

Abstract
The broad involvement of miRNAs in critical processes underlying development, tissue homoeostasis and disease has led to a surging interest among the research and pharmaceutical communities. To study miRNAs, it is essential that the quantification of microRNA levels is accurate and robust. By comparing wild-type to small RNA deficient mouse embryonic stem cells (mESC), we revealed a lack of accuracy and robustness in previous published multiplex qRT-PCR techniques. Here, we describe an optimized method, including purifying away excessive primers from previous multiplex steps before singleplex real time detection, which dramatically increases the accuracy and robustness of the technique. Furthermore, we explain how performing the technique on a microfluidic chip at nanoliter volumes significantly reduces reagent costs and permits time effective high throughput miRNA expression profiling.
Keywords: Bioengineering, Issue 54, microRNA, multiplex qRT-PCR, high throughput profiling, Fluidigm
Protocol
1. RNA-Extraction: Cells Grown in Monolayer
(Following the manufacturer's protocol using TRIzol.)
| Name of the reagent | Company | Prod/Cat # |
| TRIzol Solution | Invitrogen | 15596-018 |
| Isopropyl alcohol | Sigma-Aldrich | 1907-64 |
| Chloroform | Sigma-Aldrich | C 2432 |
| Ethanol | ||
| RNAse free water |
Homogenization
Homogenize cells directly on the culture dish by adding 1 ml Trizol Solution per 10 cm2 dish area, swirl the Trizol around and pipette up and down to ensure full coverage of the plate.
Phase Separation
Incubate the homogenized sample for 5 minutes at room temperature.
Transfer into a labeled tube, add 0.2 ml of chloroform per 1ml TrizolSolution and shake tube vigorously by hand for 15 seconds.
Incubate at room temperature for 3 minutes.
Centrifuge the sample at 12,000 x g for 15 minutes at 2 °C. After centrifugation the mixture separates into a lower red phase, an interphase and a colorless upper aqueous phase, which contains the RNA and should be 60% of the total Trizol Solution volume.
RNA Precipitation
Transfer the aqueous, RNA containing phase into a fresh, labeled tube.
Precipitate the RNA by adding 0.5 ml of isopropyl alcohol per 1 ml of Trizol Solution.
Incubate the samples at room temperature for 10 minutes.
Centrifuge at 12,000 x g for 10 minutes at 2 °C.
RNA-wash
Following centrifugation, discard the supernatant from the tubes.
Wash the RNA pellet (on the side and bottom of the tube) by adding at least 1 ml of 75% ethanol per 1 ml of Trizol Solution, vortex.
Centrifuge at 7500 x g for 5 minutes at 2 °C.
RNA Elution
Discard the supernatant and centrifuge again for 1 minute. Remove excessive fluid.
Air-dry pellet for 5-10 minutes. Note: over dried RNA is difficult to solubilize.
Redissolve the pellet in RNase free water by pipetting the solution up- and down and incubate for 10 minutes at 55 °C.
Measure RNA concentration (A260/280 ratio) using a Nanodrop™ spectrophotometer.
Store at -80 °C until ready for use.
2. RNA-Extraction: Serum
(Following mirVana PARIS Kit manufacturer's protocol modified by Mitchell et al.1)
| Name of the reagent | Company | Prod/Cat # | Comments |
| mirVana PARIS Kit 2X Denaturing Solution Acid-Phenol:Chloroform miRNA Wash Solution 1 miRNA Wash Solution 2/3 | Ambion | AM1556 | Modified protocol |
| 2-mercaptoethanol | Sigma-Aldrich | M7522 | |
| Ethanol | |||
| DEPC treated Water |
Chemical Preparation:
Add 375 μl of 2-mercaptoethanol to the 2X Denaturing Solution and mix well.
Add 21 ml of 100% ethanol to the miRNA wash Solution 1.
Add 40 ml of 100% ethanol to the miRNA wash Solution 2/3.
Sample Preparation:
Thaw serum sample on ice.
Homogenization
Mix 300 μl sample with an equal volume (300 μl) of 2X Denaturing Solution (should be at room temperature) and vortex.
Incubate mixture on ice for 5 minutes.
