

Abstract
NAD(P)H:quinone oxidoreductase 1 (NQO1), is a cytosolic flavoenzyme that catalyzes the two-electron reduction of quinones into hydroquinones. A polymorphism (NQO1*2) alters enzymatic activity of NQO1 resulting in diminished NQO1 activity. Malignancies with NQO1*2 may be resistant to radiation and chemotherapy with resulting poorer survival. NQO1 allele was evaluated in subjects enrolled in ECOG 3590, a randomized comparison of radiation (RT) vs radiation and chemotherapy with cisplatin/etoposide (RCT) in patients with completely resected stages II and IIIa NSCLC. Overall survival was estimated using the Kaplan-Meier method and compared via the log-rank test. Cox models were used to assess the impact of covariates on outcomes. Among 152 patients with assessable samples, 24 (16%) had NQO1*2. Median follow-up was 139 months. The presence of NQO1*2/*2 was associated with decreased overall survival (OS) (median in the heterozygote/wild-type group 42.3 vs. 33.5 months in the variant group, p=0.04). In a multivariable Cox model, variant NQO1 (HR=1.58, p=0.05), age <60 (HR=0.67, p=0.04), PS 1 (HR=1.47, p=0.05), cardiovascular disease (HR=1.93, p=0.003) and alkaline phosphatase <100 mg/ml (HR=0.59, p=0.005) were all significant predictors of OS. NQO1*2/*2 may be an independent predictor of poor overall survival in individuals with resected stages II and IIIa NSCLC. Although the basis for the NQO1 association with decreased survival requires additional evaluation, NQO1 may represent a biomarker for guiding individualized therapy.
Keywords: lung cancer, NAD(P)H:quinone oxidoreductase 1, polymorphism, p53
Introduction
Lung cancer is the leading cause of cancer death in the United States, with 219440 new diagnoses and 159390 deaths estimated for 2009 (1). Lung cancer is the most common cause of cancer death among both men and woman in industrialized nations (2). Both chemotherapy and radiotherapy are utilized to treat locally advanced or metastatic tumors. Moderate survival improvements are reported for patients with inoperable disease who received chemotherapy as well as for patients who have received platinum based chemotherapy following surgery (3-6). Despite treatment, overall survival remains poor, not all patients respond to therapy, and patients receiving chemotherapy experience significant toxicity (4). Genetic markers are a potential method to identify patients most likely to benefit from chemotherapy or radiotherapy.
NAD(P)H:quinone oxidoreductase 1 (NQO1), is a cytosolic flavoenzyme that catalyzes the two-electron reduction of quinones into hydroquinones. NQO1 contains one characteristic polymorphism (NQO1*2), a C→T transition at bp 609 that results in a proline to serine substitution at codon 187 (7,8). Individuals homozygous for NQO1*2 have essentially no active enzyme (9), while those who are heterozygotes have intermediate activity when compared to NQO1*1 homozygotes (10). The decreased activity associated with this polymorphism is mediated by two mechanisms: i) the polymorphism is in the active site of NQO1, and ii) the NQO1*2 protein is degraded rapidly via the ubiquitin-dependent pathway (half-life of wild-type NQO1 >18 h and the half-life of variant NQO1*2 is 1.2 h) (10).
A lack of NQO1 may increase the generation of semiquinones rather than hydroquinones, from the quinines found in cigarette smoke. Polymorphisms in NQO1 have therefore been evaluated as modifiers of lung cancer risk and have been the subject of two meta-analysis. Chao and colleagues (11) reported that in Caucasian populations, there was an increased prevelance of the variant allele among ever smokers with lung cancer (OR, 1.08; 95% CI, 0.96-1.22), but not among never smokers (OR, 0.97; 95% CI, 0.62-1.54). However, Kiyohara et al (12) showed the opposite effect in a Japanese cohort where the allele was significantly associated with decreased risk of lung cancer [random effects odds ratio (OR) = 0.70]. The role of NQO1 in lung cancer pathogenesis appears small.
