

Abstract
By virtue of advances in next generation sequencing technologies, we have access to new genome sequences almost daily. The tempo of these advances is accelerating, promising greater depth and breadth. In light of these extraordinary advances, the need for fast, parallel methods to define gene function becomes ever more important. Collections of genome-wide deletion mutants in yeasts and E. coli have served as workhorses for functional characterization of gene function, but this approach is not scalable, current gene-deletion approaches require each of the thousands of genes that comprise a genome to be deleted and verified. Only after this work is complete can we pursue high-throughput phenotyping. Over the past decade, our laboratory has refined a portfolio of competitive, miniaturized, high-throughput genome-wide assays that can be performed in parallel. This parallelization is possible because of the inclusion of DNA 'tags', or 'barcodes,' into each mutant, with the barcode serving as a proxy for the mutation and one can measure the barcode abundance to assess mutant fitness. In this study, we seek to fill the gap between DNA sequence and barcoded mutant collections. To accomplish this we introduce a combined transposon disruption-barcoding approach that opens up parallel barcode assays to newly sequenced, but poorly characterized microbes. To illustrate this approach we present a new Candida albicans barcoded disruption collection and describe how both microarray-based and next generation sequencing-based platforms can be used to collect 10,000 - 1,000,000 gene-gene and drug-gene interactions in a single experiment.
Keywords: Biochemistry, Issue 54, chemical biology, chemogenomics, chemical probes, barcode microarray, next generation sequencing
Protocol
1. Background information
There are several ways to generate mutants that carry barcode tags. The current gold standard is the Yeast KnockOut (YKO) collection created by a consortium of labs and completed in 2002 1. Since the original YKO was introduced, other yeast collections have been generated; in different strain backgrounds, using over-expression constructs, and in other microbes such as E. coli 2. In parallel, the effort to create barcoded shRNA libraries is proceeding rapidly, and in fact, many of the design principles for these mammalian collections have been adopted from yeast. To demonstrate how barcoded transposons can be a rapid, widely applicable strategy for creating systematic mutant collections, we focus on one collection we recently created in the human fungal pathogen, Candida albicans. Our work on Candida was based on the success of barcode screens in S. cerevisiae, and is used here as an example organism. The sample protocol, can with minor modifications be used to screen any organism that can be grown in suspension culture. Because few organisms have the requisite high rates of transformation and efficient mitotic recombination needed to create perfect deletion mutants, accordingly we developed a protocol that uses transposon mutagenesis in vitro to mutagenize a genomic DNA library, and then transformed these barcoded genomic fragments into Candida albicans 3, 4. Inspired by the success of the original YKO collection and its role in fundamental discoveries on the nature of gene networks 5-8, genome-wide haploinsufficiency 9, drug target and mechanism of action 10,11, and the essentiality of all genes in the genome 12 we anticipate broadening this approach to other microbes will be extremely fruitful.
The protocol below assumes that the desired mutant collection has been created (e.g. YKO or Candida albicans disruption collection) and is available as individually archived strains. For a detailed description of strain construction see 1,13,14.
2. Combine individual mutants into a single pool
Allow one week to generate pooled cell aliquots (can be stored indefinitely at -80°C).
Thaw the frozen glycerol stocks for the strains of interest completely but do not let cells remain thawed for >2hrs.
Sterilize a 96-well pin tool, dip the pin tool in water to remove any remaining cells, followed by 2 dips in 70% ethanol baths (e.g. pipette tip box lids), flame the pin tool and cool for 1 minute. Take care to flame the pin tool away from the ethanol baths. The level of the ethanol baths should exceed the level in the water bath to ensure all carry-over cells are flamed and removed. Replace water every 4-6 pinnings.
Insert the sterile 96-well pin tool into a thawed 96-well plate, swirl gently and transfer cells to a Nunc Omni Tray containing YPD-agar including the appropriate antibiotic. Grow colonies until they reach maximal size at 30°C (2-3d). To conserve plates, we find it most useful to consolidate four 96 well plates onto a single Omni-tray with ˜384 strains.
