

Abstract
The catalytic mechanism of the MgATP-dependent carboxylation of biotin in the biotin carboxylase domain of pyruvate carboxylase from R. etli (RePC) is common to the biotin-dependent carboxylases. The current site-directed mutagenesis study has clarified the catalytic functions of several residues proposed to be pivotal in MgATP-binding and cleavage (Glu218 and Lys245), HCO3− deprotonation (Glu305 and Arg301) and biotin enolization (Arg353). The E218A mutant was inactive for any reaction involving the BC domain and the E218Q mutant exhibited a 75-fold decrease in kcat for both pyruvate carboxylation and the full reverse reaction. The E305A mutant also showed a 75- and 80-fold decrease in kcat for both pyruvate carboxylation and the full reverse reaction, respectively. While Glu305 appears to be the active site base which deprotonates HCO3−, Lys245, Glu218 and Arg301 are proposed to contribute to catalysis through substrate binding interactions. The reactions of the biotin carboxylase and carboxyl transferase domains were uncoupled in the R353M-catalyzed reactions, indicating that Arg353 may not only facilitate the formation of the biotin enolate, but also assist in coordinating catalysis between the two spatially distinct active sites. The 2.5 and 4-fold increase in kcat for the full reverse reaction with the R353K and R353M mutants, respectively, suggests that mutation of Arg353 allows carboxybiotin increased access to the biotin carboxylase domain active site. The proposed chemical mechanism is initiated by the deprotonation of HCO3− by Glu305 and concurrent nucleophilic attack on the γ-phosphate of MgATP. The trianionic carboxyphosphate intermediate formed reversibly decomposes in the active site to CO2 and PO43−. PO43− then acts as the base to deprotonate the tethered biotin at the N1-position. Stabilized by interactions between the ureido oxygen and Arg353, the biotin-enolate reacts with CO2 to give carboxybiotin. The formation of a distinct salt bridge between Arg353 and Glu248 is proposed to aid in partially precluding carboxybiotin from reentering the biotin carboxylase active site, thus preventing its premature decarboxylation prior to the binding of a carboxyl acceptor in the carboxyl transferase domain.
The biotin-dependent carboxylases, including acetyl-CoA carboxylase, propionyl-CoA carboxylase, methylcrotonyl-CoA carboxylase and pyruvate carboxylase (PC1; E.C 6.4.1.1), are essential regulatory enzymes associated with various metabolic pathways (1). The MgATP-dependent carboxylation of pyruvate catalyzed by PC replenishes the oxaloacetate used in gluconeogenesis, amino acid and fatty-acid synthesis, highlighting the central importance of PC to intermediary metabolism (2). The overall reaction catalyzed by PC occurs in two steps at spatially distinct active sites (Scheme 1). Biotin is initially carboxylated in the biotin carboxylase (BC) domain by HCO3− (1). Carboxybiotin is subsequently transferred to the carboxyl transferase (CT) domain on a neighboring polypeptide chain (3), where pyruvate carboxylation occurs (2) (4). The biotin carboxyl carrier protein (BCCP) domain carries the biotin cofactor, which is covalently attached to Lys1119, between the BC and CT domains. While Mg2+ is required as an essential activator for the overall carboxylation reaction, the necessity of acetyl-CoA as an allosteric activator is highly dependent on the organism from which the enzyme originates (5). The BC domain active sites of the biotin-dependent carboxylases share significant sequence and structural homology (6), suggesting that the catalytic mechanism of biotin carboxylation may be common to all members of the family. The three individual functional domains of α4 pyruvate carboxylases, including PC from R. etli (RePC), are arranged on a single polypeptide chain, offering a unique opportunity to determine the effects of mutations incorporated in the BC domain on the overall forward and reverse reactions, as well as on the reactions of the individual BC and CT domains (3, 7).
Scheme 1.

The overall pyruvate carboxylation reaction catalyzed by RePC occurs in two distinct steps with the MgATP-dependent biotin carboxylation occurring in the BC domain (1) and the carboxyl transfer to pyruvate occurring in the CT domain (2).
The crystallographic structure of the RePC holoenzyme with ATP-γ-S bound in the active site of the BC domain revealed those residues likely to be involved in the binding and orientation of MgATP (3) and subsequent crystal structures of biotin carboxylase from E. coli (EcBC), complexed with free biotin and HCO3−, allowed for the identification of possible catalytic residues in the putative bicarbonate/biotin pocket (Figure 1) (8). In the accompanying manuscript, the 2.4 Å resolution description of the structure of the T882A RePC mutant, co-crystallized with MgADP, acetyl-CoA and an inhibitory analog of the carboxyphosphate intermediate, phosphonoacetate, discloses both the relative positioning and orientation of the covalently attached biotin cofactor and the probable location of carboxyphosphate in the BC domain active site (9)
Figure 1.

Stereo view of the biotin carboxylase active site of wild-type R. etli PC (yellow, pdb 2QF7) complexed with Mg2+ and ATP-γ-S and E. coli biotin carboxylase (blue, pdb 3G8C) containing HCO3−, MgADP and free biotin in the active site. Residues are numbered according to the R. etli sequence and all figures were generated using PyMol (14).
These structural descriptions of the BC domain active site in both RePC and EcBC have revealed the full complement of highly conserved residues proposed to have prominent catalytic functions in the mechanism of biotin carboxylation. The current work is a structure-driven, site-directed mutagenesis study of those selected residues in the BC domain of R. etli PC which are thought to be responsible for MgATP-cleavage (Glu218 and Lys245), HCO3− deprotonation (Glu305 and Arg301) and biotin enolization (Arg353). The wild-type and mutant forms of RePC were characterized with respect to their catalytic activities for the full forward and reverse reactions and the partial reactions of the individual domains. The kinetic data presented here permit the development of a more definitive description of the catalytic mechanism for the MgATP-dependent carboxylation of biotin. Additionally, in conjunction with the newly solved structure, described in the preceding report (9), of the T882A RePC mutant containing tethered biotin in the BC domain active site, this investigation revealed a possible mechanism by which the enzyme precludes carboxybiotin re-entry into the BC domain active site, thus aiding in preventing the decarboxylation of carboxybiotin prior to the coordination of a carboxyl acceptor in the CT domain. Structural data confirm the presence of a unique salt bridge, possibly formed in the presence of MgADP, at the entrance of the BC domain active site which may regulate the accessibility of carboxybiotin to the BC domain active site
Materials and Methods
Materials
IPTG, biotin, NADP, NADH, ampicillin and chloramphenicol were purchased from Research Products International Corp. (RPI). Ni2+-Profinity IMAC resin was obtained from Bio-Rad. The Pierce BCA Assay kit was purchased from Thermo Scientific and the EnzChek Phosphate Assay kit was purchased from Invitrogen. All other materials were obtained from Sigma-Aldrich and of the highest purity available.
Construction of RePC BC domain mutants
E305A, E305Q, E305D, E218Q, E218A, K245Q, R301Q, R301K, R353M and R353K mutants were generated by site-directed mutagenesis as described previously (7, 10) using oligonucleotides designed to incorporate the above substitutions (Table S1). Mutagenic reactions were performed on a 1.8 kb SacII-XhoI fragment encoding the BC domain of RePC gene, and the nucleotide sequences of the mutants were determined by automatic sequencing. The equivalent fragment of the wild-type RePC gene in the expression clone was then replaced with the mutagenic cassette. The E305A/K1119Q and R353M/K1119Q double mutants were generated using the K1119Q plasmid as a template and the primers in Table S1.
Overexpression and Purification of Protein
Preparation and purification of the wild-type and mutant forms of RePC were essentially performed as previously described (7). The wild-type and mutant RePC proteins were purified using Ni2+-affinity chromatography, concentrated to approximately 2–5 mg/mL and used without further purification or manipulation. Total protein concentrations were quantified with the Pierce BCA colorimetric assay (Thermo Scientific). The nearly complete (> 98%) biotinylation of wild-type and RePC mutant proteins that did not contain a mutation at Lys1119 was confirmed via SDS-PAGE analysis of an avidin binding gel-shift assay (7).
Sedimentation analysis of the quaternary structure of the E218Q RePC mutant
Sedimentation velocity analytical centrifugation was performed with a Beckman ProteomeLab XL-A ultracentrifuge using the absorbance optics system to visualize the protein at a wavelength of 280 nm. Two-sector cells were used, and data were acquired every 0.003 cm. Data were collected as 300 absorbance scans with a nominal time increment of 1 min at 30 °C and at a speed of 40K rpm. Enzyme samples were prepared at the concentration of 2 µM in 0.1 M Tris-HCl (pH 7.8), 20 mM NaHCO3, 5 mM MgCl2, 10 mM pyruvate, 0.1 mM acetyl CoA and 1 mM DTE. The computer-captured data were analyzed with SEDFIT (11). The partial specific volumes of the enzymes were calculated from the amino acid composition using SEDNTERP [www.bbri.org/RASMB)] (11). The density of the Tris-HCl buffer (1.005 g/mL) was assumed to be the density of the enzyme solution. The raw sedimentation and ultracentrifugation data are presented in the Supporting Information (Table S2 and Figure S1).
Enzymatic Assays
The coupled assay systems used to monitor the full forward and reverse reactions and the partial forward and reverse reactions of the individual domains are shown in Table 1. A mixed-buffer system (50 mM Bis-Tris, 25 mM Tricine and 25 mM glycine; pH 7.5) was determined to have no effect on the activities of RePC and was used for all the enzymatic assays. The methods and specific reaction conditions for the coupled assay systems used in the current study are similar to those previously described (7) and further detailed in the Supporting Information.
Table 1.
