure S1), Mg(II) is coordinated to the conserved Asp/Glu146 and water, while Co(II) is coordinated to the conserved Glu49 and His45 via coordinated water. These metal sites also are present in both H and L ferritins.6
Discussion
Ferritins combine the properties of ion-channel proteins and Fe(II)/O2 oxidoreductases. Fe(II) passes through the protein nanocage to the active sites, and the Fe(III)–O mineral precursors move to the cavity where bulk mineral grows. Until recently, the accretion of diferric oxo catalytic products into the hydrated oxide mineral containing thousands of ferric ions, coordinated water, and oxo/hydroxo bridges was considered to occur by protein-independent, hydrolytic chemistry, in spite of the differences in the mineral order of ferritins with different protein structures.50 However, NMR studies that monitored the progress of diferric products of ferritin catalysis through the protein22 identified the postcatalytic path of the Fe(III)–O mineral precursors from the active site to the cavity. The results also showed the presence of sites within the protein cage for tetrameric and possibly larger Fe(II) oxo multimers that direct the ferric oxo multimers to cavity entrances near those of other emerging ferric oxo mineral nuclei to facilitate mineral growth. By contrast, little is known about how the Fe(II) substrates reach the active sites that are in the centers of the four helix bundles in 24-subunit, eukaryotic ferritin cages.
To mark the path of sites transiently occupied by ferrous ions en route to the active sites, we crystallized frog M ferritin in the presence or in the absence of 0.1 M Co(II) ions, with the 2.0 M Mg(II) required for crystallization. While these concentrations of ions are vastly higher than are ever encountered under physiological conditions, they reveal preferred weak binding sites. Because they link the exterior to the active site and involve conserved residues, the hydrated ion sites suggest a preferred path that is sparsely and transiently occupied at lower ion concentrations. The Mg(II) and Co(II) ions, however, are not equivalent to each other, nor do they precisely mimic the binding of Fe(II) at the active site. However, the greater similarity of Co(II) to Fe(II) compared to Mg(II) explains the absence of Mg(II) in the region of the catalytic center when Co(II) is present, even at 20-fold lower concentration than Mg(II) (Figure 2D; Table S2). Such reasoning also explains the metal ion selectivity at Glu130 in the ion entry channel. Neither Co(II) nor Mg(II) ions bind to the Fe(II)-binding residues identified by effects of amino acid substitutions on catalysis and by VTVH MCD/CD spectroscopy.18,20,21,25,51–53
Hydrated Co(II) and Mg(II) ions occupy sites that trace a continuous path from the outside of the ferritin nanocage through the three-fold pores to the cavity and the oxidoreductase sites. A line of four hydrated metal ions link the external pore entrance to the inner channel exit (Figure 2). Metal–amino acid distances in the ion channel range from 5.3 Å at the external edge of the channel to 3.1 Å in the middle of the channel (Figure 2C). One of the four metal ions at the cavity opening is flanked by two other metal ions. The three metal ions that are grouped at the end of each of the eight ion channels are bound to three Asp127 residues, contributed by each of the subunits that form the channels. Such an arrangement of the three metal ions and three Asp127 residues shows how eight ion channels can deliver iron to 24 active sites: each active site is linked through a fairly short distance of polypeptide chain to one of the three Asp127 residues in the ion channels (Figures 2C and 4). Moreover, amino acid substitution of Ala for Glu130 or Asp127 inhibits formation of the diferric peroxo complex (S. Haldar, L.E. Bevers, T. Tosha, and E.C Theil, to be published). After leaving the eight ion channels, ferrous ion substrates can be guided by the negatively charged, inner cavity surface.1,54,55 Here, in the Co(II)-ferritin cocrystals, we observe Co(II) coordinated by the conserved Glu57 and Glu136 as well as active-site Asp140 in the Fe2 site (Figure 3A). The ~13 Å distance between the pore exit site and Glu136 could easily be shortened by ~5 Å if Glu136 flipped from the trans configuration to the gauche + rotamer found in crystals of other ferritins.56 Thus, Thr118 (three-fold pore entrance) → Asp127, Glu130, Ser131 → Glu135, Glu136, Lys139 (cavity surface) → Glu136, Glu57, Asp140 (oxidoreductase site) likely serve as fleeting waystations that form a favored path for Fe(II) from the outside of the ferritin cage to the active site (Figure 4). Most of the residues on the path are conserved. This path not only solves the problem of access of hydrated ions through the protein shell but also focuses directly on the oxidoreductase active site.