NQO1 has also been studied as a predictor of chemosensitivity and radiosensitivity and overexpression has typically been associated with increased chemosensitivity and radiosensitivity (13). NQO1 gene expression is induced in response to xenobiotics, antioxidants, UV light, ionizing radiation and cisplatin. Additionally, wild-type NQO1 binds to and stabilizes both mutant and wild-type p53 under conditions of oxidative stress or after exposure to γ-irradiation. Degradation of p53 is mediated by two alternative pathways, ubiquitin-independent or ubiquitin-dependent. The ubiquitin-independent pathway is regulated by NQO1 and is mediated via the 20S proteasome (13).
Song et al (14) demonstrated that NQO1 genotype significantly influenced loco-regional recurrence in Korean patients with completely resected stage II or IIIa NSCLC who were treated with adjuvant radiation and sometimes adjuvant chemotherapy [wild-type (NQO1*1/*1) 6.3% (2/32), heterozygous patients 8.1% (5/62), NQO1*2/*2 27.3% (6/22)]. Multivariate analysis revealed that NQO1 genotype and pathologic nodal stage were statistically significant predictive factors for loco-regional recurrence-free survival, but not overall survival.
Individuals with NQO1*2 polymorphisms may be both chemoresistant and radiation resistant. The current study was performed to determine the effect of the NQO1 genetic polymorphisms on overall survival in stages II and IIIa NSCLC patients treated with RT or RTC.
Materials and methods
Clinical study
ECOG 3590 was a randomized, prospective trial of adjuvant therapy in patients with resected stages II or IIIa NSCLC (15). All patients had complete resection of their tumor and mediastinal lymph node sampling or dissection. Eighty-five percent were Caucasian. Following stratification by weight loss, histology, nodal biopsy technique and lymph node status, patients were randomized to receive chemotherapy (cisplatin 60 mg/m2 IV on day 1 and etoposide 120 mg/m2 days 1-3) with concurrent RT (50.4 Gy in 28 daily fractions) or RT alone.
Patients enrolled in ECOG 3590 were also eligible for enrollment in ECOG 4592 (16), a study designed to evaluate the prognostic value of p53 mutations in exons 5-8 which were determined by single-strand confirmation polymorphism (SSCP) analysis. Genomic DNA extracted from submitted paraffin blocks were utilized. Patients enrolled in ECOG 3590 gave permission to use samples for future studies. The current study design was reviewed and approved by the University of Wisconsin Institutional Review Board.
Genotyping
Genomic DNA extracted from the ECOG 4592 samples was stored at -80°C and variations in the NQO1 gene were analyzed by pyrosequencing. Initial polymerase chain reaction (PCR) primers were designed using Primer Express version 1.5 (ABI, Foster City, CA) and consisted of forward primer (5′-CTGATGGAATTGGTTGACTTA-3′), and reverse primer [5′(Biotin)-TCTGGTGATCACCCAAGGCT-3′] to amplify a portion of the NQO1 gene. The forward pyrosequencing primer (5′-GTGGCTTCCAAGTCTTA-3′) was designed using Pyrosequencing SNP Primer Design Version 1.01 software (http://www.pyrosequencing.com). Primers were obtained from Integrated DNA Technologies (Coralville, IA). PCR was carried out using Amplitaq Gold PCR master mix (ABI), 5 pmole of each primer, and 10 ng DNA, on an MJ Research PTC-100 thermal cycler (Waltham, MA). Reactions were denatured initially at 93°C for 20 min, then cycled at 95°C for 30 sec, 55°C for 30 sec and 72°C for 30 sec for 55 cycles, followed by a final elongation step at 72°C for 5 min. Pyrosequencing was carried out as previously described using a PSQ96 instrument and software (Biotage AB, Uppsala, Sweden) (32). Genotype was called variant if it differed from the Refseq consensus sequence for the SNP position. To ensure proper amplification and the absence of contamination, each PCR and pyrosequencing run contained no template controls or cancer cell line positive controls.