Add a volume of Acid-Phenol:Chloroform equal to the total volume of the sample lysate plus the 2X Denaturing Solution (600 μl). Important: Do NOT use the aqueous buffer that lies on top of the Acid-Phenol:Chloroform.
Vortex mixture for 60 seconds.
Centrifuge at room temperature for 15 minutes at maximum speed (>10,000 x g). After centrifugation the mixture separates into an upper aqueous phase, an interphase and a lower organic phase.
Remove aqueous layer on top (should be 300 - 500 μl including any proteinacious "hazy" layer).
Re-extract aqueous layer as before by again adding chloroform. Important: The aqueous layer of the second extraction is routinely smaller in volume (250 μl). Note the volume recovered.
Preparation of Final RNA-Isolation (total RNA)
Pre-heat (95 °C) nuclease-free water.
100% Ethanol must be at room temperature.
Final RNA-Isolation (total RNA)
Add 1.25 volumes of 100% ethanol to the recovered aqueous phase (e.g. if 300 μl was recovered, add 375 μl ethanol) and mix thoroughly.
For each sample, place a filter cartridge into one of the collection tubes.
Pipette the lysate onto the filter.
Centrifuge (10,000 x g) for 30 seconds, discard the flow-through, centrifuge again for 30 seconds and replace the filter cartridge into the same collection tube.
RNA-Wash
Apply 700 μl miRNA Wash Solution 1 to the filter cartridge and centrifuge for 15 seconds, discard the flow-through from the collection tube, and replace the filter cartridge into the same collection tube.
Apply 500 μl miRNA Wash Solution 2/3, centrifuge for 15 seconds, discard the flow-through and repeat with a second 500 μl of Wash Solution 2/3.
Discard the flow-through and dry-spin for 1-2 minutes.
Place filter cartridge in a fresh collection tube, apply 100 μl RNAse free water (95 °C), close cap and incubate for 2 minutes.
Centrifuge for ˜2 minutes to recover the RNA.
Store at -80 °C until use.
3. Concentration of RNA-Solution (5X)
(Following the manufacture's protocol)
| Name of the reagent | Company | Prod/Cat # |
| MICROCON Centrifugal Filter Devices, Ultracell YM-3 | Millipore | 42404 |
| DEPC treated Water |
Device: Microcon Ultracell YM-3, Cutoff SS and DS Nucleotides: 10
Volume: Desired volume of re-concentrated RNA solution is dependent on experimental design and should include control experiments.
Insert Microcon sample reservoir into vial and pipette solution into reservoir. Note: Do not touch the membrane.
Centrifuge for 185 minutes at 4 °C with maximal 14,000 x g. Note: Position vial with crack band towards the rotor.
Pipette desired volume of RNAse free water (e.g. 20 μl) onto the reservoir, place reservoir inverted into a new vial and spin for 3 minutes at 4 °C with maximal 3000 x g.
Store at -80 °C until use.
4. Reverse Transcription
(Following a protocol by Tang et al.2 with modifications.)
| Name of the reagent | Company | Prod/Cat # | Comments |
| High capacity cDNA archive kit 10x cDNA archiving Buffer dNTP (100 mM) Moloney murine leukemia virus (MMLV) reverse transcriptase (50 U/μl) | Applied Biosystems | 4368814 | modified protocol |
| RNaseOUT (40 U/μl) | Invitrogen | 10777019 | |
| x-plex reverse stem-loop primer mix (40 nM) * | Integrated DNA Technologies | Custom | Sequences * |
| DEPC treated Water |
Reaction Volume: 5.5 μl
Note: the final concentration of the stem-loop primers should be 1-5 nM (here 2 nM).
Prepare Master-Mix (Volumes per reaction) as follows:
| 1. DEPC-Water | 1.959 μl |
| 2. 10x RT-Buffer | 0.55 μl |
| 3. RNase Inhibitor (40 U/μl) | 0.0715 μl |
| 4. dNTP (100 mM ) | 0.275 μl |
| 5. x-plex reverse stem-loop primer mix (40 nM)* | 0.275 μl |
| 6. MMLV-RT (50 U/μl) | 0.369 μl |
Mix and centrifuge briefly.
Add 3.5 μl Master-Mix to 2 μl sample.