Figure 4.

Co(II)- and Mg(II)-binding sites in the ion channels, at the inner cavity surface, and around the actives sites of ferritin–protein cages show the path from pores to active sites and indicate how eight ion channels can provide Fe(II) substrate to 24 oxidoreductase sites. (A) The inner cavity surface of three subunits at the ion channel pore; black arrows show the proposed Fe(II) path from the external pores to three active sites. (B) The same view enlarged to show a single subunit. Green, Mg(II) ions; magenta, Co(II) ions. (C) Superposition of soybean ferritin (PDB ID 3A68) on frog M ferritin to show a plausible alternative configuration of Glu136 (green stick) in frog M ferritin to assist in Fe(II) transfer from Asp127 in the ion channels to the active sites. Yellow stick, Glu173 in soybean, which corresponds to Glu136 in frog M ferritin; orange stick, Glu136; yellow sphere, Ca bound to Glu173. (D) Schematic overview of the trajectory of iron as Fe(II) enters the ferritin–protein nanocage through three-fold ion channels, moves to the active sites, and, as Fe(III)O complexes, leaves the catalytic sites to traverse the postoxidation channels for mineral nucleation and exit into the mineralization cavity.
The alternate conformations for residues at or near the active site, such as conserved Glu57 and Asp140, as well as His54, illustrate the importance of protein cage dynamics to ferritin function. Catalytic activity21 coincided with conformational flexibility of the metal substrate ligands when we analyzed the Co(II)–protein crystal structures of ferritin–proteins engineered with altered active-site ligands (Figure 3). Since the mutations created di-iron sites with the same ligands as di-iron oxygenases (e.g., methane monooxygenase, ribonucleotide reductase, and Δ9-stearoyl-acyl carrier protein desaturase (Δ9 desaturase)) that form the same diferric peroxo intermediates as the ferritins,19,45,46,57 the loss of catalytic activity in Asp140His ferritin indicates that His140 prevents Fe(II) access to a productive configuration at the ferritin active sites. The geometry of Co1 in the Asp140His ferritin–Co crystal structure is indistinguishable from the distorted square pyramid of Co1 bound to the wild-type protein. However, in contrast to Asp140 in the wild-type protein, His140 does not bind Co2 (Figure 3B,C), nor does His54 bind Co(II); His54 is flipped away from the Co2 site, suggesting a steric clash between His54 and His140. His140 also influences the orientation of Glu136, which has moved in the Asp140His variant away from the catalytic site, possibly inhibiting Fe(II) access from the entry channels. Differences in the location of the active sites–internal in 24 subunit maxi-ferritins of eukaryotes and many bacteria and on the cavity surface in 12 subunit mini-ferritins (Dps proteins) of archaea and bacteria1,54,55–indicate different postcatalytic pathways. The conformational flexibility in the ferritin–protein cage itself, observed here in relation to oxidoreductase catalysis, has been observed previously in relation to Fe(II) entry and mineralization in ferritins that lack catalytic centers26 and is also likely to play a role during diferric oxo Fe(III) dimerization to tetramers.22
The Co(II) cocrystal data in this study combined with the NMR study of Fe(III)–O in ferritin22 indicate that iron ions follow paths through the 24-subunit protein cages of eukaryoitc ferritins that are ~47 Å long. In contrast, the cage thickness is <20 Å. The long distances iron ions travel through the ferritin–protein cage permit a series of reaction steps in iron mineralization to occur. In this model, Fe(II) ions move a distance of 15.5 Å through the three-fold pores into the central cavity and are guided 12–13 Å along the inner surface of the cage by carboxylate residues to the oxidoreductase sites. After catalytic oxidation of two Fe(II) and coupling via an oxo/hydroxo bridge, the newly formed mineral precursors can follow a path of ~20 Å on the long axis of each subunit, as detected by NMR.22 Mineral nucleation begins between the active sites and the cage exits. As the ferric multimers pass through the protein cage to cavity entrances, they are guided by the protein to exits that are close enough to three others, around the four-fold axes, that bulk mineral formation is enhanced.22 The dynamics of the ferritin–protein cage structure contrasts with earlier concepts of ferritin “protein shells” that simply sequester the active sites and the stored iron mineral. Instead, a one-dimensional path of conserved residues is positioned in the structure to dynamically target ferrous ions to the catalytic sites and to promote mineral formation.