Statistical analysis
Polymorphisms were classified into two groups, heterozygous/wild-type and variant. Wild-types and heterozygotes were combined as NQO1 activity is similar. OS, defined as the time from randomization to death from any cause, was estimated using the Kaplan-Meier method. Differences between the polymorphism status groups were analyzed using the log-rank test. Fisher's exact test and the Kruskal-Wallis rank sum test were used to compare baseline variables between the two polymorphism groups. Cox models were used to assess the univariate association between baseline variables and overall survival. Variables significant at the 0.10 level were fitted to a multivariable Cox proportional hazards model and backwards selection was used to select the final model.
Results
Four hundred and eighty-eight patients were enrolled in ECOG 3590, of whom 217 entered ECOG 4592. Among 152 patients with assessable samples, 24 (16%) had variant NQO1. Most demographic variables were balanced between the two NQO1 groups (Table I). However, a greater proportion of patients with mutated p53 was present in the heterozygous/ wild-type NQO1 group, (p=0.04).
Table I.
Baseline demographic variables, stratified by NQ01 status E3590, n=152 patients.
| Variable | All (n=152) | Wild-type or heterozygous (n=128) | (%) | Variant (n=24) | (%) | P-value |
|---|---|---|---|---|---|---|
| Treatment Arm | ||||||
| RT | 78 | 65 | 50.78 | 13 | 54.17 | |
| RTC | 74 | 63 | 49.22 | 11 | 45.83 | 0.82 |
| Total | 152 | 128 | 24 | |||
| Age (median, range) | 60.5 | 35-77 | 63 | 35-81 | 0.19 | |
| Gender | ||||||
| Male | 58 | 49 | 38.28 | 9 | 37.50 | |
| Female | 94 | 79 | 61.72 | 15 | 62.50 | 1 |
| Total | 152 | 128 | 24 | |||
| p53 | ||||||
| Mutated | 66 | 61 | 48.03 | 5 | 22.73 | |
| Wild | 83 | 66 | 51.97 | 17 | 77.27 | 0.04 |
| Total | 149 | 127 | 22 | |||
| Race | ||||||
| White | 133 | 110 | 85.94 | 23 | 95.83 | |
| Other | 19 | 18 | 14.06 | 1 | 4.17 | 0.31 |
| Total | 152 | 128 | 24 | |||
| PS | ||||||
| 0 | 61 | 49 | 38.28 | 12 | 50.00 | |
| 1 | 91 | 79 | 61.72 | 12 | 50.00 | 0.36 |
| Total | 152 | 128 | 24 | |||
| Weight loss (%) | ||||||
| None | 73 | 60 | 47.24 | 13 | 54.17 | |
| <5 | 43 | 35 | 27.56 | 8 | 33.33 | |
| 5-10 | 25 | 23 | 18.11 | 2 | 8.33 | |
| >10 | 10 | 9 | 7.09 | 1 | 4.17 | 0.72 |
| Total | 151 | 127 | 24 | |||
| Cardiovascular diseases | ||||||
| Yes | 33 | 29 | 22.83 | 4 | 16.67 | |
| No | 118 | 98 | 77.17 | 20 | 83.33 | 0.6 |
| Total | 151 | 127 | 24 | |||
| Respiratory diseases | ||||||
| Yes | 26 | 22 | 17.32 | 4 | 16.67 | 1 |
| No | 125 | 105 | 82.68 | 20 | 83.33 | |
