

Abstract
Normal brain function relies not only on embryonic development when major neuronal pathways are established, but also on postnatal development when neural circuits are matured and refined. Misregulation at this stage may lead to neurological and psychiatric disorders such as autism and schizophrenia1,2. Many genes have been studied in the prenatal brain and found crucial to many developmental processes3-5. However, their function in the postnatal brain is largely unknown, partly because their deletion in mice often leads to lethality during neonatal development, and partly because their requirement in early development hampers the postnatal analysis. To overcome these obstacles, floxed alleles of these genes are currently being generated in mice 6. When combined with transgenic alleles that express Cre recombinase in specific cell types, conditional deletion can be achieved to study gene function in the postnatal brain. However, this method requires additional alleles and extra time (3-6 months) to generate the mice with appropriate genotypes, thereby limiting the expansion of the genetic analysis to a large scale in the mouse brain.
Here we demonstrate a complementary approach that uses virally-expressed Cre to study these floxed alleles rapidly and systematically in postnatal brain development. By injecting recombinant adeno-associated viruses (rAAVs)7,8 encoding Cre into the neonatal brain, we are able to delete the gene of interest in different regions of the brain. By controlling the viral titer and coexpressing a fluorescent protein marker, we can simultaneously achieve mosaic gene inactivation and sparse neuronal labeling. This method bypasses the requirement of many genes in early development, and allows us to study their cell autonomous function in many critical processes in postnatal brain development, including axonal and dendritic growth, branching, and tiling, as well as synapse formation and refinement. This method has been used successfully in our own lab (unpublished results) and others8,9, and can be extended to other viruses, such as lentivirus 9, as well as to the expression of shRNA or dominant active proteins 10. Furthermore, by combining this technique with electrophysiology as well as recently-developed optical imaging tools 11, this method provides a new strategy to study how genetic pathways influence neural circuit development and function in mice and rats.
Protocol
1. Preparing viruses for injection
rAAVs were purchased from the recommended commercial vendor, but they can also be produced in one's own lab (see discussion below). The virus solution is typically produced at a titer of ˜1x1012 genome copies per milliliter (GC/ml) and may be used at full titer to manipulate a large number of cells. Alternatively, they may be diluted to produce the desired level of sparse labeling. The appropriate dilution must be determined by the user, but a 1:10 dilution is recommended to start.
Thaw viruses on ice immediately before use. Transfer the desired pre-dilution volume of virus solution into a small (˜250μl) PCR tube. Dilute the virus solution using an appropriate volume of sterile DPBS in a tissue culture hood. Store diluted virus solution on ice until it is needed.
NOTE: handling of all viruses should be conducted in accordance with a biosafety protocol approved by your institution
2. Syringe loading and equipment assembly
Connect the loading needle to the glass syringe and draw up the virus solution from the PCR tube by gently pulling the plunger. Pull the plunger until a very small bubble of air can be seen in the syringe; this indicates that all of the solution is in the syringe and is not left in the loading needle. Alternatively, a small amount of inert dye can be added to the solution to make it visible. NOTE: A small PCR tube should be used as the loading needle is typically not long enough to reach the bottom of most other tubes.
Remove the loading needle and place the syringe on the syringe block of a syringe pump, securing it with the syringe retainer.
Gently insert one end of the connective tubing into the syringe; insert the other end into the needle holder. Insert the injection needle into the needle holder. The end of the injection needle should directly contact the end of the connective tubing inside of the needle holder.
Slowly depress the plunger manually to push the solution through the connective tubing. Push until a tiny drop of solution is visible at the tip of the injection needle.
Carefully position the plunger next to the pusher block and secure it in place with the pusher block bracket.
Set the injection parameters: injection rate should be approximately 8μl/min (˜130nl/s); injection volume is typically 1-2ul; select the appropriate syringe diameter for the syringe size you are using (see the syringe pump manual for guidelines).
Set the hotplate to ˜38°C and cover with a paper towel or other thin barrier to protect the pups during the recovery phase.