Supplementary Material
Acknowledgment
CHORI Foundation (E.C.T.), NIDDK 20251 (E.C.T., O.B., T.T.), NIH GM48958 (T.A., H.-L.N.), and JSPS (Japan Society for the Promotion of Science, postdoctoral fellowship for research abroad, T.T.) supported this research. The authors thank Drs. George Meigs and James Holton for assistance with data collection on Beamline 8.3.1 at the Lawrence Berkeley National Laboratory Advanced Light Source.
Footnotes
Supporting Information Available: Table S1, statistics for data collection and refinement; Table S2, plausible ligands for Co(II) and Co–Co distances (Å) at and around ferritin oxido-reductase sites; Table S3, plausible Co(II)–Co(II) pairs and their environment in wild-type Co(II) cocrystal; Figure S1, Co(II) binding at sites of unknown function distributed around the 24-subunit protein nanocage (1), at the four-fold opening (2), and at the two-fold axes related dimer interfaces and on the inner surface of the protein (3) cage; and Figure S2, B-factor values for the residues around oxidoreductase sites. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- (1).Liu X, Theil EC. Acc. Chem. Res. 2005;38:167. doi: 10.1021/ar0302336. [DOI] [PubMed] [Google Scholar]
- (2).Le Brun NE, Crow A, Murphy ME, Mauk AG, Moore GR. Biochim. Biophys. Acta. 2010;1800:732–744. doi: 10.1016/j.bbagen.2010.04.002. [DOI] [PubMed] [Google Scholar]
- (3).Arosio P, Ingrassia R, Cavadini P. Biochim. Biophys. Acta. 2009;1790:589. doi: 10.1016/j.bbagen.2008.09.004. [DOI] [PubMed] [Google Scholar]
- (4).Douglas T, Dickson DP, Betteridge S, Charnock J, Garner CD, Mann S. Science. 1995;269:54. doi: 10.1126/science.269.5220.54. [DOI] [PubMed] [Google Scholar]
- (5).Abe S, Niemeyer J, Abe M, Takezawa Y, Ueno T, Hikage T, Erker G, Watanabe Y. J. Am. Chem. Soc. 2008;130:10512. doi: 10.1021/ja802463a. [DOI] [PubMed] [Google Scholar]
- (6).Ueno T, Abe M, Hirata K, Abe S, Suzuki M, Shimizu N, Yamamoto M, Takata M, Watanabe Y. J. Am. Chem. Soc. 2009;131:5094. doi: 10.1021/ja806688s. [DOI] [PubMed] [Google Scholar]
- (7).Zhang L, Laug L, Munchgesang W, Pippel E, Gosele U, Brandsch M, Knez M. Nano Lett. 2010;10:219. doi: 10.1021/nl903313r. [DOI] [PubMed] [Google Scholar]
- (8).Yamashita I, Iwahori K, Kumagai S. Biochim. Biophys. Acta. 2010;1800:846–857. doi: 10.1016/j.bbagen.2010.03.005. [DOI] [PubMed] [Google Scholar]
- (9).Banyard SH, Stammers DK, Harrison PM. Nature. 1978;271:282. doi: 10.1038/271282a0. [DOI] [PubMed] [Google Scholar]
- (10).Lawson DM, Artymiuk PJ, Yewdall SJ, Smith JM, Livingstone JC, Treffry A, Luzzago A, Levi S, Arosio P, Cesareni G, Thomas CD, Shaw WD, Harrison PM. Nature. 1991;349:541. doi: 10.1038/349541a0. [DOI] [PubMed] [Google Scholar]