| Total | 151 | 127 | 24 | |||
| Other diseases | ||||||
| Yes | 41 | 37 | 30.33 | 4 | 18.18 | 0.31 |
| No | 103 | 85 | 69.67 | 18 | 81.82 | |
| Total | 144 | 122 | 22 | |||
| Analgesics | ||||||
| Yes | 47 | 44 | 34.92 | 3 | 13.04 | 0.05 |
| No | 102 | 82 | 65.08 | 20 | 86.96 | |
| Total | 149 | 126 | 23 | |||
| Steroids | ||||||
| Yes | 6 | 6 | 4.92 | 0 | 0.00 | 0.59 |
| No | 140 | 116 | 95.08 | 24 | 100.00 | |
| Total | 146 | 122 | 24 | |||
| Other medicines | ||||||
| Yes | 80 | 76 | 62.30 | 4 | 18.18 | 0.0001 |
| No | 64 | 46 | 37.70 | 18 | 81.82 | |
| Total | 144 | 122 | 22 | |||
| Albumin (median, range) | 40 | 25-53 | 39 | 31-44 | 0.16 | |
| Alkaline phosphatase | 98 | 44-284 | 112.5 | 59-416 | 0.24 | |
| (median, range) | ||||||
| Histology | ||||||
| Squamous | 53 | 43 | 33.59 | 10 | 41.67 | |
| Small cell | 0 | 0 | 0.00 | 0 | 0.00 | |
| Adenocarcinoma | 81 | 69 | 53.91 | 12 | 50.00 | |
| Large cell | 12 | 10 | 7.81 | 2 | 8.33 | |
| Other | 6 | 6 | 4.69 | 0 | 0.00 | 0.8 |
| Total | 152 | 128 | 24 | |||
| Nodal status | ||||||
| N1 | 78 | 65 | 51.59 | 13 | 54.17 | |
| N2 | 72 | 61 | 48.41 | 11 | 45.83 | |
| Total | 150 | 126 | 24 | 0.83 | ||
| Lymph node dissection | ||||||
| Complete | 74 | 60 | 47.24 | 14 | 58.33 | |
| Sampling | 77 | 67 | 52.76 | 10 | 41.67 | |
| Total | 151 | 127 | 24 | 0.38 | ||
| Stage | ||||||
| II | 71 | 61 | 47.66 | 10 | 41.67 | |
| IIIa | 81 | 67 | 52.34 | 14 | 58.33 | |
| Total | 152 | 128 | 24 | 0.66 | ||
| TNM stage | ||||||
| T1 | 38 | 32 | 25.00 | 6 | 25.00 | |
| T2 | 100 | 87 | 67.97 | 13 | 54.17 | |
| T3 | 14 | 9 | 7.03 | 5 | 20.83 | |
| Total | 152 | 128 | 24 | 0.1 | ||
| Previous chemotherapy | ||||||
| No | 151 | 128 | 100.00 | 23 | 95.83 | |
| Yes, combinations | 1 | 0 | 0.00 | 1 | 4.17 | 0.16 |
| Total | 152 | 128 | 24 |
Median follow-up was 139 months. One hundred and twenty-three (81%) patients had died. Median OS in the heterozygous/wild-type group was 42.3 months (95% CI 35.9-66.3 mos) vs. 33.5 months (95% CI 16.2-80.9 mos) in the variant group (p=0.04, Fig. 1). Among patients receiving RT only, no difference in OS was observed (45.2 mos (95% CI 35.9-91.8 mos) vs. 53.4 mos (95% CI 29.2-Inf); p=0.49, Fig. 2). However, among those patients who received chemoradiotherapy a significant difference in median OS in favor of the heterozygous/wild-type group was observed (40.0 months (95% CI 30.8-80.6 mos) vs. 16.2 months (13.6-Inf mos), p=0.007, Fig. 3).
Figure 1.

Kaplan-Meier curve comparing NQO1*1/*1 and NQO1*1/*2 to NQO1*2/*2 in all patients median OS in the heterozygous/wild-type group was 42.3 vs. 33.5 months in the variant group (p=0.04).
Figure 2.