3. Injection procedure and pup recovery
Prepare P0 or P1 mouse pups for injection by subjecting them to cold anesthesia following an IACUC approved protocol. Dampen a few paper towels with water and place on ice. Place the desired number of pups (4-5) on the paper towel (keep them well separated) and gently fold the paper towels over the pups so they are covered by damp paper towel. Gently place a small amount of crushed ice on top of the paper towels and incubate the pups for approximately 5 minutes. The pups can be kept on ice for up to 15 minutes and recover fine afterwards.
After the pup is sufficiently anesthetized, place it on a mounting block (we use a Styrofoam block that normally holds 15ml tubes) and position it for best access to the injection site. For the cortex it is easiest to lay the animal flat on its belly, while for the cerebellum it is helpful to position the pup's head over the edge of the block to provide better access to the rear of the skull. The pup may be secured in place with a band-aid, if desired.
Hold the pup's head in place with one hand while the needle is manually inserted through the skull at the desired site with the other hand. It is helpful to pull the skin tight to prevent it from moving during injection. For the cortex it is useful to make the injections relative to a well-defined anatomical landmark, such as the lambda suture of the skull, as this will aid in future injections. The cerebellum can be seen through the skull as a thin strip of tissue lying directly caudal to the colliculus. The depth to which the needle should be inserted varies slightly between individual animals and strains and can be gauged by predetermined markers on the needle, however for cortex and cerebellum we find that a depth of ˜0.5 - 1mm from the surface is usually suitable. The insertion depth for other sites should be determined experimentally by the end user. NOTE: The needle should easily penetrate the skull; if it does not, DO NOT push too hard as this can injure the animal. Reposition the needle and gently try again.
Inject the virus solution. The injection is started by pressing the START button on the pump or depressing a foot pedal, depending on the model of pressure injector used. In the absence of a foot pedal it is recommended to have a colleague assist with pressing the button to minimize unnecessary hand movements that may affect the injection.
After the selected volume has been injected, keep the needle in place for a few seconds and then gently remove the needle by sliding it out smoothly. Be sure to hold the pup's head in place to minimize extraneous movements. Since the injection equipment is cleaned and sterilized between experiments with ethanol and the wound is small, the use of antiseptic is unnecessary.
Place the pup on the covered hotplate with the temperature set at ˜38°C to warm it up for recovery, typically 5-10 minutes. During this time you may continue injecting other pups.
After all of the pups have recovered (they should be pink and moving) take a portion of the home bedding and gently rub the pups with it. Return the pups to the mother as a group.
NOTE: It is very important to make sure the pups are warm and exposed to home bedding prior to returning them to the mother and to return them as a group. Failure to do so may result in the mother injuring or killing the pups.
4. Equipment cleaning
After the injection is complete and the pups have been returned to their mother, clean the injection setup by fully loading the syringe with acetone or 70% ethanol. Flush the solution through the injection setup several times, using fresh solution each time. This will remove any remaining virus solution and precipitates. The acetone or ethanol will then evaporate. NOTE: Acetone is preferred for cleaning, but if your setup uses any acetone-sensitive components, such as cyanoacrylate adhesives, then use 70% ethanol for these steps.
Disinfect the working area with bleach and discard waste in an appropriate biohazard container.
5. Analysis
Sacrifice the animals at the desired age using an IACUC approved euthanasia method. Perfusion fixation is recommended to aid in later visualization of infected neurons, particularly in adult animals.
Standard immunostaining procedures may be used to amplify the fluorescent signal. Briefly, 100 μm floating vibratome sections are permeabilized with PBS+1% Triton X-100 and blocked in PBS+1% Triton X-100+10% normal goat serum. Sections are incubated in blocking solution with primary antibodies overnight at 4°C, followed by additional washing and blocking. Sections are then incubated in blocking solution with secondary antibodies for 2 hours at room temperature, washed thoroughly, mounted on slides and coverslipped with anti-fade mounting media. Cryosections can be also used to visualize labeled neurons, however the endogenous fluorescence might be lost.