Perform the following RT-reaction: 16 °C for 30 minutes, followed by 60 cycles at 20 °C for 30 seconds, 42 °C for 30 seconds, 50 °C for 1 second and finally 85 °C for 5 minutes to inactivate MMLV-RT, 4 °C ∞.
Store at -20 °C until use.
*List of reverse stem-loop primers can be found at http://urology.ucsf.edu/blellochlab/protocols.htm
5. Pre-Amplification (Pre-PCR)
(Following a protocol by Tang et al.2 with modifications.)
| Name of the reagent | Company | Prod/Cat # | Comments |
| dNTP (100 mM) | Applied Biosystems | 4368814 | High cap. cDNA archive Kit. |
| 2x Universal PCR Master Mix with NoAmpErase UNG | Applied Biosystems | 4324018 | |
| x-plex forward primer mix (450 nM)* | Integrated DNA Technologies | Custom | Sequences * |
| Universal Reverse Primer (100 μM) | Integrated DNA Technologies | Custom | Sequence2 |
| AmpliTaqGold polymerase (5 U/μl) | Applied Biosystems | 4311806 | |
| MgCl2 (100 mM) | |||
| DEPC treated Water |
Reaction Volume: 27.5μl (22μl MM + 5.5μl RT-Product)
Note: Pre-Amplification is necessary to enhance signal strength, especially when starting concentration of RNA is limited. The input RNA level determines the optimal number of Pre-PCR cycles. Given the same relative efficiency each PCR cycle should double the concentration of the final product. Since some miRNAs will be more highly expressed, avoid over cycling. However, under-amplification can result in a loss of detection sensitivity, especially for microRNAs expressed at low levels. In our hands 12 pre amplification cycles, using 100ng of total RNA, appeared sufficient. Using serum as starting material, 12 cycles results in detectable miRNA levels, but 15 cycles appeared to be superior. Finally, starting with 3 oocytes (roughly 400 pg total RNA per oocyte3), 16 pre-amplification cycles successfully allowed us to screen for microRNA expression levels4.
* List of forward primers can be found at http://urology.ucsf.edu/blellochlab/protocols.htm
Prepare Master-Mix (Volumes per reaction) as follows:
| 1. 2x Universal MM | 13.75 μl |
| 2. DEPC-Water | 0.792 μl |
| 3. MgCl2 (100 mM) | 0.55 μl |
| 4. dNTP (100 mM) | 1.1 μl |
| 5. x-plex forward primer mix (450 nM) | 3.06 μl |
| 6. Universal RP (100 μM) | 1.375 μl |
| 7. AmpliTaqGold polymerase (5 U/μl) | 1.375 μl |
Mix and centrifuge briefly.
Add 22 μl Master-Mix to each 5.5 μl RT-Product.
Perform the following Pre-PCR reaction: 95 °C for 10 min, 55 °C for 2 min, followed by 10 - 18 cycles of 95 °C for 1 s and 65 °C for 1 min, 4 °C ∞.
6. Purification of the Pre-PCR Product
Purification by size selection on a 10% native polyacrylamide gel
| Name of the reagent | Company | Prod/Cat # |
| 40% Acrylamide/Bis Solution | Bio-Rad | 161-0144 |
| TEMED | Bio-Rad | 161-0801 |
| 10 bp DNA ladder | Invitrogen | 10821-015 |
| SYBR Gold nucleic acid gel stain 10,000X concentrate in DMSO | Invitrogen | S11494 |
| Glycogen (20 mg/ml) | Roche | 10 901 393 001 |
| Buffer-EB | ||
| 10% Ammoniumpersulfate (APS) | ||
| TEN-Buffer | ||
| 6x Orange G | ||
| DEPC treated Water | ||
| ddH2O |
a. Gel Preparation
To make 10ml of a 10% polyacrylamide mixture add
| 1. ddH2O | 6.5 ml |
| 2. 10x TBE | 1 ml |
| 3. Polyacrylamide (40% ) | 2.5 ml |
| 4. APS (10%) | 80 μl |
| 5. TEMED | 4 μl |
- Prepare DNA ladder:
- 0.5 μl 10 bp DNA ladder
- 4.5 μl Buffer (EB)
- 1 μl 6x Orange G
Add 5.5 μl 6x Orange G to each 27.5 μl Pre-PCR product.
b. Run Gel
Run Gel with 150V for 55-60 minutes. Note: Bromophenol blue as indicator runs with 20bp region.
c. Stain Gel
Stain gel with SYBR-Gold for 25 minutes on shaker. Note: SYBR-Gold is light sensitive.
d. Cut Gel
Cut out Pre-PCR product band from 60-80 bp and transfer to labeled tube. Note: A band for low RNA input Pre-PCR products might not be visible.