After colonies have grown, note any missing or slow growing strains and repin these at ˜2X the cell mass as the rest of the strains.
Working in a microbiology environment (with flame and sterile labware, flood plate with 5-10 ml media, soak for 5 min and resuspend colonies with a cell spreader. Pour the liquid plus cells into a 50 ml conical centrifuge tube, and add glycerol to 15% or DMSO to 7% (vol/vol).
Measure the OD600 of the pool and adjust (by dilution or centrifugation) to a final concentration of 50 OD600/ml with media containing 15% glycerol or 7% DMSO.
Aliquot in 40 μl volumes in PCR strip tubes and freeze at -80°C.
3. Experimental pool growth
This procedure is outlined in Figure 1.
Thaw pool aliquots (in PCR tubes) on ice. If not using robotics, skip to step 5.
Immediately dilute (gently!) the pool into media with drug or condition of choice, inoculating at an OD600 of 0.0625 in a total volume of 700 μl in a 48-well plate. Include at least one appropriate solvent control on the plate. For experiments that extend beyond 5 generations of growth (i.e. more than 1 well), fill adjacent wells with media or condition of choice, but NO cells.
Seal with a plastic plate seal; if the condition requires aerobic growth (e.g., non-fermentable carbon sources), use a 21 gauge needle to pierce the holes in the membrane seal towards the side of each well.
Grow in a spectrophotometer, shaking at 30°C with an experimentally determined shaking regimen (e.g. shake 14 min at highest setting (or temperature-controlled shaker), read wells, resume shaking). Part of the cell suspension can be harvested by the robot and saved on a cold plate on the robotic deck at user-defined generation time points, typically 5, 10, 15 and 20 generations of growth. (http://med.stanford.edu/sgtc/technology/access.html, for details contact C. Nislow or G. Giaever).
For manual cell growth, inoculate a 50 ml culture at a starting OD600 of 0.002 in a 250 ml culture flask. Shake at 30°C at 250 rpm until cells reach a final OD600 of 2.0 (for Saccharomyces or Candida) for ˜10 generations of growth. Additional generations of growth can be obtained by diluting cells at an OD600 of 2.0 back to 0.02 in a fresh flask.
Harvest at least ˜2 OD600 units of cells for each sample/time-point into Safe-Lock microcentrifuge tubes.
Note: Always collect a starting cell sample (i.e. a "T0 time point") to assess initial strain representation in any newly created pool by adding 1-2 OD600 of pool directly from the freezer aliquots to a 1.5 ml tube and processing as described below.
4. Genomic DNA extraction, PCR and microarray hybridization or sequencing
Purify genomic DNA from ˜2 OD600 of cells with the Zymo Research YeaStar kit according to manufacturer's instructions (Protocol I if using yeast DNA), or another suitable method specific to the organism of interest (standard phenol/chloroform extraction followed by alcohol precipitation works well for diverse microbes). If using the YeaStar kit, elute the DNA with 300 μL of 0.1X TE instead of the 60 μL of 1X TE specified in the protocol. Genomic DNA can be stored indefinitely at -80°C.
Set up two PCR reactions for each sample, one for the uptags and one for the downtags, with the reaction conditions as follows: 33 μl ddH2O, 6 μl 10X PCR buffer without MgCl2, 3 μl 50 mM MgCl2, 1.2 μl 10 mM dNTPs, 1.2 μl 50 μM Up or Down primer mix, 0.6 μl 5 U/μl Taq polymerase, ˜0.1 μg genomic DNA in 15 μl. Total volume is 60 μl. Thermocycle under the following conditions: 94°C 3 min, 30 cycles of 94°C 30s, 55°C 30s, 72°C 30s; then 72°C 3min, and hold at 4°C. Check the resulting PCR products on a gel; a 60 bp product for both PCRs is expected for amplicons used for hybridization and 130bp for amplicons for barcode sequencing). The PCR products can then be stored at -80°C indefinitely.