6. Representative Results:
A successful injection procedure will produce robust infection of neurons in the region of the injection site with no observable tissue damage or other ill effects on the animal. As illustrated in Figure 1, the spread of the infection can be observed in Cre-reporter mice (ROSA26R)12 injected with rAAV8-Cre in various anatomical sites. Injections into specific regions, such as cortex and superior colliculus, typically generate local infections with minimal spread to adjacent regions (Fig. 1A-B). Use of high titer virus solution makes infection of adjacent areas, such as the hippocampus, more likely (Fig. 1A). Injection with low titer virus solution results in sparse infection, which typically remains near the injection site, as shown in the cerebellar injection of rAAV8-Cre at the midline of ROSA26R reporter mice (Figure 1C-D). We have observed recombination in ROSA26R reporter mice within 2 days after injection of rAAV8-Cre, suggesting that infection, recombination, and expression of reporter genes can all occur relatively rapidly. Additionally, we have successfully used this technique to study the effects of gene deletion at a non-ROSA locus using a floxed conditional allele, the results of which will be published elsewhere.
The number of cells infected should correlate with the titer of the injected viral solution. For example, injecting a mix of high titer solutions of two rAAV8 viruses expressing different fluorescent proteins into the cerebellum infects many Purkinje cells that are differentially labeled (Fig. 2A). Conversely, injecting a low titer virus solution can result in sparse infection to aid in visualizing single cells (Fig. 2B). Fluorescently labeled neurons can be observed directly in fresh cut, live tissue (Fig. 2B) or can be subjected to immunostaining to enhance the endogenous fluorescent signals for later image acquisition by wide-field or confocal fluorescent microscopy (Fig. 2A, Fig. 3A-C). Lastly, rAAV8 is capable of infecting a variety of neuron types in the cortex with strong labeling of fine processes (Fig. 3A-C).
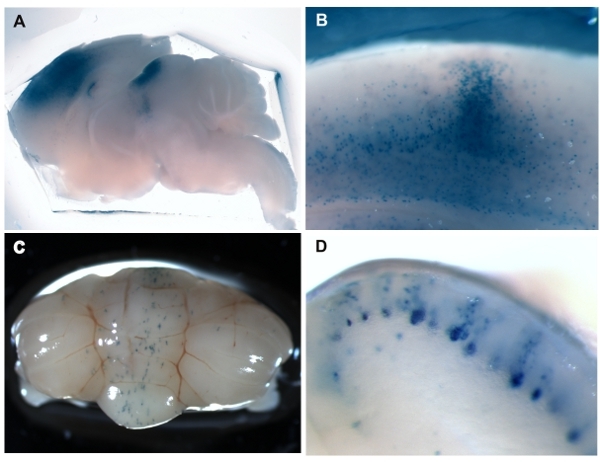 Figure 1: LacZ staining in the brain of ROSA26R reporter mice that were injected with rAAV8-Cre at P0 demonstrates Cre activity and the location of infection.
Figure 1: LacZ staining in the brain of ROSA26R reporter mice that were injected with rAAV8-Cre at P0 demonstrates Cre activity and the location of infection.
Wide-field sagittal view of a P14 brain showing high titer (1x1012 GC/ml) injections into cortex and superior colliculus. The use of high titer virus makes it more likely that adjacent areas will also be infected.
Sagittal section of P14 cortex injected with lower titer (1x1011 GC/ml) virus demonstrates a local infection with less spreading.
A wholemount view of a P28 cerebellum injected at the midline with low titer (1x109 GC/ml) virus solution shows that the infection is sparse and typically remains near the injection site.
Sagittal view of the midline of the cerebellum in C). Note that generally only Purkinje cells are infected by rAAV8.
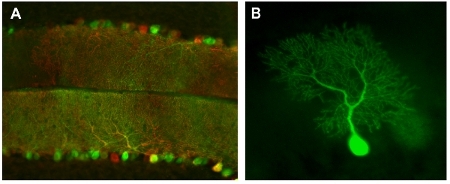 Figure 2: Cerebellar labeling by injection with high and low titers of rAAV8 expressing fluorescent proteins.