- (11).Stillman TJ, Connolly PP, Latimer CL, Morland AF, Quail MA, Andrews SC, Treffry A, Guest JR, Artymiuk PJ, Harrison PM. J. Biol. Chem. 2003;278:26275. doi: 10.1074/jbc.M207354200. [DOI] [PubMed] [Google Scholar]
- (12).d’Estaintot B. Langlois, Santambrogio P, Granier T, Gallois B, Chevalier JM, Precigoux G, Levi S, Arosio P. J. Mol. Biol. 2004;340:277. doi: 10.1016/j.jmb.2004.04.036. [DOI] [PubMed] [Google Scholar]
- (13).Toussaint L, Bertrand L, Hue L, Crichton RR, Declercq JP. J. Mol. Biol. 2007;365:440. doi: 10.1016/j.jmb.2006.10.010. [DOI] [PubMed] [Google Scholar]
- (14).Pereira AS, Small W, Krebs C, Tavares P, Edmondson DE, Theil EC, Huynh BH. Biochemistry. 1998;37:9871. doi: 10.1021/bi980847w. [DOI] [PubMed] [Google Scholar]
- (15).Moenne-Loccoz P, Krebs C, Herlihy K, Edmondson DE, Theil EC, Huynh BH, Loehr TM. Biochemistry. 1999;38:5290. doi: 10.1021/bi990095l. [DOI] [PubMed] [Google Scholar]
- (16).Hwang J, Krebs C, Huynh BH, Edmondson DE, Theil EC, Penner-Hahn JE. Science. 2000;287:122. doi: 10.1126/science.287.5450.122. [DOI] [PubMed] [Google Scholar]
- (17).Jameson GN, Jin W, Krebs C, Perreira AS, Tavares P, Liu X, Theil EC, Huynh BH. Biochemistry. 2002;41:13435. doi: 10.1021/bi026478s. [DOI] [PubMed] [Google Scholar]
- (18).Bou-Abdallah F, Papaefthymiou GC, Scheswohl DM, Stanga SD, Arosio P, Chasteen ND. Biochem. J. 2002;364:57. doi: 10.1042/bj3640057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Logan DT, Su XD, Aberg A, Regnstrom K, Hajdu J, Eklund H, Nordlund P. Structure. 1996;4:1053. doi: 10.1016/s0969-2126(96)00112-8. [DOI] [PubMed] [Google Scholar]
- (20).Schwartz JK, Liu XS, Tosha T, Theil EC, Solomon EI. J. Am. Chem. Soc. 2008;130:9441. doi: 10.1021/ja801251q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Tosha T, Hasan MR, Theil EC. Proc. Natl. Acad. Sci. U.S.A. 2008;105:18182. doi: 10.1073/pnas.0805083105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Turano P, Lalli D, Felli IC, Theil EC, Bertini I. Proc. Natl. Acad. Sci. U.S.A. 2010;107:545. doi: 10.1073/pnas.0908082106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Ueno T, Yokoi N, Abe S, Watanabe Y. J. Inorg. Biochem. 2007;101:1667. doi: 10.1016/j.jinorgbio.2007.06.025. [DOI] [PubMed] [Google Scholar]
- (24).Crow A, Lawson TL, Lewin A, Moore GR, Le Brun NE. J. Am. Chem. Soc. 2009;131:6808. doi: 10.1021/ja8093444. [DOI] [PubMed] [Google Scholar]