Enzymatic reactions and assaysa
| Reactionsb | Assay |
|---|---|
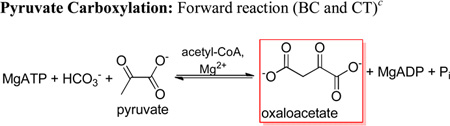 |
malate dehydrogenase |
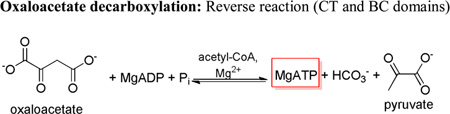 |
hexokinase/glucose-6-phosphate dehydrogenase |
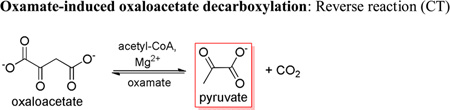 |
lactate dehydrogenase |
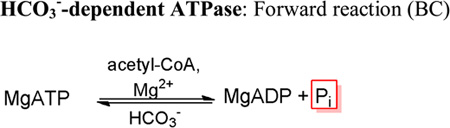 |
phosphorylase/phosphoglucomutase/glucose-6-phosphate dehydrogenase or PNP/MESGd |
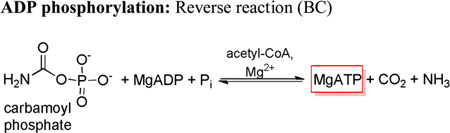 |
hexokinase/glucose-6-phosphate dehydrogenase |
Products highlighted are those assayed to determine specific activities and initial rates.
Detailed reaction and assay conditions are described in the Supplementary Information.
Domains involved in the reaction, BC = biotin carboxylase, CT = carboxyl transferase.
PNP/MESG = purine nucleoside phosphorylase and 2-amino-6-mercapto-7-methyl purine riboside.
Determination of the extent of coupling between the rates of Pi release and oxaloacetate formation
The initial rates of oxaloacetate formation and Pi release were determined at varying concentrations of pyruvate (0.09–5.25 mM) in order to determine the degree of coupling between the two reactions. For the determination of the rate of Pi release in these experiments, the EnzChek Phosphate (Invitrogen) assay system was used. The PNP-catalyzed phosphorylation of MESG to ribose-1-phosphate and 2-amino-6-mercapto-7-methyl purine with Pi was used as an effective coupled assay system to measure the initial rate of Pi release (12) during the RePC-catalyzed HCO3−-dependent ATPase reaction. Initial rates were determined by monitoring the corresponding increase in absorbance at 360 nm due to the formation of 2-amino-6-mercapto-7-methyl purine. Under the conditions used for the HCO3−-dependent ATPase reaction (50 mM Bis-Tris, 25 mM Tricine, 25 mM glycine pH 7.5, 25° C), the extinction coefficient for the purine product was determined to be 6,300 cm−1 M−1 and used for all subsequent calculations. To ensure that the initial rates of the two reactions were determined under nearly identical conditions, two 1 mL reactions containing 50 mM Bis-Tris, 25 mM Tricine, 25 mM glycine (pH 7.5), 15 mM HCO3−, 5.0 mM MgCl2, 2.5 mM MgATP and 0.25 mM acetyl-CoA were prepared at each concentration of pyruvate (0.09–5.25 mM). For the determination of oxaloacetate formation, 0.24 mM NADH and malate dehydrogenase (10 U) were added and the reduction of NADH to NAD+ was determined at 340 nm. 3 U of PNP and 0.2 mM of MESG were added to the reaction mixtures where the initial rates of Pi release were being determined and the background rate of MESG decomposition was monitored for approximately 2 min at 360 nm. Reactions were then initiated with the addition of RePC and the corrected rate was used to determine the initial rate of Pi release. Data for the wild-type catalyzed reactions and the ratios of the initial rates of oxaloacetate formation/initial rates of Pi release at varying pyruvate concentrations were fitted to eqn (1). Ratios of the initial rate of oxaloacetate formation to Pi release for the R353M-catalyzed reactions were fitted to eqn (4). For the E305A mutant-catalyzed reaction, data were fitted to eqn (1). Standard errors reported for all kinetic parameters were determined from the fits of these data to the corresponding equation.
Data Analysis
kcat (min−1) and kcat/Km (min−1 mM−1) values were determined by fitting velocity versus substrate concentration data to eqn (1) using least-squares nonlinear regression, where A is the substrate concentration. All least-square fits were performed using FORTRAN programs (13) and best-fit lines were plotted using SigmaPlot v. 12.0.
| (1) |
Substrate inhibition data were fitted to eqn (2), where A is the concentration of substrate and Ki is the inhibition constant for the substrate.
| (2) |
Inhibition data for the R353M/K1119Q catalyzed HCO3−-dependent ATPase reaction by free biotin were fitted to eqn (3) where A is the concentration of MgATP, I is the concentration of free biotin and Ki is the inhibition constant for biotin.
| (3) |
To determine the extent of coupling between MgATP-cleavage and oxaloacetate formation, the ratios of the initial rate of oxaloacetate formation to Pi release in the R353M catalyzed reactions at varying concentrations of pyruvate were fitted to a two/one function, eqn (4),
| (4) |
where A is the concentration of pyruvate, V is the limiting ratio of oxaloacetate formation to Pi release at saturating concentrations of pyruvate and b, c, and d are kinetic constants associated with the extent of coupling between oxaloacetate formation and Pi release at varying pyruvate concentrations.
Results
Reactions involving the carboxyl transferase domain
The kcat (min−1) and kcat/Km MgATP (min−1 mM−1) for pyruvate carboxylation were determined by measuring the initial rates of oxaloacetate formation for the RePC mutant-catalyzed reactions (Table 2). Most mutations of the proposed catalytic residues in the BC domain had pronounced effects on both the rate and catalytic efficiency for pyruvate carboxylation. Of those enzymes which retained pyruvate carboxylation activity, the R301K and R353K mutant RePCs exhibited the greatest decrease in kcat as compared to the wild-type rate (180 and 160-fold reduction, respectively). Unlike the conservative E305D mutation, which had a relatively small effect on kcat only, the mutation of Glu305 to Ala resulted in a 75-fold decrease in kcat and almost 30-fold decrease in kcat/Km MgATP. The sizable increases in the Km for MgATP resulted in an 1850-fold decrease in kcat/Km for the R301K mutant and a 170-fold decrease for the K245Q mutant. Interestingly, the E218Q, R353M and R301Q mutant forms of RePC showed noticeable MgATP substrate inhibition of the full forward reaction. The apparent Km (0.03 mM) and Ki (1.5 mM) values for MgATP determined for each of the three mutant-catalyzed reactions were virtually identical within experimental error. While the E218Q mutant retained a small amount of pyruvate carboxylating ability, the E218A mutant RePC enzyme lacked catalytic activity for both pyruvate carboxylation and the full reverse reaction. Although decreased activities and catalytic efficiencies were observed in the E305A and R353M-catalyzed reactions, pyruvate carboxylation activity was not observed with the E305A/K1119Q and R353M/K1119Q double mutants.
Table 2.
Activities of the biotin carboxylase domain mutants for the pyruvate carboxylation reactiona.
| kcat (min−1)b |
% wild- type rate |
Km MgATP (mM) |
kcat/Km MgATP (min−1 mM−1) |
% wild-type kcat/Km MgATP |
|
|---|---|---|---|---|---|
| wild-type | 700 ± 80 | (100) | 0.27 ± 0.04 | 2590 ± 15 | (100) |
| E218A | NAc | --- | --- | --- | --- |
| E218Q | 8.0 ± 0.3 | 1.1 | 0.03 ± 0.005d | 270 ± 10 | 10 |
| K245Q | 71 ± 4 | 10 | 4.82 ± 0.07 | 15 ± 1 | 0.6 |
| E305A | 9.2 ± 0.2 | 1.3 | 0.09 ± 0.01 | 96 ± 12 | 4 |
| E305Q | 87 ± 1 | 12 | 1.32 ± 0.06 | 64 ± 1 | 2.5 |
| E305D | 187 ± 1 | 27 | 0.32 ± 0.08 | 580 ± 20 | 22 |
| R301Q | 18 ± 1 | 2.5 | 0.05 ± 0.02e | 340 ± 10 | 13 |
| R301K | 3.8 ± 0.4 | 0.5 | 2.8 ± 0.2 | 1.4 ± 0.9 | 0.05 |
| R353K | 4.3 ± 0.1 | 0.6 | 0.12 ± 0.01 | 35 ± 4 | 1.4 |
| R353M | 28 ± 1 | 4 | 0.034 ± 0.009f | 760 ± 10 | 30 |
| E305A/K1119Q | NA | --- | --- | --- | --- |
| R353M/K1119Q | NA | --- | --- | --- | --- |
Reaction conditions: 50 mM Bis-Tris, 25 mM Tricine, 25 mM glycine (pH 7.5), 25° C, 25 mM HCO3−, 12 mM pyruvate, 7.0 mM MgCl2, MgATP (0.075–3.0 mM), 0.25 mM acetyl-CoA.
Data fitted to eqn (1) unless otherwise indicated.
NA = No activity detected.
Substrate inhibition with respect to MgATP data fitted to eqn (2), Ki = 1.1 ± 0.2 mM.
Substrate inhibition, Ki = 1.2 ± 0.3 mM.
Substrate inhibition, Ki = 1.8 ± 0.2 mM.
The effects of the BC domain mutations on the specific activities for the full reverse reaction (Table 3) were similar to those on the full forward reaction. A 75-fold decrease in kcat for both the full forward and full reverse reactions were observed with the E305A RePC mutant whereas the effects of the R301K (7-fold decrease), E218Q (10-fold decrease) and E305Q (22-fold decrease) mutations on the activities of the full reverse reaction were not as pronounced as those observed for the full forward reaction. Unexpectedly, the specific activities determined for reactions catalyzed by the R353K and R353M mutants, both of which showed marked decreases in the kcat for pyruvate carboxylation, were 2.5 and 4-fold greater than the rate of the wild-type catalyzed reaction, respectively. In order to determine if the observed rate acceleration was due to increased activation by the allosteric effector, acetyl-CoA, specific activities for the full reverse reaction were determined in the absence of acetyl-CoA (Table 4). Unlike the wild-type catalyzed reaction, which showed only 18% activity in the absence of acetyl-CoA, the presence or absence of acetyl-CoA appeared to have little effect on the specific activities for the full reverse reaction catalyzed by the R353M and R353K RePC mutants.