Crush gel band (e.g. tube in tube centrifugation).
e. Re-Extract cDNA
Add 300 μl TEN-Buffer and incubate at 37 °C for 4 hours on a rotator.
Transfer fluid to a labeled collection tube, add another 300 μl TEN-Buffer to the gel-containing tube and incubate all at 4 °C overnight on a rotator.
Aspirate remaining liquid, transfer to the labeled collection tube as before and note the total recovered volume.
f. Ethanol Precipitation
Add 3 volumes of room temperature 100% ethanol.
Add 0.5 μl Glycogen (20 mg/ml).
Vortex.
Incubate on dry ice for at least 2 hours or at -80 °C overnight.
Spin in microcentrifuge at 4 °C for 30 minutes to pellet cDNA.
Dump fluid, add 700 μl room temperature 75% ethanol and spin for 10 minutes.
Dump fluid and spin again for 5 minutes.
Suck off supernatant without disturbing pellet and air dry for 10 minutes.
Redissolve the pellet in clean water.
Note: Desired volume of concentrated cDNA Solution is dependent on experimental design and should include control experiments.
7. Purification of the Pre-PCR Product
Purification by enzymatic digestion of primers followed by spin column (following the manufactures protocols)
| Name of the reagent | Company | Prod/Cat # |
| ExoSAP-IT | USB-Affymetrix | 78250 |
| MinElute PCR Purification Kit | Qiagen | 28004 |
Note: In our hands the exclusive enzymatic clean-up successfully removed primers but also led to partial degradation of the pre-amplified product, possibly due to partial denaturing of the short products as temperature rises up to 80 °C during the inactivation step of the enzyme. Avoiding heat inactivation and directly purifying the PCR products from the enzymatic reaction using spin columns removes any primer without product degradation.
Mix 5 μl post-PCR product with 2 μl ExoSAP-IT.
Incubate at 37 °C for 15 minutes to degrade remaining primers and nucleotides.
Add 5 volumes of Buffer PB to 1 volume of the PCR reaction and mix, if pH indicator I has been added to Buffer PB, check that the color of the mixture is yellow.
Place a MinElute column in a provided 2 ml collection tube in a suitable rack.
Apply the sample to the MinElute column and centrifuge for 1 minute.
Discard flow-through, place the MinElute column back into the same tube and add 750 μl Buffer PE to the MinElute column to wash and centrifuge for 1 minute.
Discard flow-through and place the MinElute column back in the same tube. Centrifuge the column for an additional 1 minute at maximum speed.
Place the MinElute column in a clean 1.5 ml microcentrifuge tube.
To elute DNA, add 10 μl DEPC treated water to the center of the membrane, let the column stand for 1 minute, and then centrifuge for 1 minute.
8. Fludigim 96.96 qRT-PCR Profiling
| Name of the reagent | Company | Prod/Cat # | Comments |
| Fluidigm 96.96 Dynamic Array Kit 96.96 dynamic array 96.96 control line fluid 20x Loading Reagent | Fluidigm Corporation | ||
| Mix of Universal Reverse Primer (1 μM) Forward Primer (1 μM) TaqMan Probe (0.2 μM) | Integrated DNA Technologies | Custom | Sequence (1) |
| 2x Universal PCR Master Mix with NoAmpErase UNG | Applied Biosystems | 4324018 | |
| Tween |
1. Priming the 96.96 Dynamic Array IFC
Inject control line fluid (150 μl) into each of the accumulators on the chip.
Place chip into the IFC (integrated fluidic circuit) controller and run the Prime (136x) script to prime control line fluid into the chip.