Prewarm the hybridization oven temperature to 42°C and set up a boiling water bath and ice bucket containing an ice-water slurry.
Pre-wet the arrays by slowly filling with 120 μl 1X hybridization buffer.
Incubate in the hybridization buffer at 42°C and 20 rpm for 10 minutes.
Prepare 90 μl of hybridization mix per sample, plus one extra as a buffer, as follows: 75 μl 2X hybridization buffer, 0.5 μl B213 control oligonucleotide (0.2 fm/μl), 12 μl mixed oligonucleotides (12.5 pm/μl), 3 μl 50X Denhardt's solution) in lock-top 0.5 ml tubes.
Add 30 μl uptag PCR and 30 μl downtag PCR to 120 μl hybridization mix for a total volume of 150 μL. Boil for 2 minutes and set in ice-water for at least 2 minutes. Briefly spin the tubes prior to use.
Remove the pre-hybridization buffer from the arrays and add 90 μl hybridization/PCR mix. To prevent evaporation, cover the array gaskets with a Tough-Spot. Hybridize for 16 hours at 42°C, 20 rpm.
Freshly prepare 600 μl biotin labeling mix per sample plus one extra, as follows: 180 μl 20X SSPE, 12 μl 50X Denhardt's, 6 μl 1% Tween 20 (vol/vol), 1 μl 1 mg/ml streptavidin-phycoerythrin, 401 μl ddH2O. Store all streptavidin-PE samples in the dark. Aliquot 600 μl into 2 ml tubes. Remove Tough-Spots from chips.
Slowly remove hybridization mix from the arrays by pipette and fill microarrays with 120 μl Wash A. Prime the Affymetrix fluidics station.
Wash the arrays using an Affymetrix fluidics station according to the manufacturer's instructions, using "Gene-Flex_Sv3_450" protocol with the following modifications: 1 extra step with Wash A (1 cycle, 2 mixes) before staining, Wash B temperature 42°C instead of 40°C, stain at 42°C instead of 25°C. It is also possible to perform the post hybridization wash, the biotin staining, and the post staining wash manually (see p. 396 in reference 15). Following fluidics operations, run the fluidics station "SHUTDOWN_450" protocol.
Following the wash, ensure no air bubbles are present. If necessary add 90 μl Wash A and pipette slowly until the bubbles disappear. If there are any marks or smudges on the array surface, clean the glass window with isopropanol and a lint-free tissue. Apply fresh Tough-Spots over the gaskets/septa and place the arrays in the scanner.
Scan in an Affymetrix GeneArray scanner at an emission wavelength of 560 nm.
5. Array analysis (see Figure 2 for an example obtained using the Candida albicans disruption collection)
Outlier masking: Because the Affymetrix TAG4 array contains 5 replicates of each barcode complement dispersed randomly any barcode probe that deviates from the replicates can be masked and discarded. To do this, for each array feature that appears to be an outlier based on its signal compared to the 4 other replicates of this feature, our software first examines the 5 features that surround the suspect outlier, producing a matrix of 25 features with the suspect feature at the center. If >13/25 probes in this region differ from each of their individual their trimmed replicate mean (the mean of the middle three replicates, excluding the highest and lowest replicates) by more than 10%, this probe is then discarded from further analysis. Because such outliers are most often the result of post-hybridization washing inconsistencies, we expand or "pad" the region containing suspect probes. Pad such probes by including all probes within a 5-probe radius, as defined by ((x1-x2)2 + (y1-y2)2)½ < 6 where x1, x2, y1, and y2are the x and y coordinates for the two features. Finally, discard features for which standard deviation (included in the .cel file for Affymetrix arrays) of feature pixels/mean feature pixels. After removing outliers, average the intensity values for all remaining replicates.