Figure 2: Cerebellar labeling by injection with high and low titers of rAAV8 expressing fluorescent proteins.
A sagittal section of P21cerebellum infected with a mix of two viruses expressing EGFP (green) and DsRed (red). A high titer mix of viruses was used to infect a large number of Purkinje cells.
A purkinje cell in live P14 cerebellum from very low titer viral infection imaged immediately after dissection.
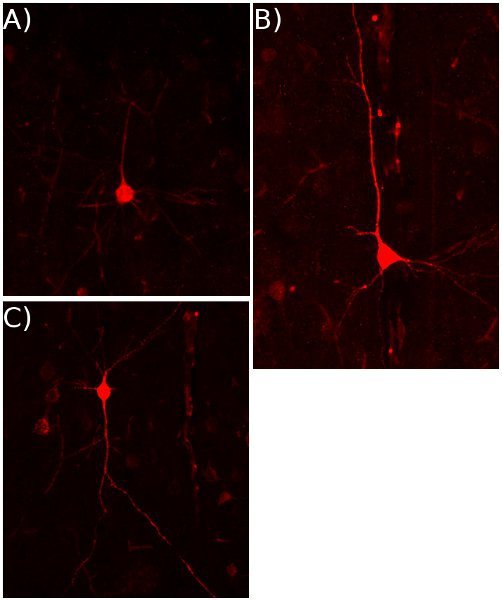 Figure 3: Cortical labeling by rAAV8-DsRed.
Examples of the variety of cortical neuron types in the coronal sections of a P21 cortex that was infected by rAAV8-DsRed at P0.
Figure 3: Cortical labeling by rAAV8-DsRed.
Examples of the variety of cortical neuron types in the coronal sections of a P21 cortex that was infected by rAAV8-DsRed at P0.
Discussion
The neonatal viral injection method presented here provides a simple and rapid way to generate in vivo mosaics for the study of postnatal brain development. The method takes advantage of floxed alleles that are currently available as well as those that are being made through the High Throughput Gene Targeting project6. Compared to the use of transgenic expression of Cre, this method provides a rapid way to test gene function in various cell types, as mice carrying the floxed alleles can be used directly for experiments, and the entire virus injection procedure from start to finish can be accomplished in under an hour. In addition, the number of cells infected can be easily controlled by altering the titer of the virus, which allows sparse labeling of individual neurons. Although we only described the neuronal morphology in the sample images, the analysis of infected neurons can be expanded to many important developmental processes, including axonal and dendritic growth, branching and tiling, as well as synaptic development such as spine morphogenesis.
The three most critical parts of the procedure are the accuracy of targeting the injection site, the reliability of the injection equipment, and the survival of the pups. The first aspect – accuracy – is best learned through experience. The reliability of the injection equipment refers to the fact that one must make sure there are no clogs, leaks, or other problems with the equipment prior to injection. Lastly, survival of the pups is typically not a problem if the tips mentioned in Step 3.7 are followed.
An obvious modification of this procedure is to use a mixture of viruses expressing different genes. For example, a mixture of two viruses, rAAV-Cre-GFP and rAAV-RFP, can be injected to produce a mosaic of sparsely labeled knockout (green) and control (red) cells in a single animal. This provides a distinct advantage over traditional gene knockouts for the analysis of single cells, making it particularly useful for morphological analysis of neurons with wild type or mutant alleles in the same mouse brain. This can also be a limitation, however, if one wishes to perform behavioral studies, in which it may be necessary to manipulate a large number of neurons to produce an observable effect. We find that we can generally perform injections up to postnatal age 3 (P3), however from age P4 and on the skull is often too thick to easily puncture with the injection needle. The thickness of the skull at a particular age can vary from animal to animal, however, so this issue must be assessed by the experimenter.