Note: Chip needs to be used 24 hours after opening the package. Make sure not to spill the control line fluid since control line fluid on the chip or in the inlets makes the chip unusable. Chip needs to be loaded within 60 minutes of priming.
2. Preparing the samples
2.1 Pre-Sample Preparation
In a disposable plastic tube, combine the components in the table below to make the Sample-Pre-Mix (Volumes per inlet).
| 2x TaqMan Universal Master Mix | 2.5 μl |
| 20x Loading Reagent | 0.25 μl |
Note: Final Volume is 2.75 μl, as overage for pipette dispense losses
2.2 Sample-Preparation
Combine in 96 well format each sample with pre-sample mix (Volumes per inlet).
| Pre-Sample-Mix | 2.75 μl |
| Sample | 2.25 μl |
Note: Vortex and centrifuge 96 well plate briefly to collect fluid. Final Volume per inlet 5 μl, take pipetting loss into account by preparing slightly higher volume.
2.3 Preparing the 13x Assays
Use a 96 well plate to prepare 13x assays. 1x concentrations in the table below, scale up for 13x.
| Universal Reverse Primer | 1 μm (1x) | Sequence in2 |
| Forward Primer* | 1 μm (1x) | * |
| Gene-specific TaqMan Probe # | 0.2 μm | # |
* List of forward primers can be found at http://urology.ucsf.edu/blellochlab/protocols.htm
# List of TaqMan Probes can be found at http://urology.ucsf.edu/blellochlab/protocols.htm
Combine in a new 96 well plate aliquots of the 13x assays with Tween for a 0.25% final Tween concentration Note: Final Volume per inlet is 5 μl, prepare sufficient volume (5.5 μl) to take loss during volume transfer into account.
Mix well, avoiding bubbles and centrifuge briefly to collect fluid.
3. Loading the chip
Note: In order to allow for correct loading of samples and assays ensure the correct positioning of the chip. It is convenient to align the notch to the top left hand corner.
Ensure all assay and sample solutions are mixed before loading the chip. It is convenient to do this in a 96 well plate and spin the plate down before loading the chip. Be aware of the chip pipetting map for later reference.
Unused sample inlets should be filled with 2.75 μl sample Mix and 2.25 μl DNA free water.
Unused assay inlets should be filled with 2.5 μl assay loading reagent and 2.5 μl water.
Do not go past the first stop while pipetting to avoid introducing air bubbles.
After loading the samples and assays (Volume per inlet: 5 μl) run the Load Mix script (136x) script to load the samples and assays into the chip.
After the script has finished, remove the chip from the ICF controller.
Peel the blue protective film from the underside of the loaded chip.
Remove any dust particles or debris from the chip surface.
4. 96.96 qRT-PCR
Transfer the chip to the BioMark system and use the following the amplification conditions: 55 °C for 2 min, 95 °C for 10 min, followed by 40 cycles of 95 °C for 15 s and 55 °C for 1 min.
Follow manufacture's protocol for data collecting software.
5. Using the qPCR analysis software
After RT- PCR the proprietary software provides amplification curves, heat maps and Ct values for each well. Please refer to the software manual for data analysis.
9. Representative Results:
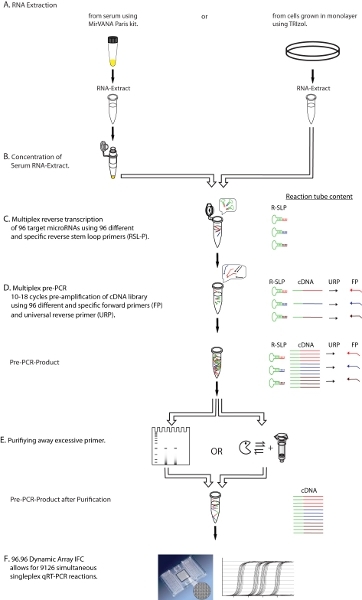 Figure 1. Diagrammatic representation of the experimental workflow. Shown is a step-by-step description of the multiplex qRT-PCR protocol, including the purification (Step E) of excessive primers from multiplex reverse transcription (Step C) and multiplex preamplification (Step D) before real time detection (Step F), resulting in a purified cDNA template for qRT-PCR. FP: microRNA specific forward primer, URP: universal reverse primer, R-SLP: microRNA specific reverse stem-loop primer. Click here for a larger image.