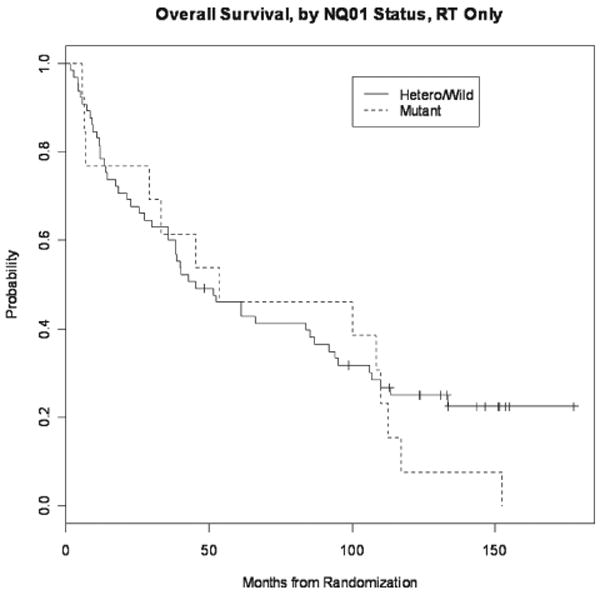
Kaplan-Meier curve comparing NQO1*1/*1 and NQO1*1/*2 to NQO1*2/*2 in patients receiving RT only, no difference in OS was observed (p=0.49).
Figure 3.

Kaplan-Meier curve comparing NQO1*1/*1 and NQO1*1/*2 to NQO1*2/*2 in patients receiving CRT, demonstrating a significant difference in median OS in favor of the heterozygous/wild-type group was observed (40.0 vs. 16.2 months, p=0.007).
Univariate Cox proportional hazards models (Table II) revealed that overall survival was associated with: NQO1 status, age, race, performance status, cardiovascular disease, alkaline phosphatase (mg/ml) and TNM stage. Treatment and NQO1 status were not statistically significant when adjusted for these two factors alone (p=0.16). A multivariable Cox model revealed variant NQO1 (HR=1.58, p=0.05), age <60 (HR 0.67, p=0.04), PS 1 (HR=1.47, p=0.05), cardiovascular disease (HR=1.93, p=0.003) and alkaline phosphatase <100 mg/ml (HR= 0.59, p=0.005) were all significant predictors of OS. A treatment by NQ01 status interaction test was not statistically significant (p=0.16).
Table II.
Univariate Cox model results.
| Variable | No. of deaths | Median OS | HR for death | 95% CI | P-value |
|---|---|---|---|---|---|
| NQ01 | |||||
| Wild-type or heterozygote | 99 | 42.3 | ref | 0.046 | |
| Variant | 24 | 33.5 | 1.58 | 1.01-2.47 | |
| Treatment Arm | |||||
| RT | 62 | 48.5 | ref | ||
| RT + chemotherapy | 61 | 37.7 | 1.13 | 0.79-1.62 | 0.49 |
| Age (years) | |||||
| ≥60 | 74 | 35.9 | ref | ||
| <60 | 49 | 62.2 | 0.624 | 0.43-0.9 | 0.011 |
| Gender | |||||
| Male | 77 | 37.1 | 1.2 | 0.83-1.74 | 0.32 |
| Female | 46 | 55.1 | ref | ||
| p53 | |||||
| Mutated | 57 | 39.9 | ref | ||
| Wild-type | 63 | 50.7 | 0.863 | 0.6-1.23 | 0.42 |
| Race | |||||
| White | 110 | 38.2 | 1.64 | 0.92-2.92 | 0.091 |
| Other | 13 | 80.6 | ref | ||
| PS | |||||
| 0 | 46 | 63.7 | ref | ||
| 1 | 77 | 38.3 | 1.37 | 0.95-1.98 | 0.091 |
| Weight loss (%) | |||||
| <5 | 94 | 44 | 0.972 | 0.6-1.41 | 0.9 |
| ≥5 | 28 | 38.3 | ref | ||
| Cardiovascular diseases | |||||
| Yes | 33 | 30.8 | 1.93 | 1.29-2.89 | 0.0015 |
| No | 89 | 52.7 | ref | ||
| Respiratory diseases | |||||
| Yes | 23 | 53.2 | 0.964 | 0.61-1.52 | 0.88 |
| No | 99 | 39.8 | ref | ||
| Other diseases | |||||
| Yes | 32 | 65.4 | 0.782 | 0.52-1.18 | 0.24 |
| No | 83 | 38.2 | ref | ||
| Analgesics | |||||
| Yes | 38 | 40.2 | 1.02 | 0.7-1.51 | 0.91 |
| No | 82 | 43.5 | ref | ||
| Steroids | |||||
| Yes | 5 | 55.5 | 0.993 | 0.4-2.44 | 0.99 |
| No | 112 | 41 | ref | ||
| Other medicins | |||||
| Yes | 67 | 41.8 | 1.15 | 0.81-1.65 | 0.45 |
| No | 49 | 41 | ref | ||
| Albumin | |||||
| ≥40 | 60 | 39.8 | ref | ||
| <40 | 60 | 40.2 | 1.11 | 0.76-1.59 | 0.57 |
| Alkaline phosphatase | |||||
| ≥100 | 65 | 28 | ref | ||
| <100 | 56 | 66.3 | 0.59 | 0.41-0.84 | 0.004 |
| Histology | |||||
| Squamous | 45 | 45.2 | 1.01 | 0.7-1.46 | 0.96 |
| Non-squamous | 78 | 39.9 | ref | ||
| Nodal status | |||||
| N1 | 60 | 43.5 | ref | ||
| N2 | 61 | 40 | 1.19 | 0.83-1.69 | 0.35 |
| Lymph node | |||||
| Complete | 62 | 61 | ref | ||
| Dissection | |||||
| Sampling | 60 | 40 | 1.04 | 0.73-1.49 | 0.83 |