- Removing unusable tags: Tags with low-intensity values will give poor-quality results and must be removed. An exclusion cutoff for these low intensity probes can be calculated as follows:
- For any treatment-control pair of arrays, calculate log2((ic-bg)/(it-bg)) for each tag, where ic is the control intensity, it is the treatment intensity, and bg is the mean intensity of the unassigned tag probes.
- Pair the uptag and downtag ratios by strain and for each tag pair, take the minimum intensity for the two tags in the two samples. Sort the ratio pairs by this minimum intensity.
- Use a sliding window size of 50 on the ranked ratio pairs, calculate the correlation of uptag and downtag ratio pairs within the window. Also calculate the average of the minimum intensities calculated in the previous step.
- Slide the window by 25 pairs, and repeat the previous step until all pairs have been crossed.
- Plot the average minimum intensity versus the uptag-downtag correlation for all windows.
- Finally, choose an intensity threshold; generally we use the intensity value where the correlation first reaches 80% of its maximum level. Flag and remove from further analysis any tags below this cutoff.
Saturation correction: Because each feature on the barcode microarray can become saturated, the signal on the TAG4 array is not linearly related to tag concentration. To correct for this saturation follow the protocol described in reference 16.
Array normalization: For each array, normalize the uptags and downtags separately. To quantile normalize, rank the values obtained from each array for uptags and downtags in order of increasing intensity. To mean normalize for each set of uptags and downtags, divide by the mean. The mean normalization of all arrays is a second step following transforming the raw data to the mean of each array.
Calculating sensitivity scores for control-treatment comparisons: To use log2 ratios as a metric of sensitivity: For each strain, calculate log2((μc-bg)/(μt-bg)), where μc is the mean intensity for the control samples, μt is the mean intensity for the treatment samples, and bg is the mean intensity of the unassigned probes. Strains with a positive log2 ratio are sensitive to the treatment, and those that are resistant have negative log2 ratios.
6. Assessing fitness of barcoded yeast strains by sequencing
Isolate DNA from the deletion pools as described for microarrays.
Amplify each 20-mer uptag barcode with composite primers comprised of the sequences of the common barcode primers and the sequences required for hybridization to the Illumina flowcell (See Table of Illumina primers and Figure 3 for a diagram of the amplicon). These primers can be used desalted without additional purification. PCR is performed in 100μL, using Invitrogen Platinum PCR Supermix (Cat. No. 11306-016) with the following conditions: 95°C/3 min; 25 cycles of 94°C/30 sec, 55°C/30 sec, 68°C/30 sec; followed by 68°C/10 min.
Purify the PCR product (˜130bp) with Qiagen MinElute 96 UF PCR Purification Kit (Cat. No. 28051).
Following PCR purification, quantify DNA with the Invitrogen Quant-iT dsDNA BR Assay Kit (Cat No. Q32853). Do not rely on 260/280 readings!
Normalize DNA concentration to 10μg/ml and pool equal volumes of normalized DNA.
Separate pooled DNA on a 12% polyacrylamide TBE gel for 3-4 hours depending on voltage used.
Stain gels with ethidium bromide (Sybr Green should work as well) for 30 minutes.
Locate the band of interest on a longwave UV lightbox (wearing appropriate face protection), cut it out and extracted the DNA using the crush and soak method17 followed by ethanol precipitation.
Confirm that the appropriate size DNA (130bp) has been isolated and that primers have been removed using the Agilent Bioanalyzer High Sensitivity DNA kit (Cat No. 5067-4626).
- Sample sequencing:
- Illumina GAIIx platform:
- Generate clusters on a Single-Read flowcell using the cBOT and Single-Read Cluster Generation Kit (Cat No. GD-300-1001). For Read 1, UP and DOWN-tag modified sequencing primers are pooled at a 100uM stock concentration and added to a strip-tube (0.6μL of each 100uM sequencing primer in 120 μl HT1). Recipe SR_Amp_Block_StripTubeHyb_v7.0 is used to generate R1 clusters.