Table 3.
Specific activities of the biotin carboxylase domain mutants for the full reverse reactiona.
| kcat (min−1)b |
% wild- type rate |
|
|---|---|---|
| wild-type | 0.835 ± 0.005 | (100) |
| E218A | NAc | ---- |
| E218Q | 0.077 ± 0.001 | 9 |
| K245Q | 0.23 ± 0.03 | 27 |
| E305A | 0.011 ± 0.002 | 1 |
| E305Q | 0.037 ± 0.001 | 4 |
| E305D | 0.42 ± 0.02 | 50 |
| R301Q | 0.45 ± 0.02 | 50 |
| R301K | 0.12 ± 0.01 | 14 |
| R353K | 2.10 ± 0.05 | 250 |
| R353M | 3.28 ± 0.02 | 390 |
| E305A/K1119Q | NA | --- |
| R353M/K1119Q | NA | --- |
Reaction conditions: 50 mM Bis-Tris, 25 mM Tricine, 25 mM glycine (pH 7.5), 25° C, 7.5 mM MgCl2, 3.0 mM ADP, 2.5 mM phosphate, 0.95 mM oxaloacetate, 0.25 mM acetyl-CoA.
Number of determinations = 3, error reported is standard deviations of 3 trials
NA = No activity detected.
Table 4.
Specific activities of the biotin carboxylase domain mutants for the full reverse reaction in the absence of acetyl-CoAa.
| kcat (min−1)b |
% of rate with acetyl-CoA |
|
|---|---|---|
| wild-type | 0.152 ± 0.003 | 18 |
| R301Q | 0.376 ± 0.004 | 85 |
| R353K | 2.75 ± 0.02 | 130 |
| R353M | 3.09 ± 0.08 | 94 |
Reaction conditions: 50 mM Bis-Tris, 25 mM Tricine, 25 mM glycine (pH 7.5), 25° C, 7.5 mM MgCl2, 3.0 mM ADP, 2.5 mM phosphate, 0.95 mM oxaloacetate.
Number of determinations = 3, error reported is standard deviations of 3 trials
NA = No activity detected.
While the oxamate-induced decarboxylation of oxaloacetate is proposed to occur solely within the confines of the CT domain (15), previous studies suggest that mutations in one domain can have an effect on reactions occurring in the neighboring domain (7). Mutations in the BC domain appeared to affect the rates of the oxamate-induced decarboxylation of oxaloacetate to varying degrees (Table 5). Most notable was the 96% and 94% loss in activity, as compared to wild-type, with the E218A and E218Q mutants, respectively. Sedimentation analysis of the quaternary structure of the E218Q RePC mutant indicates that this mutant exists predominately as a tetramer in solution (Table S2 and Figure S1).
Table 5.
Specific activities of the biotin carboxylase domain mutants for the oxamate induced decarboxylation of oxaloacetatea.
| kcat (min−1)b |
% wild- type rate |
|
|---|---|---|
| wild-type | 9.03 ± 0.04 | (100) |
| E218A | 0.42 ± 0.01 | 4 |
| E218Q | 0.59 ± 0.01 | 6 |
| K245Q | 2.01 ± 0.01 | 22 |
| E305A | 3.28 ± 0.07 | 36 |
| E305Q | 1.27 ± 0.02 | 14 |
| E305D | 7.45 ± 0.05 | 82 |
| R301Q | 6.0 ± 0.1 | 66 |
| R301K | 5.67 ± 0.07 | 63 |
| R353K | 4.23 ± 0.09 | 47 |
| R353M | 5.8 ± 0.2 | 64 |
| E305A/K1119Q | NAc | --- |
| R353M/K1119Q | NA | --- |
Reaction conditions: 50 mM Bis-Tris, 25 mM Tricine, 25 mM glycine (pH 7.5), 25° C, 1 mM oxamate, 0.95 mM oxaloacetate, 0.25 mM acetyl-CoA.
Number of determinations = 3, error reported is standard deviations of 3 trials
NA = No activity detected.
Reactions involving only the BC domain
RePC catalyzes the HCO3−-dependent ATPase reaction in the absence of carboxyl acceptors, releasing MgADP and Pi as products (7, 16). The initial rates of Pi release were measured using the phosphorylase a/phosphoglucomutase/glucose-6-phosphate dehydrogenase coupled assay system to determine the kcat and kcat/Km MgATP values for the HCO3−-dependent ATPase reaction. While this assay system had proven to be reliable for the determination of the kcat with saturating concentrations of MgATP, MgATP Km values determined using this coupled assay system were at least 2-orders of magnitude higher than those reported for the R. etli wild-type catalyzed reaction (17) and those subsequently determined with the PNP/MESG coupled assay system (5). The discrepancy is most likely due to the ability of phosphorylase a to bind nucleotides, thereby reducing the overall concentrations of MgATP in solution (18). Consequently, while the kcat/Km MgATP values in Tables 6 and 7 cannot be considered absolute values, the apparent Km values for MgATP determined with this assay system were used to establish the general effects of these mutations relative to the wild-type enzyme assayed under the same conditions.
Table 6.
Activities of the biotin carboxylase domain mutants for the HCO3−-dependent ATPase reactiona.
| kcat (min−1)b |
% wild- type rate |
Km MgATP (mM) |
kcat/Km MgATP (min−1 mM−1) |
% wild-type kcat/Km MgATP |
|
|---|---|---|---|---|---|
| wild-type | 0.68 ± 0.03 | (100) | 0.89 ± 0.09 | 0.76 ± 0.05 | (100) |
| E218A | NAc | --- | --- | --- | --- |
| E218Q | 0.034 ± 0.002 | 5 | 2.8 ± 0.4 | 0.012 ± 0.009 | 1.6 |
| K245Q | 1.0 ± 0.4 | 150 | 0.3 ± 0.2d | 3.06 ± 0.05 | 400 |
| E305A | 0.32 ± 0.01 | 48 | 0.82 ± 0.03 | 0.39 ± 0.02 | 50 |
| E305Q | 0.19 ± 0.02 | 30 | 0.98 ± 0.01 | 0.19 ± 0.08 | 25 |
| E305D | 0.55 ± 0.04 | 82 | 0.7 ± 0.1 | 0.79 ± 0.09 | 100 |
| R301Q | 0.13 ± 0.04 | 20 | 0.12 ± 0.02 | 1.1 ± 0.1 | 145 |
| R301K | 0.14 ± 0.03 | 20 | 0.053 ± 0.006e | 2.5 ± 0.5 | 330 |
| R353K | 0.33 ± 0.07 | 50 | 0.10 ± 0.01 | 3.2 ± 0.4 | 420 |
| R353M | 0.58 ± 0.01 | 85 | 0.30 ± 0.02 | 1.9 ± 0.1 | 250 |
| E305A/K1119Q | 0.336 ± 0.007 | 50 | 0.068 ± 0.007 | 4.9 ± 0.5 | 640 |
| R353M/K1119Q | 1.16 ± 0.06 | 170 | 0.22 ± 0.03 | 5.5 ± 0.6 | 720 |
Reaction conditions: 50 mM Bis-Tris, 25 mM Tricine, 25 mM glycine (pH 7.5), 25° C, 15 mM HCO3−, 7.5 mM MgCl2, MgATP (0.09–3.0 mM), 0.25 mM acetyl-CoA.
Data fitted to eqn 1 unless otherwise indicated.
NA = No activity detected.
Substrate inhibition with respect to MgATP data fitted to eqn (2), Ki = 0.4 ± 0.2 mM.
Substrate inhibition, Ki = 4.1 ± 0.4 mM.
Table 7.
Activities of the biotin carboxylase domain mutants for the HCO3−-dependent ATPase reaction in the presence of free biotina.
| kcat (min−1)b |
% wild- type rate |
Km MgATP (mM) |
kcat/Km MgATP (min−1 mM−1) |
% wild-type kcat/Km MgATP |
|
|---|---|---|---|---|---|
| wild-type | 0.93 ± 0.04 | (100) | 1.2 ± 0.1 | 0.75 ± 0.05 | (100) |
| E218A | NAc | --- | --- | --- | --- |
| E218Q | 0.015 ± 0.005 | 2 | 1.0 ± 0.2 | 0.015 ± 0.006 | 2 |
| K245Q | 0.032 ± 0.005 | 3 | 1.81 ± 0.02 | 0.018 ± 0.04 | 2.5 |
| E305A | 0.41 ± 0.02 | 44 | 0.05 ± 0.01 | 8.2 ± 0.9 | 1090 |
| E305Q | 0.28 ± 0.03 | 30 | 1.7 ± 0.5 | 0.16 ± 0.01 | 21 |
| E305D | 0.88 ± 0.07 | 95 | 0.5 ± 0.1 | 1.7 ± 0.2 | 225 |
| R301Q | 0.11 ± 0.04 | 12 | 0.25 ± 0.03 | 0.47 ± 0.02 | 63 |
| R301K | 0.23 ± 0.01 | 25 | 0.98 ± 0.07 | 0.234 ± 0.02 | 31 |
| R353K | 0.62 ± 0.06 | 67 | 0.18 ± 0.03d | 3.4 ± 0.4 | 450 |
| R353M | 0.481 ± 0.006 | 52 | 0.050 ± 0.004 | 9.5 ± 0.6 | 1260 |
| E305A/K1119Q | 0.41 ± 0.02 | 45 | 0.21 ± 0.04 | 1.9 ± 0.3 | 250 |
| R353M/K1119Qe | 1.29 ± 0.02 | 140 | 0.257 ± 0.009 | 5.01 ± 0.08 | 670 |
Reaction conditions: 50 mM Bis-Tris, 25 mM Tricine, 25 mM glycine (pH 7.5), 25° C, 15 mM HCO3−, 10 mM biotin, 7.5 mM MgCl2, MgATP (0.09–3.0 mM), 0.25 mM acetyl-CoA.