| Stage | |||||
| II | 53 | 41.8 | ref | ||
| IIIa | 70 | 40.2 | 1.26 | 0.88-1.81 | 0.2 |
| TNM stage | |||||
| T1/T2 | 109 | 44 | ref | ||
| T3 | 14 | 18.6 | 1.93 | 1.1-3.37 | 0.022 |
Discussion
Our study is the first to report that NQO1*2/*2 may be an independent predictor of poor overall survival in individuals with resected stages II and IIIa NSCLC. These results are in contrast with Song and colleagues who identified a decrease in locoregional recurrence-free survival in NQO1*2/*2 patients, but not in overall survival. This may be related to the different length of median follow-up (49.2 months compared to 139 months), and chemotherapy differences (cisplatin/vinorelbine allowed vs. patients randomized to cisplatin/etoposide or no chemotherapy). Locoregional recurrence-free survival was not evaluated as part of E3590. These results suggest that NQO1*2/*2 may be considered an independent predictor of poor survival for patients with NSCLC. Individuals with NQO1*2/*2 have no NQO1 activity, and subsequently lose NQO1 stabilization of three tumor suppressors p53 (17-19), p73a (20) and p33 (21), by inhibiting their breakdown via the 20S proteosome. Lack of stabilization may lead to a more aggressive tumor biology and lack of responsiveness to chemotherapy and radiation therapy (22-25).
These data suggest that RT is the optimal therapy for individuals with NQO1*2/*2. The lack of a difference in OS when analyzed by treatment and NQO1 status (p=0.016) may be related to inadequate power.
In this study, patients who were NQO1*2/*2 were more likely to be p53 wild-type. Since NQO1*1 is known to bind to and stabilize both mutant and wild-type p53 (and these genotypes should have opposite prognostic effects), we hypothesize that that p53 status becomes important only in the absence of NQO1 activity. That the role of p53 in lung cancer prognosis has been the subject of dozens of paper and two meta-analyses (25,26) without resolution of the question, lends support to this hypothesis. The association between NQO1*2/*2 and p53 wild-type suggests that these genes may be important in lung cancer pathogenesis.
Our study has several limitations, including that is was as unplanned retrospective analysis, had limited in power to detect treatment effects and used of SSCP for identifying p53 mutations. While p53 mutations were evaluated in exons 5-8, the specific mutation is not identifiable by SSCP. Therefore, the effect of the mutation on p53 protein expression and the interaction between NQO1 and the mutant p53 is unknown. However, this study does have the advantage of utilizing data from a large, randomized multi-institutional trial with mature survival data.
In summary, NQO1*2/*2 independently predicts poor survival in completely resected NSCLC stages II and III patients. In addition, NQO1*2 is associated with p53 wild-type. Prospective trials and determination of specific p53 mutations are needed to confirm and define the relationship of NQO1, p53 and NSCLC. The results of the current study demonstrate that evaluation of NQO1 polymorphisms may be an important consideration in individualizing the treatment of lung cancer.