- Sequencing on the Genome Analyzer IIx. Following 18 sequencing cycles, the Paired-end module is used to strip the synthesized first strand and rehybridize the flowcell, using the Illumina R1 (below). Clusters are regenerated and sequenced for 5 cycles to capture the index tag.
The Index tag sequence is used to bin sequences into experimental bins.
Within each experimental bin, the yeast barcode sequences are tallied to give a total number of counts for each barcode.
Counts are quantile normalized so each experiment has the same count distribution. By analogy with barcode microarray fitness experiments, fitness defect ratios for each strain are calculated and expressed as the log2 ratio (control/treatment). Positive fitness defect scores signify a decrease in strain abundance during drug treatment and suggest that the wild-type version of the gene deleted in that strain is required for resistance to that drug or inhibitor.
Note: Considering Bar-seq as an alternative to array hybridization. With the costs of high-throughput sequencing decreasing, using high-throughput sequencing as a readout of tag abundance is becoming feasible and in many cases, is more cost effective 18. In this way, amplified PCR product is measured directly as 'counts' rather than as signal intensity as hybridized to an array. This eliminates false negatives and positives that arise from tag cross-contamination, saturation, or issues arising from very high or very low signal intensities. Furthermore, multiple experiments can be combined prior to sequencing by the addition of a 4-8 base DNA index19. Because the yeast barcodes are 20 bp, one single, 2-step read of 26-28 bases captures both multiplex index and unique barcode , allowing for extreme 100+ multiplexing. At the time of this writing, Bar-seq offers a cost advantage over Bar-code microarrays, and furthermore, Bar-seq is inherently flexible such that as the number of reads/run increases, the level of multiplexing can increase to further decrease costs. Several "mid-capacity" sequencers from all of the major platform manufacturers will further democratize Bar-seq, with sequencing likely to become the readout of choice.
This protocol has also been validated on the Illumina HiSeq2000.
An excellent demonstration of the use of Bar-seq to address a fundamental biological question in Saccharomyces cerevisiae growth control is presented in a recent study by Gresham et al.20 who outline several important experimental design and interpretation guidelines.
7. Validation of pooled screening data
The results from any functional genomics screen should be verified using the individual strains in isolated culture. Because each experiment will differ in terms of the number of sensitive strains, selecting the number of candidate strains to confirm is somewhat arbitrary. As a guide, ranking the most sensitive strains by the log2 ratio or z-score and testing the top 25-50% of the candidates (which typically translates to 2-3 standard deviations from the mean of all strains in the pool) is a good balance between costs and benefits. Individual confirmations can be performed in any flask but we perform these tests for 5 generations of growth in 96 well plates using a starting inoculum of 0.06 OD600 in 100 μl of media in a shaking spectrophotometer, taking measurements every 15 minutes (See Figure 4).
8. Representative Results
Once a genome-wide screen is complete, and the arrays have been normalized and the behavior of each strain compared to a control treatment (e.g. by comparing microarray intensities or sequencing counts/strain) the data are most easily manipulated in an excel file with genes ranked by the log2 ratios of control/experiment. In this manner, the greater the negative log2 ratio, the more sensitive that particular strain is to the test condition. These excel files can be plotted in a variety of graphing software packages. We find it simplest to plot the log2 ratios on the Y axis and the gene or ORF names on the X axis. In the example shown in Figure 2a, such a plot of clotrimazole treatment (a known antifungal agent) is shown. All strain that are significantly sensitive to treatment with a log2 ratio of 2 are highlighted in red, and we would typically verify many of such strains in individual growth assays of each mutant in the presence of the same concentration of drug. In this example, 4 strains are highlighted, NCP1, ERG2 and 2 independent alleles of ERG11, the known protein target of clotrimazole. Each of these 4 genes is directly involved in ergosterol biosynthesis, the yeast equivalent of cholesterol. For example, NCP1 encodes a NADP-cytochrome P450 reductase that is involved in ergosterol biosynthesis and which is associated and coordinately regulated with Erg11. This example highlights the fact that the known drug target (Erg11) is identified in this unbiased screen, as well as several other key components of the target pathway. Finally, several of the highlighted genes in red represent genes that may be involved in ergosterol biosynthesis or in distinct biological processes. As mentioned above, each strain detected as sensitive in a pooled screen should be verified as to its sensitivity in individual growth assay. In the example shown in Figure 2b, four strains are confirmed to be sensitive to clotrimazole based on their decreased growth relative to the wild-type parent strain, BWP17. These individual growth curves highlight an important feature of such pooled gene-drug screens; that is the absolute rank of a particular strain does not necessarily reflect its exact level of sensitivity. Furthermore, Figure 2b also shows the value of having multiple alleles for each gene, in this case, the two erg11 disruption mutants have slightly differing sensitivities. Correlating the nature of these disruptions with the degree of sensitivity can provide additional insight into the drugs mechanism of action.