Data fitted to eqn 1 unless otherwise indicated.
NA = No activity detected.
Substrate inhibition with respect to MgATP data fitted to eqn 2, Ki = 2.8 ± 0.7 mM.
Values determined from global fit of biotin inhibition data to eqn 4.
While the E218A mutant had proven to be inactive for any reaction that involved the BC domain, incorporation of a Gln mutation at Glu218 resulted in a 20-fold decrease in kcat and a 60-fold decrease in the kcat/Km MgATP relative to the wild-type catalyzed reaction. The K245Q mutant-catalyzed reaction exhibited a slight increase in kcat as compared to wild-type, but the strong MgATP substrate inhibition (Ki = 0.4 ± 0.2 mM) made the determination of an accurate kcat/Km value problematic. Nonetheless, it is clear that mutations of those residues which reside in the MgATP-binding pocket of the BC domain resulted in substantial decreases in both kcat and the apparent kcat/Km MgATP for the HCO3−-dependent ATPase reaction.
The series of Glu305 mutants were moderately active for the HCO3−-dependent ATPase reaction (1.2–3.5 fold decrease in kcat) and there was no significant effect on the apparent Km for MgATP. Interestingly, the E305A/K1119Q mutant exhibited a 40-fold increase in the kcat for the ATPase reaction as compared to the K1119Q-apoenzyme (5). When compared to the wild-type catalyzed reaction, the E305A/K1119Q mutant was significantly slower, but an accompanying 13-fold decrease in the apparent Km for MgATP resulted in a 6-fold increase in the kcat/Km. Mutations of Arg301 and Arg353 mostly resulted in decreases in kcat for MgATP-hydrolysis and significant increases in kcat/Km as compared to the wild-type enzyme. Surprisingly, the R353M/K1119Q double mutant showed a nearly 2-fold increase in kcat.
While the addition of 10 mM free biotin had little effect on the kcat for reactions catalyzed by the RePC BC domain mutants (Table 7), its presence did alleviate the observed MgATP substrate inhibition for the K245Q and R301K mutants. Most notable were the effects of free biotin on those mutant enzyme forms whose mutations were contained within the putative biotin binding pocket of the BC domain (Figure 1). With the exception of the E305A, R353M and R353M/K1119Q mutants, free biotin increased the apparent Km MgATP for the BC domain mutants, resulting in enzyme catalyzed reactions with only marginal decreases in kcat/Km compared to the wild-type enzyme. Significant decreases in the apparent Km MgATP were observed for the E305A and R353M-catalyzed reactions in the presence of free biotin, giving rise to an order of magnitude increase in catalytic efficiency compared to the wild-type catalyzed reaction with 10 mM biotin. Free biotin also induced MgATP substrate inhibition (Ki = 2.8 ± 0.7 mM) in the R353K mutant catalyzed reaction. In contrast to the activating effect free biotin has on the K1119Q-catalyzed HCO3−-dependent ATPase reaction (5, 7), biotin was determined to be a linear uncompetitive inhibitor of the R353M/K1119Q-catalyzed reaction with respect to MgATP (Ki = 4.5 ± 0.1 mM, Figure 2). Further, only a minimal effect on the apparent Km MgATP was observed with the E305A/K1119Q mutant in the presence of 10 mM biotin (Table 7).
Figure 2.

Reciprocal plots demonstrating the linear, uncompetitive inhibition with respect to MgATP by free biotin for the bicarbonate-dependent ATPase reaction catalyzed by the R353M/K1119Q RePC double mutant. Solid lines indicate the least-square fits to eqn (3) at varying concentrations of MgATP (0.09–3.0 mM) and fixed concentrations of biotin (0 mM, black; 1 mM, red; 3 mM, blue; 5 mM, green; 7 mM, pink; 10 mM, cyan).
The kcat and kcat/Km for the phosphorylation of MgADP using carbamoyl phosphate as the phosphoryl donor was also determined for all of the BC domain mutants (Table 8). The E218A mutant was not active for the phosphorylation reaction, while the apparent carbamoyl phosphate substrate inhibition detected in the K245Q mutant-catalyzed reaction was so strong that, again, reasonable values for kcat and kcat/Km for carbamoyl phosphate could not be determined. The E218Q mutant exhibited the greatest measurable decrease in kcat and an overall 14-fold decrease in the kcat/Km. Similar patterns of reactivity were observed for the E305Q, R301Q and R301K mutants which had significant decreases in kcat (4–16% of wild-type activities) and kcat/Km (7–10% that of wild-type RePC). Conversely, the E305D, E305A/K1119Q and R353K RePC mutants exhibited slightly increased kcat values for MgADP phosphorylation with carbamoyl phosphate as the phosphoryl donor. Remarkably, the R353M-catalyzed phosphorylation of MgADP was 10-fold faster than the wild-type-catalyzed phosphorylation reaction, partially accounting for the 30-fold increase in catalytic efficiency. While the kcat of the E305A single mutant catalyzed reaction was only 30% of that of the wild-type rates, an accompanying decrease in the Km for carbamoyl phosphate gave rise to a kcat/Km value similar to that observed for MgADP phosphorylation with the E305A/K1119Q double mutant. The R353M/K1119Q mutant displayed an unexpectedly low activity for the phosphorylation reaction when compared to the kcat determined for the R353M single RePC mutant (1% of kcat). As with the E305A/K1119Q RePC mutant, a considerable decrease in the Km for carbamoyl phosphate with the R353M/K1119Q mutant catalyzed reaction resulted in kcat/Km values that were almost 90% of that determined for wild-type RePC.
Table 8.
Activities of the biotin carboxylase domain mutants for the ADP phosphorylation reaction using carbamoyl phosphate as the phosphoryl donora.
| kcat (min−1)b |
% wild- type rate |
Km CPc (mM) |
kcat/Km CP (min−1 mM−1) |
% wild-type kcat/Km CP |
|
|---|---|---|---|---|---|
| wild-type | 2.4 ± 0.1 | (100) | 2.6 ± 0.5 | 0.93 ± 0.02 | (100) |
| E218A | NAd | --- | --- | --- | --- |
| E218Q | 0.10 ± 0.01 | 4 | 1.5 ± 0.5 | 0.067 ± 0.006 | 7 |
| K245Qe | --- | --- | --- | --- | --- |
| E305A | 0.79 ± 0.02 | 33 | 0.31 ± 0.02 | 2.6 ± 0.1 | 280 |
| E305Q | 0.255 ± 0.006 | 10 | 3.2 ± 0.2 | 0.079 ± 0.002 | 8 |
| E305D | 5.4 ± 0.1 | 225 | 0.35 ± 0.05 | 16 ± 3 | 1720 |
| R301Q | 0.39 ± 0.06 | 16 | 0.89 ± 0.02 | 0.45 ± 0.02 | 50 |
| R301K | 0.108 ± 0.004 | 4.5 | 1.2 ± 0.3 | 0.092 ± 0.003 | 10 |
| R353K | 3.4 ± 0.1 | 140 | 5.3 ± 0.5 | 0.79 ± 0.05 | 85 |
| R353M | 24.9 ± 0.8 | 1000 | 0.9 ± 0.2 | 28 ± 4 | 3010 |
| E305A/K1119Q | 2.6 ± 0.2 | 100 | 1.4 ± 0.1 | 1.8 ± 0.1 | 190 |
| R353M/K1119Q | 0.30 ± 0.01 | 12 | 0.37 ± 0.09 | 0.8 ± 0.1 | 86 |
Reaction conditions: 50 mM Bis-Tris, 25 mM Tricine, 25 mM glycine (pH 7.5), 25° C, 7.5 mM MgCl2, 3.5 mM MgADP, carbamoyl phosphate (1.0–20 mM), 0.25 mM acetyl-CoA.
Data fitted to eqn 1 unless otherwise indicated.
CP = carbamoyl phosphate
NA = No activity detected.
Due to the extreme substrate inhibition with respect to carbamoyl phosphate reasonable values could not be determined.
Acetyl phosphate was discovered to be a suitable substitute for carbamoyl phosphate in the MgADP phosphorylation reaction catalyzed by wild-type RePC (5). The kcat and kcat/Km for MgADP phosphorylation using acetyl phosphate as the phosphoryl donor were determined for the RePC mutants where the incorporated mutations were located near the putative HCO3−/biotin binding site (Table 9). With the exception of the E305A/K1119Q RePC double mutant, the kcat and Km values with acetyl phosphate were considerably less than those determined for carbamoyl phosphate. Even so, the kcat/Km determined for the R353K and R353M-catalyzed reactions with acetyl phosphate were comparable to those determined with carbamoyl phosphate, while the E305A RePC mutant exhibited a nearly 7-fold decrease in kcat/Km with acetyl phosphate as the phosphoryl donor. In contrast to the previously established activating effects of free biotin on the rate of the wild-type-catalyzed phosphorylation of MgADP (5, 7, 19), 10 mM biotin had little effect on the BC domain mutant-catalyzed phosphorylation, either with acetyl phosphate (Table 9) or carbamoyl phosphate (data not shown) as the phosphoryl donor.
Table 9.