In addition to using fluorescent proteins expressed from the Cre-expressing virus, many fluorescent reporter mice with the loxP-STOP-loxP alleles have been generated13 and can be incorporated with the floxed allele. Although including the new allele adds the time needed for obtaining the mice with appropriate genotypes, these reporter mice can simultaneously monitor Cre activity and label individual neurons.
Because of the selective tropism of recombinant rAAVs, different neuronal cell types can be labeled in anatomical regions by choosing a particular rAAV serotype (Figure 1D). The specificity can be also achieved by using different promoters. In addition, other viruses, such as lentivirus, can be used to infect neurons10. The advantage here is that these viruses can carry larger DNA inserts to express not only Cre but also other genetic materials, such as shRNA and dominant active genes. Furthermore, the method can be also used in the newly developed transgenic rats14, where conditionally expressed Cre lines are not well established. Finally, once this technique is established in the lab, it can be further combined with other analytical tools, such as in vivo two-photon imaging11 and/or electrophysiology, to study the function of each gene in neural circuit development and function.
Disclosures
No conflicts of interest declared.
Acknowledgments
This work is supported by an RO1 grant from NIH (NINDS).
References
- Geschwind DH, Levitt P. Autism spectrum disorders: developmental disconnection syndromes. Curr Opin Neurobiol. 2007;17:103–111. doi: 10.1016/j.conb.2007.01.009. [DOI] [PubMed] [Google Scholar]
- Giedd JN, Rapoport JL. Structural MRI of pediatric brain development: what have we learned and where are we going? Neuron. 2010;67:728–734. doi: 10.1016/j.neuron.2010.08.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chedotal A, Richards LJ. Wiring the brain: the biology of neuronal guidance. Cold Spring Harb Perspect Biol. 2010;2 doi: 10.1101/cshperspect.a001917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen K, Cowan CW. Guidance molecules in synapse formation and plasticity. Cold Spring Harb Perspect Biol. 2010;2 doi: 10.1101/cshperspect.a001842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen SY, Cheng HJ. Functions of axon guidance molecules in synapse formation. Curr Opin Neurobiol. 2009;19:471–478. doi: 10.1016/j.conb.2009.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guan C, Ye C, Yang X, Gao J. A review of current large-scale mouse knockout efforts. Genesis. 2010;48:73–85. doi: 10.1002/dvg.20594. [DOI] [PubMed] [Google Scholar]
- Tenenbaum L. Recombinant AAV-mediated gene delivery to the central nervous system. J Gene Med. 2004;6:212–222. doi: 10.1002/jgm.506. [DOI] [PubMed] [Google Scholar]
- Broekman ML, Comer LA, Hyman BT, Sena-Esteves M. Adeno-associated virus vectors serotyped with AAV8 capsid are more efficient than AAV-1 or -2 serotypes for widespread gene delivery to the neonatal mouse brain. Neuroscience. 2006;138:501–510. doi: 10.1016/j.neuroscience.2005.11.057. [DOI] [PubMed] [Google Scholar]
- Pilpel N, Landeck N, Klugmann M, Seeburg PH, Schwarz MK. Rapid reproducible transduction of select forebrain regions by targeted recombinant virus injection into the neonatal mouse brain. J Neurosci Methods. 2009;182:55–63. doi: 10.1016/j.jneumeth.2009.05.020. [DOI] [PubMed] [Google Scholar]
- Szulc J, Aebischer P. Conditional gene expression and knockdown using lentivirus vectors encoding shRNA. Methods Mol Biol. 2008;434:291–309. doi: 10.1007/978-1-60327-248-3_18. [DOI] [PubMed] [Google Scholar]
- Pan F, Gan WB. Two-photon imaging of dendritic spine development in the mouse cortex. Dev Neurobiol. 2008;68:771–778. doi: 10.1002/dneu.20630. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat Genet. 1999;21:70–701. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Madisen L. A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat Neurosci. 2010;13:133–1340. doi: 10.1038/nn.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong C, Li P, Wu NL, Yan Y, Ying QL. Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature. 2010;467:211–213. doi: 10.1038/nature09368. [DOI] [PMC free article] [PubMed] [Google Scholar]