Acknowledgments
This study was supported by the ACCP Oncology Research Award, the NIH Pharmacogenetics Research Network (GM63340), and the American Cancer Society.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59:225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Pisani P, Parkin DM, Ferlay J. Estimates of the worldwide mortality from eighteen major cancers in 1985. Int J Cancer. 1993;55:891–903. doi: 10.1002/ijc.2910550604. [DOI] [PubMed] [Google Scholar]
- 3.Keller SM, Adak S, Wagner H, et al. A randomized trial of postoperative adjuvant therapy in patients with completely resected stage II or IIIA non-small cell lung cancer. N Engl J Med. 2000;343:1217–1222. doi: 10.1056/NEJM200010263431703. [DOI] [PubMed] [Google Scholar]
- 4.Rube C, Fleckenstein J. Combined-modality treatment of non-small cell lung cancer stages I-III (take home messages) Lung Cancer. 2004;45:S139–S141. doi: 10.1016/j.lungcan.2004.07.980. [DOI] [PubMed] [Google Scholar]
- 5.Belani CP. Adjuvant and neoadjuvant therapy in non-small cell lung cancer. Semin Oncol. 2005;32:S9–S15. doi: 10.1053/j.seminoncol.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 6.Bradley JD, Paulus R, Graham MV, et al. Phase II trial of postoperative adjuvant paclitaxel/carboplatin and thoracic radiotherapy in resected stage II and IIIa non-small cell lung cancer: promising long-term results of the radiation therapy oncology group-RTOG 9705. J Clin Oncol. 2005;23:3480–3487. doi: 10.1200/JCO.2005.12.120. [DOI] [PubMed] [Google Scholar]
- 7.Traver RD, Horikoshi T, Danenberg KD, et al. NAD(P)H:quinone oxidoreductase gene expression in human colon carcinoma cells: characterization of a mutation which modulates DT-diaphorase activity and mitomycin sensitivity. Cancer Res. 1992;52:797–802. [PubMed] [Google Scholar]
- 8.Ross D, Traver RD, Siegel D, et al. A polymorphism in NAD(P) H:quinone oxidoreductase (NQO1): relationship of a homozygous mutation at position 609 of the NQO1 cDNA to NQO1 activity. Br J Cancer. 1996;74:995–996. doi: 10.1038/bjc.1996.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Traver RD, Siegel D, Beall HD, et al. Characterization of a polymorphism in NAD(P)H: quinone oxidoreductase (DT-diaphorase) Br J Cancer. 1997;75:69–75. doi: 10.1038/bjc.1997.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Siegel D, McGuinness SM, Winski SL, et al. Genotype-phenotype relationships in studies of a polymorphism in NAD(P)H:quinone oxidoreductase 1. Pharmacogenetics. 1999;9:113–121. doi: 10.1097/00008571-199902000-00015. [DOI] [PubMed] [Google Scholar]
- 11.Chao C, Zhang ZF, Berthiller J, Boffetta P, Hashibe M. NAD(P) H:quinone oxidoreductase 1 (NQO1) Pro187Ser polymorphism and the risk of lung, bladder, and colorectal cancers: a meta-analysis. Cancer Epidemiol Biomarkers Prev. 2006;5:979–987. doi: 10.1158/1055-9965.EPI-05-0899. [DOI] [PubMed] [Google Scholar]
- 12.Kiyohara C, Yoshimasu K, Takayama K, Nakanishi Y. NQO1, MPO, and the risk of lung cancer: a HuGE review. Genet Med. 2005;7:463–478. doi: 10.1097/01.gim.0000177530.55043.c1. [DOI] [PubMed] [Google Scholar]