Activities of the biotin carboxylase domain mutants for the ADP phosphorylation reaction using acetyl phosphate as the phosphoryl donor.
| (+) 10 mM biotin | ||||||||
|---|---|---|---|---|---|---|---|---|
| kcat (min−1)b |
Km AcPc (mM) |
kcat/Km AcP (min−1 mM−1) |
% wild-type kcat/Km |
kcat (min−1) |
Km (mM) |
kcat/Km AcP (min−1 mM−1) |
% wild-type kcat/Km AcP |
|
| wild-type | 0.58 ± 0.01 | 0.34 ± 0.02 | 1.70 ± 0.005 | (100) | 0.29 ± 0.002 | 0.14 ± 0.04 | 2.1 ± 0.2 | (100) |
| E305A | 0.189 ± 0.003 | 0.61 ± 0.05 | 0.309 ± 0.005 | 18 | 0.105 ± 0.001 | 0.98 ± 0.02 | 0.107 ± 0.006 | 5 |
| R353K | 0.37 ± 0.03 | 0.49 ± 0.04 | 0.75 ± 0.02 | 44 | 0.21 ± 0.01 | 0.43 ± 0.03 | 0.48 ± 0.02 | 23 |
| R353M | 14.2 ± 0.3 | 0.48 ± 0.03 | 30 ± 2 | 1760 | 13.7 ± 0.1 | 0.48 ± 0.02 | 28.4 ± 0.2 | 1350 |
| E305A/K1119Q | 2.66 ± 0.06 | 0.8 ± 0.1 | 3.40 ± 0.05 | 200 | 2.90 ± 0.09 | 0.9 ± 0.1 | 3.3 ± 0.5 | 160 |
| R353M/K1119Q | 0.55 ± 0.01 | 1.5 ± 0.2 | 0.37 ± 0.04 | 22 | 0.602 ± 0.003 | 0.68 ± 0.05 | 0.85 ± 0.05 | 40 |
Reaction conditions: 50 mM Bis-Tris, 25 mM Tricine, 25 mM glycine (pH 7.5), 25° C, 7.5 mM MgCl2, 3.5 mM MgADP, acetyl phosphate (0.1–20 mM), 0.25 mM acetyl-CoA.
Data fitted to eqn 1 unless otherwise indicated.
AcP = acetyl phosphate
Coupling between the reactions of the biotin carboxylase and carboxyl transferase domains
The extent of coupling between MgATP-cleavage in the BC domain and pyruvate carboxylation in the CT domain was established by monitoring the initial rates of Pi release and oxaloacetate formation in the presence of varying concentrations of pyruvate under identical reaction conditions for the wild-type, R353M and E305A-catalyzed reactions (Figure 3). Plots of the ratio of the initial rates of oxaloacetate formation/initial rates of Pi release vs. pyruvate concentration were hyperbolic for both the wild-type and E305A-catalyzed reactions. The ratio of vOAA formation/vPi release approaches the limiting value of 1.08 ± 0.02 for the wild-type catalyzed reaction in the presence of saturating acetyl-CoA and free Mg2+, indicating a near stoichiometric coupling between MgATP-cleavage and oxaloacetate formation at saturating concentrations of pyruvate. While a similar limiting value of 0.983 ± 0.01 was determined for the E305A-catalyzed reactions, the Kd determined for pyruvate (0.031 ± 0.003 mM) was 8-fold lower than that determined for the wild-type catalyzed reactions (0.25 ± 0.003 mM). Unlike in the wild-type and E305A catalyzed reactions, the limiting ratio of the vOAA formation/vPi release in the R353M-catalyzed reactions was determined to be only 0.83 ± 0.02 (eqn (4)).
Figure 3.

Ratios of the initial rates of oxaloacetate formation to Pi release at varying concentrations of pyruvate (0.09–5.25 mM) for the wild-type (○), E305A (▲) and R353M (●) mutant RePC-catalyzed reactions. Both the E305A and wild-type data were fitted to eqn (1) while data for the R353M catalyzed reaction were fitted to eqn (4).
Discussion
The MgATP-dependent carboxylation of biotin in the BC domain by HCO3− is proposed to occur through the formation of carboxyphosphate and subsequent carboxylation of the tethered-biotin enolate (4). The observed transfer of 18O from HCO3− to phosphate in the forward reaction (20) and ability of PC to catalyze the phosphorylation of MgADP with carbamoyl phosphate supports the existence of a carboxyphosphate intermediate. Based on this two-step mechanism of biotin carboxylation and their general locations in the BC domain active site, the catalytic residues selected for the current site-directed mutagenesis study can be loosely categorized as those responsible for the formation of carboxyphosphate and those responsible for the formation of carboxybiotin. These residues were determined to be pivotal in MgATP-binding and cleavage (Glu218 and Lys245), HCO3− deprotonation (Glu305 and Arg301) and biotin enolization (Arg353) and the kinetic data presented here are consistent with the proposed mechanism (Scheme 2).
Scheme 2.

Proposed catalytic mechanism of the HCO3−-dependent MgATP cleavage and biotin carboxylation.
Lys245 is essential to the binding and orientation of MgATP
Part of the conserved catalytic triad (Glu218-Lys245-Glu305) which forms a hydrogen-bonded bridging network between the MgATP and HCO3−-binding pockets in the BC domain active site (Figure 4A), Lys245 is positioned 3.6 Å from the phosphoryl-oxygen of the γ-phosphate of ATP-γ-S in the RePC holoenzyme structure (3). While previous kinetic (21, 22) and isotope effect studies (23) suggested that the Lys245-Cys237 pair was responsible for biotin enolization and carboxylation, the thiolate of Cys237 is buried away from the active site in RePC and is not involved in catalysis (3). The extremely strong MgATP substrate inhibition observed for the HCO3−-dependent ATPase reaction catalyzed by the RePC K245Q mutant, which results in an apparent kcat/Km greater than wild-type RePC, is indicative of increased amounts of non-productive substrate binding, possibly due to the presence of several MgATP molecules in a single BC domain active site. Structural evidence has shown that the EcBC domain active site can not only accommodate a single MgATP in multiple binding modes (24) but can also accommodate the binding of more than one nucleotide in a single active site (25).
Figure 4.

(A) Hydrogen-bonding interactions of the highly conserved catalytic triad, Glu218-Lys245-Glu305, HCO3− and MgATP-γ-S determined from the superposition of EcBC (blue, 8) and RePC holoenzyme (yellow, 3). The position of Glu218 in both the RePC and EcBC structures suggest that its main catalytic function is to position Lys245 and lower the pKa of Glu305. (B) The actual (light green) and modeled (dark green) positions of tethered biotin in the BC domain active site of the T882A RePC mutant crystal structure (pdb 3WT6) relative to the positioning of HCO3− in the BC domain active site from EcBC (blue). Hydrogen bonding interactions between key residues are indicated by dashed lines.
If the observed substrate inhibition in the K245Q RePC mutant is due to the coordination of multiple nucleotides in a single BC domain active site, then the presence of 10 mM free biotin is most likely prohibiting the binding of a second nucleotide, thus eliminating the observed MgATP substrate inhibition without affecting the kcat of the K245Q-catalyzed MgATP hydrolysis. The 27-fold decrease in the kcat for MgATP-hydrolysis, even in the presence of free biotin, and significant decreases in kcat and kcat/KmMgATP observed in the K245Q-catalyzed carboxylation of pyruvate can be attributed to the abolishment of interactions between Lys245 and the γ-phosphate of MgATP. While a significant amount of the substrate binding energy comes from interactions between the BC active site and the adenosine-ring of MgATP (26), interactions between Lys245 and the γ-phosphate of MgATP would still have substantial contributions to catalysis by ensuring the optimal alignment of the γ-phosphate for the nucleophilic attack by HCO3−.
Glu305 is the proposed active site base which deprotonates HCO3−
In the EcBC active site (8), Glu305 is ideally positioned near the oxygen-atom of HCO3− (2.9 Å) to act as the active site base needed to deprotonate HCO3− and initiate the nucleophilic attack of bicarbonate on the γ-phosphate of MgATP (Figure 4A). A conservative Glu305 to Asp mutation resulted in slight decreases in the rates of pyruvate carboxylation, MgATP-cleavage and the full reverse reaction. While differing only in their magnitude, a similar decrease in kcat for the MgATP-cleavage and phosphorylation of MgADP with carbamoyl phosphate was observed with the E305Q mutant. In contrast, drastic decreases in the kcat and Km MgATP for pyruvate carboxylation were observed with the E305A mutant (75-fold decrease in kcat, 30-fold decrease in the Km for MgATP). Further, the similar rates of MgATP-hydrolysis observed with the E305A single mutant, which contains tethered biotin, and the E305A/K1119Q-apoenzyme suggest that the rate of the HCO3−-dependent cleavage has now become partially rate-limiting compared to biotin carboxylation.
In wild-type RePC, incomplete coupling between the HCO3−-dependent ATPase reaction and carboxyl transfer was observed at pyruvate concentrations less than approximately 1.5 mM, indicating that concentrations lower than or near the determined Km for pyruvate (1.1 mM) (7) facilitates MgATP-cleavage and the subsequent release of the BC domain products (MgADP and Pi) to a greater extent than the overall reaction (Figure 3). The structure of the T882A RePC mutant in the preceding manuscript (9) lends structural evidence to a possible mechanism where the translocation of the BCCP-domain, located on top of the B-subdomain lid of the BC domain, to the CT domain in the presence of a carboxyl acceptor would stimulate the moderately rate-limiting release of BC domain products (27). From a mechanistic perspective, these results suggest that while concentrations of pyruvate below the Km will adequately stimulate the movement of carboxybiotin from the BC domain to the CT domain (27), they are not sufficient to saturate the enzyme in the full forward reaction. Thus, the disassociation of pyruvate prior to carboxylation occurs more often (28).
It is expected, then, that the extent of coupling between the reactions of the BC and CT domains at sub-saturating concentrations of pyruvate in the E305A-catalyzed reaction would increase since the initial deprotonation of HCO3− is now partially rate-limiting. In fact, while the rates of both Pi release and oxaloacetate formation are significantly lower in the RePC E305A mutant, a tighter coupling between the reactions of the individual domains is observed at considerably lower concentrations of pyruvate (Ka pyruvate = 0.03 mM) than in the wild-type enzyme (Figure 3). Invoking a similar mechanistic rationale, the E305A RePC enzyme requires a lower concentration of pyruvate to fully saturate the CT domain active site for the full forward reaction, most likely due to the deprotonation of HCO3− in the BC domain becoming more rate-limiting relative to BCCP-carboxybiotin translocation and carboxyl transfer in the CT domain.