- (25).Liu X, Theil EC. Proc. Natl. Acad. Sci. U.S.A. 2004;101:8557. doi: 10.1073/pnas.0401146101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Trikha J, Theil EC, Allewell NM. J. Mol. Biol. 1995;248:949. doi: 10.1006/jmbi.1995.0274. [DOI] [PubMed] [Google Scholar]
- (27).Ha Y, Shi D, Small GW, Theil EC, Allewell NM. J. Biol. Inorg. Chem. 1999;4:243. doi: 10.1007/s007750050310. [DOI] [PubMed] [Google Scholar]
- (28).Holton J, Alber T. Proc. Natl. Acad. Sci. U.S.A. 2004;101:1537. doi: 10.1073/pnas.0306241101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Leslie AG. Acta Crystallogr. D: Biol. Crystallogr. 2006;62:48. doi: 10.1107/S0907444905039107. [DOI] [PubMed] [Google Scholar]
- (30).Evans P. Acta Crystallogr. D: Biol. Crystallogr. 2006;62:72. doi: 10.1107/S0907444905036693. [DOI] [PubMed] [Google Scholar]
- (31).Kissinger CR, Gehlhaar DK, Fogel DB. Acta Crystallogr. D: Biol. Crystallogr. 1999;55:484. doi: 10.1107/s0907444998012517. [DOI] [PubMed] [Google Scholar]
- (32).Murshudov GN, Vagin AA, Dodson EJ. Acta Crystallogr. D: Biol. Crystallogr. 1997;53:240. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- (33).Emsley P, Cowtan K. Acta Crystallogr. D: Biol. Crystallogr. 2004;60:2126. doi: 10.1107/S0907444904019158. [DOI] [PubMed] [Google Scholar]
- (34).Perrakis A, Harkiolaki M, Wilson KS, Lamzin VS. Acta Crystallogr. D: Biol. Crystallogr. 2001;57:1445. doi: 10.1107/s0907444901014007. [DOI] [PubMed] [Google Scholar]
- (35).Hooft RW, Vriend G, Sander C, Abola EE. Nature. 1996;381:272. doi: 10.1038/381272a0. [DOI] [PubMed] [Google Scholar]
- (36).Delano W. PyMOL. Delano Scientific; San Carlos, CA: 2002. [Google Scholar]
- (37).Ha Y, Theil EC, Allewell NM. Acta Crystallogr. D: Biol. Crystallogr. 1997;53:513. doi: 10.1107/S0907444997003983. [DOI] [PubMed] [Google Scholar]
- (38).Hempstead PD, Yewdall SJ, Fernie AR, Lawson DM, Artymiuk PJ, Rice DW, Ford GC, Harrison PM. J. Mol. Biol. 1997;268:424. doi: 10.1006/jmbi.1997.0970. [DOI] [PubMed] [Google Scholar]
- (39).Takagi H, Shi D, Ha Y, Allewell NM, Theil EC. J. Biol. Chem. 1998;273:18685. doi: 10.1074/jbc.273.30.18685. [DOI] [PubMed] [Google Scholar]
- (40).Gallois B, d’Estaintot B. Langlois, Michaux MA, Dautant A, Grainer T, Precigoux G, Soruco JA, Roland F, ChavasAlba O, Herbas A, Crichton RR. J. Biol. Inorg. Chem. 1997;2:360. [Google Scholar]
- (41).Granier T, Gallois B, d’Estaintot B. Langlois, Dautant A, Chevalier JM, Mellado JM, Beaumont C, Santambrogio P, Arosio P, Precigoux G. Acta Crystallogr. D: Biol. Crystallogr. 2001;57:1491. doi: 10.1107/s0907444901008897. [DOI] [PubMed] [Google Scholar]