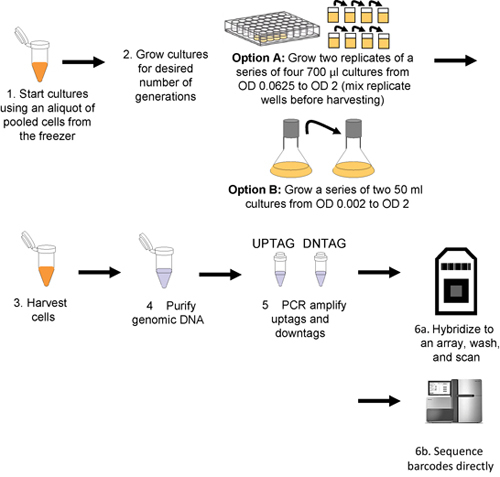 Figure 1. Workflow for pooled growth assay and barcode detection. Cultures are inoculated with thawed aliquots of pooled cells (step 1), and then grown for the desired number of generations (step 2) either robotically (Option A) or manually (Option B). Cells are harvested by centrifugation (step 3) and genomic DNA is then isolated from the harvested cells (step 4), uptags and downtags are independently amplified (step 5), and hybridized to an array (step 6a or sequenced directly step 6b).
Figure 1. Workflow for pooled growth assay and barcode detection. Cultures are inoculated with thawed aliquots of pooled cells (step 1), and then grown for the desired number of generations (step 2) either robotically (Option A) or manually (Option B). Cells are harvested by centrifugation (step 3) and genomic DNA is then isolated from the harvested cells (step 4), uptags and downtags are independently amplified (step 5), and hybridized to an array (step 6a or sequenced directly step 6b).
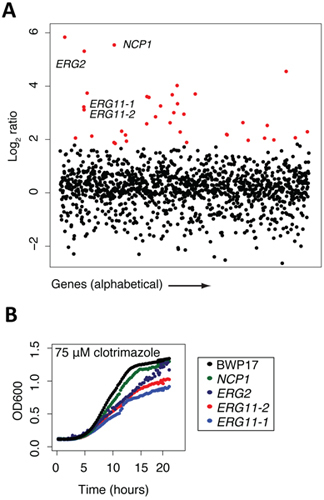 Figure 2. Sample data collected at certain points of the protocol. (A) Sample data from screening results (adapted from 13). The pool of tagged mutants was grown for 20 generations in the presence of clotrimazole and DMSO (control). Log2 ratio (control intensity/treatment intensity) was calculated and plotted as a function of gene. Highly sensitive strains (red) included the known target of clotrimazole, ERG11p. Note that this assay frequently uncovers other sensitive mutants in addition to the compound's actual target. Generally, these mutants are those that act synthetically with the target, those that are part of a general stress/treatment response, or are false positives that fail to confirm. (B) Example of confirmation data (adapted from 13). Results from the pooled growth assays can be validated by growing the strain in individual culture and compared against wild-type growth (black).