Analysis of the pH profiles of the full forward reaction with wild-type RePC and MgATP as the variable substrate and of the full reverse reactions with MgADP as the variable substrate (5) revealed a pKa on the acid side in the V/K profiles that could possibly be assigned to Glu305. In the pyruvate carboxylation reaction, Glu305 must be ionized to allow for the initial deprotonation of HCO3− while in the reverse reaction, a proton transfer from Glu305 to the carboxyphosphate intermediate concurrent with the phosphoryl transfer to MgADP would form a more reasonable leaving group than carbonate. In contrast to the phosphorylation of MgADP by carboxyphosphate in the full reverse reaction, the partial reverse reaction of the BC domain using either carbamoyl phosphate or acetyl phosphate as the phosphoryl donor is pH independent since both the carbamate and acetate formed during the phosphoryl transfer are good leaving groups (5). This also suggests that the E305A RePC mutant would catalyze the phosphorylation of MgADP with the non-natural substrates at least as, if not more, efficiently than the wild-type enzyme. In fact, the kcat/Km for the E305A and the E305A/K1119Q catalyzed phosphorylation of MgADP with carbamoyl phosphate is nearly 10 and 3-times that of wild-type RePC, respectively. Interestingly, while acetyl phosphate is also predicted to be a better substrate in the E305A single and double mutant-catalyzed reactions, the observed kcat/Km was only 18% of wild-type values.
Glu218 interacts with residues in the catalytic triad to facilitate catalysis
Located nearly 6 Å away from the γ-phosphate of ATP-γ-S in the RePC holoenzyme structure (3) and ~ 7 Å from HCO3− in the EcBC structure (8), it is difficult to envision how Glu218 can have such a marked effect on catalysis (Figure 4A). While the possibilty exists that a single point mutation in the active site may affect the overall structure of the enzyme, sedimentation analysis of the quaternary structure revealed that the E218Q RePC mutant exists predominately as a tetramer in solution. Analogous to the effects of the K245Q mutation, the strong MgATP substrate inhibition observed in the RePC E218Q catalyzed MgATP-cleavage reaction is partially alleviated by the presence of 10 mM free biotin. Rather than directly assisting in substrate binding, the position of Glu218 in the highly conserved catalytic triad (Fig 4A) indicates that hydrogen-bonding interactions with Glu305 and Lys245 are pivotal to catalysis. The close proximity (2.4–2.8 Å) of Glu218 to Glu305 is suggestive of the presence of a low-barrier hydrogen bond formed between these two residues which would aid in the deprotonation of HCO3− and the partial stabilization of the carboxyphosphate intermediate. Interactions between the carboxylate of Glu218 and the ε-amino group of Lys245 (4 Å) would also raise the pKa of Lys245, thus favoring its protonation at physiological pH. Retaining the positive charge on Lys245 in the forward reaction would not only allow for the proper alignment of MgATP (24), but would also assist the migration of PO43− from the MgATP-binding region to the biotin/bicarbonate binding pocket (29).
Arg301 bridges the HCO3− and biotin binding pockets
Previous kinetic studies of the corresponding R301A mutant in EcBC (30) suggested that Arg301 has no direct role in MgATP binding. The Km MgATP values determined for the Arg301 RePC mutants were significantly lower than wild-type values for the HCO3−-dependent ATPase reaction and the accompanying decrease in kcat suggests that either HCO3−-deprotonation or carboxyphosphate formation has now become more rate-limiting in the catalytic reaction. The positioning of Arg301 at the interface of the proposed biotin/bicarbonate/MgATP-binding regions of the BC domain in RePC (3) and EcBC (8) suggests that Arg301 not only aids in the alignment and deprotonation of HCO3− in the initial steps of the catalytic mechanism, but also may facilitate the formation of the biotin enolate by bringing PO43− in close proximity to the N1-position of biotin. In conjunction with the protonated ε-amino group of Lys245, the guanidinium groups of Arg301 and Arg353 are positioned such that a small positively charged pocket is formed near the N1-position of both free biotin in EcBC (8) and tethered biotin (modeled in the BC domain active site) in the T882A RePC mutant structure (dark green, Figure 4B) (9). Disruption of the positively charged binding pocket in EcBC through the introduction of an equivalent K245Q mutation resulted in an enzyme form that not only catalyzed MgATP-hydrolysis at a slower rate than the wild-type enzyme, but was also incapable of producing carboxybiotin (22).
A superposition of the EcBC structure complexed with HCO3− and the BC domain of the T882A RePC mutant disclosed the distance between HCO3− and the N1-positon of the tethered biotin in the proposed, catalytically active conformation to be 3.4 Å (Figure 4B) (9). Interestingly, the location of the electron density of HCO3− in the recent EcBC structure (8) overlaps with density for HPO32− observed in the previously determined apo-structure of EcBC (29), lending structural support to the proposed role of PO43− as the base responsible for deprotonating biotin at the N1-positon. While interactions with Arg301 are expected to decrease the pKa of PO43− (~12.7 for HPO32−), hydrogen-bonding interactions between Arg353 and the ureido-oxygen of the tethered biotin would also result in a decrease of the pKa of the proton at the N1-position of biotin (pKa 17.4) (31).
Arg353 aids in biotin enolization and carboxybiotin synthesis
In the crystal structure of the T882A RePC mutant with tethered biotin modeled in the BC domain active site (Figure 4B, dark green), Arg353 is ideally positioned to facilitate biotin enolization through the stabilization of the developing negative charge at the ureido-oxygen located 3.8 Å away. The decrease in kcat and accompanying decrease in Km MgATP and MgATP substrate inhibition observed for the R353M and R353K -catalyzed carboxylation of pyruvate and HCO3−-dependent ATPase reaction suggests that there is a partial uncoupling between MgATP-hydrolysis and carboxybiotin formation in the BC domain but MgATP-cleavage is mostly unaffected (32). The inability of the R353M RePC mutant to effectively catalyze the carboxylation of biotin also results in the uncompetitive inhibition of the R353M/K1119Q catalyzed HCO3−-dependent ATPase reaction by free biotin (Figure 2). Free biotin is proposed to bind to the BC domain after MgATP and HCO3− (33), inducing conformational changes in the active site which aid in preventing the release of Pi and MgADP prior to carboxylation of either free or tethered biotin (8). Since the R353M/K1119Q mutant cannot effectively carboxylate free biotin, the BC domain active site remains in a tighter, closed conformation until the random motions of the B-subdomain allows for the release of MgADP and Pi (34). While hydrogen-bonding interactions with Arg353 are proposed to be the dominant stabilizing effect for the anionic-biotin enolate intermediate, several other residues are in close proximity to the ureido oxygen of the modeled biotin in the T882A RePC structure which may aid in the formation of the enolate (9).
The extent of coupling between the overall reaction of the BC domain and carboxyl transfer in the CT domain for the R353M RePC mutant catalyzed reactions was determined at varying concentrations of pyruvate (Figure 3). The limiting ratio of the vOAA formation/vPi release in the R353M-catalyzed reactions was determined to be approximately 0.8, signifying incomplete coupling between the reactions of the two active sites. Accordingly, at saturating concentrations of pyruvate there is a 5-fold difference between the partitioning of the HCO3−-dependent ATPase reaction through the productive catalytic cycle to form oxaloacetate in the CT domain and through the abortive pathways of the BC domain, resulting in the nonproductive cleavage of MgATP. Undeniably, the rates of pyruvate carboxylation and MgATP-cleavage and inhibition of the R353M-mutant by free biotin indicate that biotin enolization is partially rate-limiting relative to both the movement of the BCCP-carboxybiotin from the BC domain to the CT domain and the actual carboxyl transfer in the CT domain in this mutant. If the only role of Arg353 were to promote carboxybiotin formation in the BC domain, then the ratios of oxaloacetate formation and Pi release would be expected to eventually reach a limiting value of one, similar to that observed for the E305A RePC mutant. Thus, the uncoupling of the BC and CT domain reactions even with saturating concentrations of pyruvate and 4-fold increase in the rate of the full reverse reaction catalyzed by the R353M RePC mutant strongly suggest that carboxybiotin may have less restricted access to the BC domain active site in this mutant. Further, the 10-fold increase in the rate of MgADP phosphorylation with carbamoyl phosphate in the R353M-catalyzed reaction is analogous to that previously observed in the T882A RePC mutant catalyzed reactions, where the increased amount of biotin occupancy in the BC domain was proposed to result in accelerated rates of MgADP phosphorylation compared to the wild-type rates (7).
The T882A RePC mutant structure shows a catalytically inactive, but relevant, conformation of the tethered-biotin moiety where the N1-postion of biotin is removed from the active site (light green, Figure 4B). The formation of a salt bridge between Arg353 and Glu248, possibly promoted by the presence of MgADP in the active site, creates a ridge at the opening of the BC domain which may partially preclude carboxybiotin reentry into the active site. Previous kinetic studies of sheep and chicken liver PC (27, 35) have described the existence of two different enzyme-carboxybiotin complexes that are relatively stable in the absence of carboxyl acceptors. Free Mg2+ decreases the rate of the decay of the enzyme-carboxybiotin in both sheep and chicken liver PC (t1/2 at 0° C in the absence of any substrates is ~ 340 min) and is proposed to facilitate the movement of carboxybiotin to a more stable position near the BC domain active site. Based on the increased relative stability of the second enzyme-carboxybiotin complex and small accompanying conformational change upon its formation, it was proposed that carboxybiotin is expelled from the interior of the BC domain active site and positioned in the vicinity of the active site where the premature decarboxylation of carboxybiotin is less likely to occur (27, 35). In fact, the T882A RePC mutant structure may be representative of these two possible positions of the tethered carboxybiotin, one directly threaded into the BC active site (dark green, Figure 4B) and the second adjacent to the active site (light green, Figure 4B). Preventing carboxybiotin from returning to the BC domain prior to the binding of a carboxyl acceptor in the CT domain is one way in which the enzyme reduces the amount of nonproductive MgATP-cleavage. In this way, Arg353 is not only important to the catalytic mechanism of the BC domain, but is also key in coordinating the activity between the two spatially distinct active sites.