Figure 1. Diagrammatic representation of the experimental workflow. Shown is a step-by-step description of the multiplex qRT-PCR protocol, including the purification (Step E) of excessive primers from multiplex reverse transcription (Step C) and multiplex preamplification (Step D) before real time detection (Step F), resulting in a purified cDNA template for qRT-PCR. FP: microRNA specific forward primer, URP: universal reverse primer, R-SLP: microRNA specific reverse stem-loop primer. Click here for a larger image.
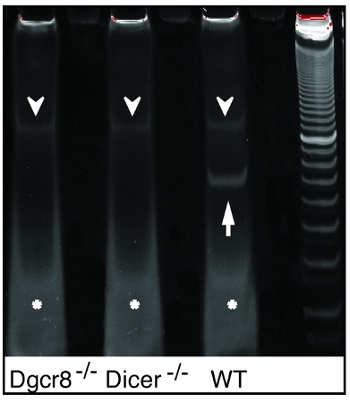 Figure 2. Polyacryamide gel purification of the qRT-PCR template after pre-PCR. Bands of pre-PCR product size are exclusively seen in WT samples, but not in microRNA deficient DGCR8-/- or Dicer-/- samples (arrows) after 96-plex RT and 12 cycles of preamplification. Note that non-specific products of unknown origin (arrowheads) and excessive primers are found in all samples (stars).
Figure 2. Polyacryamide gel purification of the qRT-PCR template after pre-PCR. Bands of pre-PCR product size are exclusively seen in WT samples, but not in microRNA deficient DGCR8-/- or Dicer-/- samples (arrows) after 96-plex RT and 12 cycles of preamplification. Note that non-specific products of unknown origin (arrowheads) and excessive primers are found in all samples (stars).
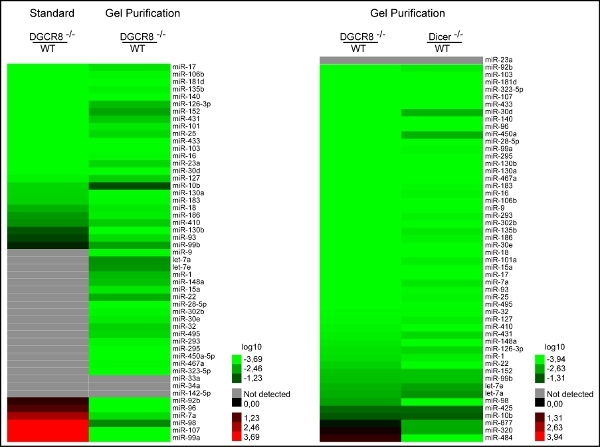 Figure 3. Purification after multiplex preamplification vastly improves accuracy of qRT-PCR results. a) Comparison of relative expression levels of WT mESC to Dgcr8-/- (canonical microRNA deficient) mESC reveals 1) the detection of more microRNAs and 2) the loss of false positive signals in Dgcr8-/- backgrounds after purifying away primers of the pre-PCR product. b) After purification, relative expression levels of both DGCR8 -/-/WT and Dicer -/-/WT show a loss of false positive signals and allow for proper categorization of rare Dgcr8 independent /Dicer dependent small RNAs (miR-320, -484, -877).5
Figure 3. Purification after multiplex preamplification vastly improves accuracy of qRT-PCR results. a) Comparison of relative expression levels of WT mESC to Dgcr8-/- (canonical microRNA deficient) mESC reveals 1) the detection of more microRNAs and 2) the loss of false positive signals in Dgcr8-/- backgrounds after purifying away primers of the pre-PCR product. b) After purification, relative expression levels of both DGCR8 -/-/WT and Dicer -/-/WT show a loss of false positive signals and allow for proper categorization of rare Dgcr8 independent /Dicer dependent small RNAs (miR-320, -484, -877).5