- 13.Dinkova-Kostova AT, Talalay P. NAD(P)H:quinone acceptor oxidoreductase 1 (NQO1), a multifunctional antioxidant enzyme and exceptionally versatile cytoprotector. Arch Biochem Biophys. 2010;1:116–123. doi: 10.1016/j.abb.2010.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Song SY, Jeong SY, Park HJ, et al. Clinical significance of NQO1 C609T polymorphisms after postoperative radiation therapy in completely resected non-small cell lung cancer. Lung Cancer. 2010;2:278–282. doi: 10.1016/j.lungcan.2009.06.009. [DOI] [PubMed] [Google Scholar]
- 15.Schiller JH, Harrington D, Belani CP, Langer C, Sandler A, Krook J, Zhu J, Johnson DH Eastern Cooperative Oncology Group. Comparison of four chemotherapy regimens for advanced non-small cell lung cancer. N Engl J Med. 2002;346:92–98. doi: 10.1056/NEJMoa011954. [DOI] [PubMed] [Google Scholar]
- 16.Schiller JH, Adak S, Feins RH, Keller SM, Fry WA, Livingston RB, Hammond ME, Wolf B, Sabatini L, Jett J, Kohman L, Johnson DH. Lack of prognostic significance of p53 and K-ras mutations in primary resected non-small cell lung cancer on E4592: a Laboratory Ancillary Study on an Eastern Cooperative Oncology Group Prospective Randomized Trial of Postoperative Adjuvant Therapy. J Clin Oncol. 2001;19:448–457. doi: 10.1200/JCO.2001.19.2.448. [DOI] [PubMed] [Google Scholar]
- 17.Asher G, Lotem J, Sachs L, et al. Mdm-2 and ubiquitin-independent p53 proteasomal degradation regulated by NQO1. Proc Natl Acad Sci USA. 2002;99:13125–13130. doi: 10.1073/pnas.202480499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Asher G, Lotem J, Kama R, et al. NQO1 stabilizes p53 through a distinct pathway. Proc Natl Acad Sci USA. 2002;99:3099–3104. doi: 10.1073/pnas.052706799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Asher G, Lotem J, Cohen B, et al. Regulation of p53 stability and p53-dependent apoptosis by NADH quinine oxidoreductase 1. Proc Natl Acad Sci USA. 2001;98:1188–1193. doi: 10.1073/pnas.021558898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Asher G, Tsvetkov P, Kahana C, Shaul Y. A mechanism of ubiquitin-independent proteasomal degradation of the tumor suppressors p53 and p73. Genes Dev. 2005;19:316–321. doi: 10.1101/gad.319905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garate M, Wong RP, Campos EI, Wang Y, Li G. NAD(P)H quinone oxidoreductase 1 inhibits the proteasomal degradation of the tumour suppressor p33(ING1b) EMBO J. 2008;6:576–581. doi: 10.1038/embor.2008.48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsutsumi K, Yasuda M, Nishioka T. X-ray irradiation altered chemosensitivity of a p53-null non-small cell lung cancer cell line. Cell Struct Funct. 2006;31:47–52. doi: 10.1247/csf.31.47. [DOI] [PubMed] [Google Scholar]
- 23.Lai SL, Perng RP, Hwang J. p53 gene status modulates the chemosensitivity of non-small cell lung cancer cells. J Biomed Sci. 2000;7:64–70. doi: 10.1007/BF02255920. [DOI] [PubMed] [Google Scholar]
- 24.Rusch V, Klimstra D, Venkatramen E, et al. Aberrant p53 expression predicts clinical resistance to cisplatin-based chemotherapy in locally advanced non-small cell lung cancer. Cancer Res. 1995;55:5038–5042. [PubMed] [Google Scholar]
- 25.Tammemagi MC, McLaughlin JR, Bull SB. Meta-analyses of p53 tumor suppressor gene alterations and clinicopathological features in resected lung cancers. Cancer Epidemiol Biomarkers Prev. 1999;8:625–634. [PubMed] [Google Scholar]
- 26.Mitsudomi T, Hamajima N, Ogawa M, et al. Prognostic significance of p53 alterations in patients with non-small cell lung cancer: a meta-analysis. Clin Cancer Res. 2000;6:4055–4063. [PubMed] [Google Scholar]