An attractive alternative to the mechanism by which RePC couples the formation of carboxybiotin in the BC domain to carboxyl transfer in the CT domain would require the initial coordination of pyruvate to the CT domain as the driving force for the MgATP-cleavage/biotin carboxylation reaction in the BC domain. While this mechanism would theoretically abate some non-productive cleavage of MgATP in that carboxylation of biotin would only occur when the second substrate is poised in the active site, the particularly rapid decay of the isolated (sheep kidney) enzyme-carboxybiotin complex in the presence of competitive inhibitors of pyruvate indicates that biotin carboxylation occurs prior to the coordination of any CT domain active site substrates or substrate analogs (27). While the non-productive cleavage of MgATP to form carboxybiotin when pyruvate concentrations are less than saturating is an inefficient utilization of ATP, several bacteria species, including Zymomonas mobilus (36) and Lactococcus cremoris (37), have been shown to consume ATP in a futile cycle that is not directly related to cell growth or viability when concentrations of various metabolites are limited. Given that Rhizobium etli contains phosphoenolpyruvate carboxylase and an α4 PC, both of which are allosterically regulated by acetyl-CoA (38), the consequences of the ineffective use of ATP at low concentrations of pyruvate by RePC is most likely mediated by a several complex regulatory mechanisms. For example, the formation of the distinct salt bridge between Arg353 and Glu248, hindering the return of carboxybiotin to the BC domain active site, does increase the coupling between the reactions of the two domains (Figure 3) despite the inherent level of enzyme inefficiency at low concentrations of pyruvate.
In summary, this detailed structure-driven site directed mutagenesis study offers convincing evidence supporting the proposed catalytic functions of several highly conserved residues in the BC domain of RePC. The proposed catalytic mechanism for biotin carboxylation is initiated through the deprotonation of HCO3− by the active site general base, Glu305, during a concerted nucleophilic attack by bicarbonate on the γ-phosphate of MgATP, resulting in the formation of a trianionic carboxyphosphate intermediate (Scheme 2). Hydrogen-bonding interactions within the Lys245-Glu218-Glu305 catalytic triad increase the pKa of Glu305, facilitating the deprotonation of HCO3−, and stabilize Lys245 in an active conformation, allowing for the formation of key electrostatic interactions between Lys245 and the γ-phosphate of MgATP. Once formed, the carboxyphosphate intermediate will reversibly decompose in the active site to CO2 and PO43−. Guided by interactions with Lys245 and Arg301, PO43− migrates towards the tethered biotin in the BC domain active site and deprotonates biotin at the N1-position. The developing negative charge at the ureido oxygen is partially stabilized by Arg353, thus promoting the formation of the nucleophilic biotin enolate. Addition of CO2 at N1-position of the biotin enolate produces carboxybiotin which is rapidly excluded from the active site of the BC domain via the formation of a discrete salt bridge between Arg353 and Glu248. The newly formed carboxybiotin remains in the vicinity of the BC domain active site, protected from spontaneous decarboxylation, until the binding of pyruvate, or similar carboxyl acceptor, in the CT domain active site prompts the translocation of the BCCP-carboxybiotin to the CT domain.
Supplementary Material
Footnotes
This work was supported by the National Institute of Health grant GM070455 to WWC, MStM, JCW and PVA and an NIH award F32DK083898 from the National Institute of Diabetes and Digestive And Kidney Diseases to TNZ.
All residues are numbered according to their position in R. etli pyruvate carboxylase, including those mutations in E. coli biotin carboxylase. Abbreviations: PC, pyruvate carboxylase; BC, biotin carboxylase; CT, carboxyl transferase; BCCP, biotin carboxyl carrier protein; ATP, adenosine triphosphate; ADP, adenosine diphosphate; acetyl-CoA, acetyl-Coenzyme A; RePC, Rhizobium etli PC; hPC, human PC; SaPC, Staphylococcus aureus PC; BirA, biotin protein ligase; IPTG, isopropyl-beta-D-thiogalactopyranoside; NADH, nicotinamide adenine dinucleotide; acetyl-CoA, acetyl-coenzyme A; NADP+, nicotinamide adenine dinucleotide phosphate; Pi, inorganic phosphate; PNP, purine nucleoside phosphorylase; MESG, 2-amino-6-mercapto-7-methyl purine riboside
Supporting Information Available. Detailed methods for the kinetic assays, primer sequences used to incorporate the biotin carboxylase domain mutations (Table S1) and sedimentation analysis of the quaternary structure of the E218Q RePC mutant (Table S2 and Figure S1) are available. This material is available free of charge at http://pubs.acs.org.
REFERENCES
- 1.(a) Jitrapakdee S, Wallace JC. The biotin enzyme family: conserved structural motifs and domain rearrangements. Curr. Prot. Pept. Sci. 2003;4:217–229. doi: 10.2174/1389203033487199. [DOI] [PubMed] [Google Scholar]; (b) Tong L. Acetyl-coenzyme A carboxylase: crucial metabolic enzyme and attractive target for drug discovery. Cell. Mol. Life. Sci. 2005;62:1784–1803. doi: 10.1007/s00018-005-5121-4. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Moss J, Lane DM. Biotin-dependent enzymes. Adv. Enzymol. Molec. Biolo. 1971;35:321–442. doi: 10.1002/9780470122808.ch7. [DOI] [PubMed] [Google Scholar]; (d) Jitrapakdee S, Wallace JC. Structure, function and regulation of pyruvate carboxylase. Biochem. J. 1999;340:1–16. doi: 10.1042/bj3400001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.(a) Garcia-Cazorla A, Rabier D, Touati G, Chadefaux-Vekemans B, Marsac C, de Lonlay P, Saudubray J-M. Pyruvate carboxylase deficiency: metabolic characteristics and new neurological aspects. Ann. of Neurology. 2006;59:121–127. doi: 10.1002/ana.20709. [DOI] [PubMed] [Google Scholar]; (b) Jitrapakdee S, Vidal-Puig A, Wallace JC. Anaplerotic roles of pyruvate carboxylase in mammalian tissues. Cell. Mol. Life. Sci. 2006;63:843–854. doi: 10.1007/s00018-005-5410-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Marin-Valencia I, Roe CR, Pascual JM. Pyruvate carboxylase deficiency: Mechanisms, mimics and anaplerosis. Mol. Gene. and Metab. 2010;101:9–17. doi: 10.1016/j.ymgme.2010.05.004. [DOI] [PubMed] [Google Scholar]; (d) Jitrapakdee S, Wutthisathapornchai A, Wallace JC, MacDonald MJ. Regulation of insulin secretion: role of mitochondrial signaling. Diabetologia. 2010;53:1019–1032. doi: 10.1007/s00125-010-1685-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.St. Maurice M, Reinhardt L, Surinya KH, Attwood PV, Wallace JC, Cleland WW, Rayment I. Domain architecture of pyruvate carboxylase, a biotin-dependent multifunctional enzyme. Science. 2007;317:1076–1079. doi: 10.1126/science.1144504. [DOI] [PubMed] [Google Scholar]
- 4.(a) Knowles JR. The mechanism of biotin-dependent enzymes. Ann. Rev. Biochem. 1989;58:195–221. doi: 10.1146/annurev.bi.58.070189.001211. [DOI] [PubMed] [Google Scholar]; (b) Attwood PV, Wallace JC. Chemical and catalytic mechanism of carboxyl transfer reaction in biotin-dependent enzymes. Acc. Chem. Res. 2002;35:113–120. doi: 10.1021/ar000049+. [DOI] [PubMed] [Google Scholar]; (c) Jitrapakdee S, St. Maurice M, Rayment I, Cleland WW, Wallace JC, Attwood PV. Structure, mechanism and regulation of pyruvate carboxylase. Biochem. J. 2008;413:369–387. doi: 10.1042/BJ20080709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zeczycki TN, Menefee AL, Jitrapakdee S, Wallace JC, Attwood PV, St. Maurice M, Cleland WW. Activation and inhibition of reactions occurring in the biotin carboxylase domain of pyruvate carboxylase from Rhizobium etli. Biochemistry. 2011 doi: 10.1021/bi2012788. (accompanying manuscript in this series) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.(a) Lim F, Morris CP, Occhiodoro F, Wallace JC. Sequence and domain structure of yeast pyruvate carboxylase. J. Biol. Chem. 1988;263:11493–11497. [PubMed] [Google Scholar]; (b) Wallace JC, Jitrapakdee S, Chapman-Smith A. Pyruvate carboxylase. Inter. J. Biochem. Cell. Biol. 1998;30:1–5. doi: 10.1016/s1357-2725(97)00147-7. [DOI] [PubMed] [Google Scholar]