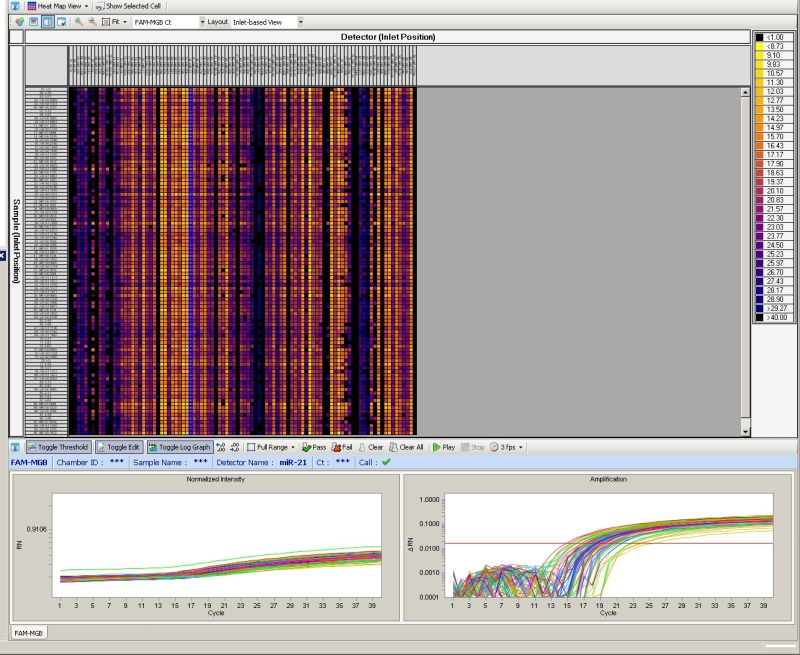 Figure 4. Fluidigm high-throughput qRT-PCR miRNA profiling. Example screen shot of 96.96 Fluidigm qRT-PCR mircoRNA profiling to screen for alteration in miRNA levels of prostate cancer patient sera. The real-time qPCR analysis software provides amplification curves, color-coded heat maps and cycle threshold (Ct).
Figure 4. Fluidigm high-throughput qRT-PCR miRNA profiling. Example screen shot of 96.96 Fluidigm qRT-PCR mircoRNA profiling to screen for alteration in miRNA levels of prostate cancer patient sera. The real-time qPCR analysis software provides amplification curves, color-coded heat maps and cycle threshold (Ct).
Discussion
miRNAs are short (18-24 nucleotides), non-coding RNAs, which regulate gene expression post-transcriptionally by both destabilizing messenger RNAs (mRNA) and inhibiting their translation.6 The fact that at least one-third of human genes contain conserved miRNA binding-sites in their 3'UTR and the evident interactions of miRNAs with genes for pluripotency, proliferation and apoptosis suggests a critical contribution in cell fate decisions, tissue homeostasis and diseases such as cancer.6,7 Therefore accurate microRNA expression profiles are of broad interest.
To test the published multiplex qRT-PCR miRNA expression profiling protocol2 we used small RNA deficient mESC as negative controls. DGCR8-/- cells are deficient of all canonical microRNAs, while Dicer-/- cells lack both canonical and non-canoncial micoRNAs.8,9
Comparing levels in wild type mES- to DGCR8-/- cells, we uncovered a lack of accuracy as several expressed10 microRNAs were not detected in the wild-type cells11. Some even showed lower expression levels relative to the knockout cells (Figure 3a). We hypothesized that the lack of accuracy might be caused by the massive primer carry over of the two consecutive multiplex steps (RT and Pre-PCR) before singleplex RT-quantification. However, multiplex pre-amplification is necessary to enhance signal strength, especially when starting concentration of RNA is limited. By purifying away pre-PCR products from primers by size selection on native polyacrylamide gels (Figure 2), we could demonstrate a substantial improvement in accuracy by detecting more microRNAs and a loss of false positive signals in both Dgcr8-/- and Dicer-/- backgrounds (Figures 3a and 3b) In addition the modified qRT-PCR approach allowed for the proper categorization of rare Dgcr8 independent / Dicer dependent small RNAs (Figure 3b)11. Therefore an additional step to purify excessive primers away from pre-PCR product is clearly advantageous, especially when using large multiplex primer sets per sample.
The optimal number of pre-amplification cycles is determined by the input RNA concentrations. The balance to not under- or overamplify should be guided by particulars of the specific experiment.