- (42).Granier T, d’Estaintot B. Langlois, Gallois B, Chevalier JM, Precigoux G, Santambrogio P, Arosio P. J. Biol. Inorg. Chem. 2003;8:105. doi: 10.1007/s00775-002-0389-4. [DOI] [PubMed] [Google Scholar]
- (43).Hamburger AE, West AP, Jr., Hamburger ZA, Hamburger P, Bjorkman PJ. J. Mol. Biol. 2005;349:558. doi: 10.1016/j.jmb.2005.03.074. [DOI] [PubMed] [Google Scholar]
- (44).Fabelo O, Canadillas-Delgado L, Pasan J, Delgado FS, Lloret F, Cano J, Julve M, Ruiz-Perez C. Inorg. Chem. 2009;48:11342. doi: 10.1021/ic901843r. [DOI] [PubMed] [Google Scholar]
- (45).Rosenzweig AC, Nordlund P, Takahara PM, Frederick CA, Lippard S. J. Chem. Biol. 1995;2:409. [PubMed] [Google Scholar]
- (46).Shu L, Nesheim JC, Kauffmann K, Munck E, Lipscomb JD, Que L., Jr. Science. 1997;275:515. doi: 10.1126/science.275.5299.515. [DOI] [PubMed] [Google Scholar]
- (47).Cho KJ, Shin HJ, Lee JH, Kim KJ, Park SS, Lee Y, Lee C, Park SS, Kim KH. J. Mol. Biol. 2009;390:83. doi: 10.1016/j.jmb.2009.04.078. [DOI] [PubMed] [Google Scholar]
- (48).Wade VJ, Levi S, Arosio P, Treffry A, Harrison PM, Mann S. J. Mol. Biol. 1991;221:1443. doi: 10.1016/0022-2836(91)90944-2. [DOI] [PubMed] [Google Scholar]
- (49).Bou-Abdallah F, Biasiotto G, Arosio P, Chasteen ND. Biochemistry. 2004;43:4332. doi: 10.1021/bi0498813. [DOI] [PubMed] [Google Scholar]
- (50).St Pierre T, Tran KC, Webb J, Macey DJ, Heywood BR, Sparks NH, Wade VJ, Mann S, Pootrakul P. Biol. Met. 1991;4:162. doi: 10.1007/BF01141308. [DOI] [PubMed] [Google Scholar]
- (51).Bauminger ER, Harrison PM, Hechel D, Hodson NW, Nowik I, Treffry A, Yewdall SJ. Biochem. J. 1993;296:709. doi: 10.1042/bj2960709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Treffry A, Zhao Z, Quail MA, Guest JR, Harrison PM. Biochemistry. 1995;34:15204. doi: 10.1021/bi00046a028. [DOI] [PubMed] [Google Scholar]
- (53).Levi S, Corsi B, Bosisio M, Invernizzi R, Volz A, Sanford D, Arosio P, Drysdale J. J. Biol. Chem. 2001;276:24437. doi: 10.1074/jbc.C100141200. [DOI] [PubMed] [Google Scholar]
- (54).Zeth K, Offermann S, Essen LO, Oesterhelt D. Proc. Natl. Acad. Sci. U.S.A. 2004;101:13780. doi: 10.1073/pnas.0401821101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Bellapadrona G, Stefanini S, Zamparelli C, Theil EC, Chiancone E. J. Biol. Chem. 2009;284:19101. doi: 10.1074/jbc.M109.014670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Masuda T, Goto F, Yoshihara T, Mikami B. J. Biol. Chem. 2010;285:4049. doi: 10.1074/jbc.M109.059790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Broadwater JA, Ai J, Loehr TM, Sanders-Loehr J, Fox BG. Biochemistry. 1998;37:14664. doi: 10.1021/bi981839i. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.