- 7.Zeczycki TN, St. Maurice M, Jitrapakdee S, Wallace JC, Attwood PV, Cleland WW. Insight into the carboxyl transferase domain mechanism of pyruvate carboxylase from Rhizobium etli. Biochemistry. 2009;48:4305–4313. doi: 10.1021/bi9003759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chou C-YY, Yu LPC, Tong L. Structure of biotin carboxylase in complex with substrates and implications for its catalytic mechanism. J. Biol. Chem. 2009;285:11690–11697. doi: 10.1074/jbc.M805783200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lietzan AD, Menefee AL, Zeczycki TN, Kumar S, Attwood PV, Wallace JC, Cleland WW, St. Maurice M. Interaction between the biotin carboxyl carrier domain and the biotin carboxylase domain in the asymmetrical pyruvate carboxylase tetramer from Rhizobium etli. Biochemistry. 2011 doi: 10.1021/bi201277j. (accompanying manuscript in this series) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Duangpan S, Jitrapakdee S, Adina-Zada A, Byrne L, Zeczycki TN, St. Maurice M, Cleland WW, Wallace JC, Attwood PV. Probing the catalytic roles of Arg548 and Gln552 in the carboxyl-transferase domain of the Rhizobium etli pyruvate carboxylase by site-directed mutagenesis. Biochemistry. 2010;49:3296–3304. doi: 10.1021/bi901894t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and lamm equation modeling. Biophys J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Webb MR. A continuous spectrophotometric assay for inorganic phosphate and for measuring phosphate release kinetics in biological systems. Proc. Nat. Acad. Sci. 1992;89:4884–4887. doi: 10.1073/pnas.89.11.4884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cleland WW. Statistical analysis of enzyme kinetic data. Methods in Enzymology. 1979;63:103–138. doi: 10.1016/0076-6879(79)63008-2. [DOI] [PubMed] [Google Scholar]
- 14.Schrödinger LLC. The PyMOL molecular graphics system. V. 1.3r1. 2010 [Google Scholar]
- 15.Attwood PV, Tipton PA, Wallace JC. Carbon-13 and deuterium isotope effects on oxaloacetate decarboxylation by pyruvate carboxylase. Biochemistry. 1986;25:8197–8205. doi: 10.1021/bi00373a012. [DOI] [PubMed] [Google Scholar]
- 16.Attwood PV, Graneri BDLA. Bicarbonate-dependent ATP-cleavage catalyzed by pyruvate carboxylase in the absence of pyruvate. Biochem. J. 1992;287:1011–1017. doi: 10.1042/bj2871011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Adina-Zada A, Hazra R, Sereeruk C, Jitrapakdee S, Zeczycki TN, St. Maurice M, Cleland WW, Wallace JC, Attwood PV. Probing the allosteric activation of pyruvate carboxylase using 2′,3′-O-(2,4,6-trinitrophenyl) adenosine 5′-triphosphate as a fluorescent mimic of the allosteric activator acetyl CoA. Arch. Biochem. Biophys. 2011;509:117–126. doi: 10.1016/j.abb.2011.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sprang S, Fletterick R, Stern M, Yang D, Madsen N, Sturtevant J. Analysis of an allosteric binding site: the nucleoside inhibitor site of phosphorylase a. Biochemistry. 1982;21:2036–2048. doi: 10.1021/bi00538a010. [DOI] [PubMed] [Google Scholar]
- 19.Attwood PV, Graneri BDLA. Pyruvate carboxylase catalysis of phosphate transfer between carbamoyl phosphate and ADP. Biochem. J. 1991;273:443–448. doi: 10.1042/bj2730443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kaziro Y, Hass LF, Boyer PD, Ochoa S. Mechanism of the propionyl carboxylase reaction. II. Isotopic exchange and tracer experiments. J. Biol. Chem. 1962;237:1460–1468. [PubMed] [Google Scholar]
- 21.(a) Kazuta Y, Tokunaga E, Aramaki E, Kondo H. Identification of lysine-238 of Escherichia coli biotin carboxylase as an ATP-binding residue. FEBS Lett. 1998;427:377–380. doi: 10.1016/s0014-5793(98)00472-4. [DOI] [PubMed] [Google Scholar]; (b) Tipton PA, Cleland WW. Catalytic mechanism of biotin carboxylase: steady-state kinetics investigations. Biochemistry. 1988;27:4317–4325. doi: 10.1021/bi00412a019. [DOI] [PubMed] [Google Scholar]
- 22.Levert KL, Lloyd RB, Waldrop GL. Do Cys230 and Lys 238 of biotin carboxylase play a key role in the activation of biotin. Biochemistry. 2000;39:4122–4128. doi: 10.1021/bi992662a. [DOI] [PubMed] [Google Scholar]
- 23.Tipton PA, Cleland WW. Carbon-13 and deuterium isotope effects on the catalytic reactions of biotin carboxylase. Biochemistry. 1988;27:4325–4331. doi: 10.1021/bi00412a020. [DOI] [PubMed] [Google Scholar]
- 24.Molchalkin I, Miller RJ, Evodokimov A, Lightle S, Chan S, Stover CK, Waldrop GL. Structural evidence for substrate-induced synergism and half-sites reactivity in biotin carboxylase. Prot. Science. 2008;17:1706–1718. doi: 10.1110/ps.035584.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chou C-YY, Tong L. Structural and biochemical studies on the regulation of biotin carboxylase by substrate inhibition and dimerization. J. Biol. Chem. 2011 doi: 10.1074/jbc.M111.220517. (published as ASAP on web) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.(a) Geeves MA, Branson JP, Attwood PV. Kinetics of nucleotide binding to pyruvate carboxylase. Biochemistry. 1995;34:11846–11854. doi: 10.1021/bi00037a024. [DOI] [PubMed] [Google Scholar]; (b) Attwood PV, Coates JH, Wallace JC. Interaction of formycin A-5’-triphosphate with pyruvate carboxylase. Febs Lett. 1984;175:45–50. doi: 10.1016/0014-5793(84)80566-9. [DOI] [PubMed] [Google Scholar]; (c) Zeczycki TN, St. Maurice M, Attwood PV. Inhibitors of pyruvate carboxylase. Open Enzy. Inhib. J. 2010;3:8–26. doi: 10.2174/1874940201003010008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Goodall GJ, Baldwin GS, Wallace JC, Keech DB. Factors that influence the translocation of the N-carboxybiotin moiety between the two sub-sites of pyruvate carboxylase. Biochem. J. 1981;199:603–609. doi: 10.1042/bj1990603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cheung Y-F, Walsh C. Studies on the intramolecular and intermolecular kinetic isotope effects on pyruvate carboxylase catalysis. Biochemistry. 1976;17:3749–3754. doi: 10.1021/bi00662a017. [DOI] [PubMed] [Google Scholar]
- 29.(a) Waldrop GL, Rayment I, Holden HM. Three-dimensional structure of the biotin carboxylase subunit of acetyl-CoA carboxylase. Biochemistry. 1994;33:10249–10256. doi: 10.1021/bi00200a004. [DOI] [PubMed] [Google Scholar]; (b) Thoden JB, Blanchard CZ, Holden HM, Waldrop GL. Movement of the biotin carboxylase B-domain as a result of ATP binding. J. Biol. Chem. 2000;21:16183–16190. doi: 10.1074/jbc.275.21.16183. [DOI] [PubMed] [Google Scholar]
- 30.Blanchard CZ, Lee YM, Frantom PA, Waldrop GL. Mutations at four active site residues of biotin carboxylase abolish substrate-induced synergism by biotin. Biochemistry. 1999;38:3393–3400. doi: 10.1021/bi982660a. [DOI] [PubMed] [Google Scholar]
- 31.Fry DC, Fox TL, Lane MD, Mildvan AS. Exchange characteristics of the amide protons of d-biotin and derivatives: implications for the mechanism of biotin enzymes and the role of sulfur in biotin. J. Am. Chem. Soc. 1985;107:7659–7665. [Google Scholar]
- 32.Sloane V, Waldrop GL. Kinetic characterization of mutations found in propionic acidemia and methylcrotonylglycinuira. Evidence for cooperativity in biotin carboxylase. J. Biol. Chem. 2004;279:15772–15778. doi: 10.1074/jbc.M311982200. [DOI] [PubMed] [Google Scholar]
- 33.Blanchard CZ, Amspacher D, Strongin R, Waldrop GL. Inhibition of biotin carboxylase by a reaction intermediate analog: implications for the kinetics mechanism. Biochem. Biophys. Res. Comm. 1999;266:466–471. doi: 10.1006/bbrc.1999.1844. [DOI] [PubMed] [Google Scholar]
- 34.Novak BR, Moldovan D, Waldrop GL, de Queiroz MS. Umbrella sampling simulations of biotin carboxylase: is a structure with an open ATP grasp domain stable in solution? J. Phys. Chem. B. 2009;113:10097–10103. doi: 10.1021/jp810650q. [DOI] [PubMed] [Google Scholar]
- 35.(a) Attwood PV, Wallace JC, Keech DB. The carboxybiotin complex of pyruvate carboxylase. A kinetic analysis of the effects of Mg2+ ions on its stability and on its reaction with pyruvate. Biochem. J. 1984;219:243–251. doi: 10.1042/bj2190243. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Attwood PV, Wallace JC. The carboxybiotin complex of chicken liver pyruvate carboxylase. A kinetic analysis of the effects of acetyl-CoA, Mg2+ ions and temperature on its stability and on its reaction with 2-oxobutyrate. Biochem. J. 1986;235:359–364. doi: 10.1042/bj2350359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benthin S, Schulze U, Nielsen J, Villadsen J. Growth energetics of of Lactococcus cremoris FD1 during energy-, carbon- and nitrogen-limitation in steady-state and transient cultures. Chem. Enger. Sci. 1994;49:589–609. [Google Scholar]
- 37.Haughney HA, Dziewulski DM, Nauman EB. On material balance and maintenance coefficents in CSTRs with various extents of biomass recycle. J. Biotech. 1988;7:113–130. [Google Scholar]
- 38.(a) Encarnacion S, Dunn M, Willms K, Mora J. Fermentative and aerobic metabolism in Rhizobium etli. J. Bacteriol. 1995;177:3058–3066. doi: 10.1128/jb.177.11.3058-3066.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Dunn MF, Araicza G, Cevallos MA, Mora J. Regulation of pyruvate carboxylase in Rhizobium etli. FEMS Micro. Lett. 2006;157:301–306. doi: 10.1111/j.1574-6968.1997.tb12789.x. [DOI] [PubMed] [Google Scholar]; (c) Sauer U, Eikmanns BJ. The PEP-pyruvate-oxaloacetate node as the switch point for carbon flux distribution in bacteria. FEMS Micro. Rev. 2005;29:765–794. doi: 10.1016/j.femsre.2004.11.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.