Figure 2. Sample data collected at certain points of the protocol. (A) Sample data from screening results (adapted from 13). The pool of tagged mutants was grown for 20 generations in the presence of clotrimazole and DMSO (control). Log2 ratio (control intensity/treatment intensity) was calculated and plotted as a function of gene. Highly sensitive strains (red) included the known target of clotrimazole, ERG11p. Note that this assay frequently uncovers other sensitive mutants in addition to the compound's actual target. Generally, these mutants are those that act synthetically with the target, those that are part of a general stress/treatment response, or are false positives that fail to confirm. (B) Example of confirmation data (adapted from 13). Results from the pooled growth assays can be validated by growing the strain in individual culture and compared against wild-type growth (black).
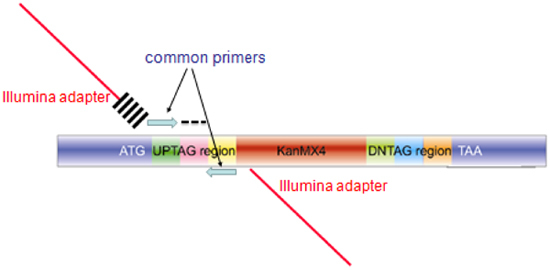 Figure 3. Structure of the amplicon produced from pooled barcode assays for microarray hybridization or Barcode sequencing. The amplicon produced for each mutant in the collection contains homology to the genome for integration (blue regions labeled ATG and TAA), unique barcodes (labeled AG and indicated by a black dash). For microarray hybridization, the blue common primers are used to amplify a 60bp probe for microarray hybridization. For barcode sequencing, extended primers are used in the PCR reaction, comprised of sequences encoding the Illumina adapter (red bar), and index of 6bases cross-hatches) and the blue common primer for the upstream primer, and the same composite primer (minus the 6 base index) for the second primer.
Figure 3. Structure of the amplicon produced from pooled barcode assays for microarray hybridization or Barcode sequencing. The amplicon produced for each mutant in the collection contains homology to the genome for integration (blue regions labeled ATG and TAA), unique barcodes (labeled AG and indicated by a black dash). For microarray hybridization, the blue common primers are used to amplify a 60bp probe for microarray hybridization. For barcode sequencing, extended primers are used in the PCR reaction, comprised of sequences encoding the Illumina adapter (red bar), and index of 6bases cross-hatches) and the blue common primer for the upstream primer, and the same composite primer (minus the 6 base index) for the second primer.
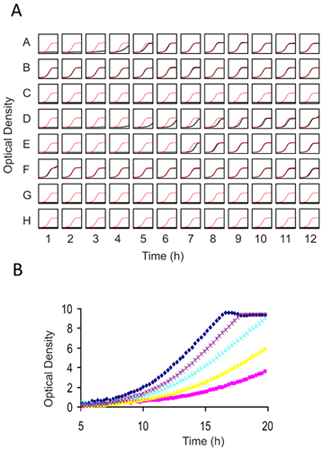 Figure 4. Individual Growth assays for 1) Prescreening compounds against wild-type yeast to determine an appropriate dose for genome-wide screening and 2) Confirming results from genome wide screens. (A) A 96 well flat bottom plate is filled with 100 μl of cell suspension at an OD of 0.062. Each well can contain the same strain (for dose-determination) or different strain and drug combinations (for confirmation assays). 2 μl of compound (typically dissolved in DMSO) is added and cells are grown with constant shaking for 16-20 h at 30°C. The final concentration of DMSO should not exceed 2%. In this example, in each well of the plate the growth curve is plotted in black against a plot of the control growth curve in red. (B) Higher resolution image of several prescreens obtained with an example drug overlaid on top of one another. In this titration series, an IC10-15 is obtained with the purple dose and would be appropriate for deletion profiling (HIP and HOP). Due to the non-linearity at higher optical densities, Tecan (or any similar plate reader) ODs must be calibrated using those obtained with a "traditional" 1mm path-length cuvette.