With more than a thousand known microRNAs, use of standard 384-well plates may not be optimal for extensive microRNA profiling, especially when comparing multiple samples. The Fluidigm Dynamic Array IFC enables, with a significant decrease of pipetting steps and needed chemistry, testing up to 96 individual samples against 96 different microRNAs in a single experiment (9216 reactions) at nanoliter scale (6.7 nL). For each run the analysis software provides amplification curves, color-coded heat maps and cycle threshold values (Ct). The simultaneous large-scale profiling reduces experimental variances and allows for mean expression value normalization, which out-performs other normalization strategies that make use of small RNA controls.12
Compared to commercially available 384 well platforms with pre-assigned TaqMan probes, the combination of a high-throughput profiling platform with custom-made primer sets offers high experimental flexibility.
The optimized multiplex qRT-PCR approach in combination with the Dynamic Array platform successfully allowed us to simultaneously screen 48 prostate cancer patient sera for alterations in levels of 384 miRNA (Figure 4) and to normalize data without spike in controls (i.e. synthetic microRNAs).11
Despite the increase of steps from samples preparation to profiling results, the described approach is a time- and cost-effective high throughput method to profile large sample sets for miRNA expression levels, even with limited starting material.
Disclosures
Alan Mir is an employee of Fluidigm Corporation. Otherwise, we have no financial interests to disclose.
Acknowledgments
We would like to thank the Blelloch lab for commenting on the text. This work was supported by funds to RB from NIH (K08 NS48118 and R01 NS057221), California Institute of Regenerative Medicine (CIRM) (Seed Grant RS1-00161, New Faculty Award RN2-00906) and the Pew Charitable Trust and to F.M. from the Wissenschaftlich Urologische Gesellschaft eV.
References
- Mitchell PS. Circulating microRNAs as stable blood-based markers for cancer detection. Proc Natl Acad Sci U S A. 2008;105:10513–10518. doi: 10.1073/pnas.0804549105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang F. 220-plex microRNA expression profile of a single cell. Nat Protoc. 2006;1:1154–1159. doi: 10.1038/nprot.2006.161. [DOI] [PubMed] [Google Scholar]
- Gertsenstein M, Vintersten K, Behringer R. Manipulating the Mouse Embryo: A Laboratory Manual. Vol. 3. Cold Spring Harbor Laboratory Press; 2003. pp. 644–645. [Google Scholar]
- Suh N. MicroRNA function is globally suppressed in mouse oocytes and early embryos. Curr Biol. 2010;20:271–277. doi: 10.1016/j.cub.2009.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiarz JE, Ruby JG, Wang Y, Bartel DP, Blelloch R. Mouse ES cells express endogenous shRNAs, siRNAs, and other Microprocessor-independent, Dicer-dependent small RNAs. Genes Dev. 2008;22:2773–2785. doi: 10.1101/gad.1705308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babiarz JE. Small RNAs: their biogenesis, regulation and function in embryonic stem cells. StemBook. 2009. [PubMed]
- Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome Res. 2008;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Medvid R, Melton C, Jaenisch R, Blelloch R. DGCR8 is essential for microRNA biogenesis and silencing of embryonic stem cell self-renewal. Nat Genet. 2007;39:380–385. doi: 10.1038/ng1969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanellopoulou C. Dicer-deficient mouse embryonic stem cells are defective in differentiation and centromeric silencing. Genes Dev. 2005;19:489–501. doi: 10.1101/gad.1248505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomson JM, Parker J, Perou CM, Hammond SM. A custom microarray platform for analysis of microRNA gene expression. Nat Methods. 2004;1:47–53. doi: 10.1038/nmeth704. [DOI] [PubMed] [Google Scholar]
- Moltzahn F. Microfluidic based multiplex qRT-PCR identifies diagnostic and prognostic microRNA signatures in sera of prostate cancer patients. Cancer Res. 2011;71:550–560. doi: 10.1158/0008-5472.CAN-10-1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mestdagh P. A novel and universal method for microRNA RT-qPCR data normalization. Genome Biol. 2009;10:R64–R64. doi: 10.1186/gb-2009-10-6-r64. [DOI] [PMC free article] [PubMed] [Google Scholar]