Figure 4. Individual Growth assays for 1) Prescreening compounds against wild-type yeast to determine an appropriate dose for genome-wide screening and 2) Confirming results from genome wide screens. (A) A 96 well flat bottom plate is filled with 100 μl of cell suspension at an OD of 0.062. Each well can contain the same strain (for dose-determination) or different strain and drug combinations (for confirmation assays). 2 μl of compound (typically dissolved in DMSO) is added and cells are grown with constant shaking for 16-20 h at 30°C. The final concentration of DMSO should not exceed 2%. In this example, in each well of the plate the growth curve is plotted in black against a plot of the control growth curve in red. (B) Higher resolution image of several prescreens obtained with an example drug overlaid on top of one another. In this titration series, an IC10-15 is obtained with the purple dose and would be appropriate for deletion profiling (HIP and HOP). Due to the non-linearity at higher optical densities, Tecan (or any similar plate reader) ODs must be calibrated using those obtained with a "traditional" 1mm path-length cuvette.
Discussion
Here, we outlined a protocol that, with modest modification, can easily be adapted to a wide range of existing collections of barcoded mutant collections of different microorganisms to create tagged mutant collections. We emphasize that while we have reported a protocol on the tagged transposon mutagenesis for the pathogenic yeast C. albicans, a very similar protocol could be adapted to a wide variety of unicellular fungi. Modified, this protocol works well in bacteria 13, and currently collections for a number of additional fungal and bacterial genomes are under construction. At present, this assay provides the only comprehensive, genome-wide unbiased screen for gene-small molecule interactions. One particularly compelling feature of the assay is that no prior knowledge of the gene or small molecule is required. Despite the scope and power of these assays, their transferability to other labs has been hindered somewhat by the initial capital investment and informatics tools for the analysis of the results. With the adoption of a next generation sequence readout combined with robust tools for analysis, we expect their adoption to increase.
Disclosures
No conflicts of interest declared.
Acknowledgments
We thank Ron Davis, Adam Deutschbauer, and the entire HIP HOP laboratory at the University of Toronto for discussions and advice. C.N. is supported by grants from the National Human Genome Research Institute (Grant Number HG000205), RO1 HG003317, CIHR MOP-84305, and Canadian Cancer Society (#020380). J.O. was supported by the Stanford Genome Training Program (Grant Number T32 HG00044 from the National Human Genome Research Institute) and the National Institutes of Health (Grant Number P01 GH000205). GG is supported by the NHGRI RO1 HG003317 and the Canadian Cancer Society, Grant # 020380, TD and the Donnelly Sequencing Centre is supported in part by grants from the Canadian Foundation for Innovation to Drs. Brenda Andrews and Jack Greenblatt. A.M.S. is supported by a University of Toronto Open Fellowship.
References
- Giaever G. Functional profiling of the Saccharomyces cerevisiae genome. Nature. 2002;418:387–391. doi: 10.1038/nature00935. [DOI] [PubMed] [Google Scholar]
- Baba T. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Molecular systems biology. 2006;2:2006.0008–2006.0008. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claus H, Frosch M, Vogel U. Identification of a hotspot for transformation of Neisseria meningitidis by shuttle mutagenesis using signature-tagged transposons. Mol Gen Genet. 1998;259:363–371. doi: 10.1007/s004380050823. [DOI] [PubMed] [Google Scholar]
- Hava DL, Camilli A. Large-scale identification of serotype 4 Streptococcus pneumoniae virulence factors. Molecular microbiology. 2002;45:1389–1406. [PMC free article] [PubMed] [Google Scholar]
- Costanzo M. The Genetic Landscape of a Cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong AH. Systematic genetic analysis with ordered arrays of yeast deletion mutants. Science. 2001;294:2364–2368. doi: 10.1126/science.1065810. [DOI] [PubMed] [Google Scholar]
- Pan X. A robust toolkit for functional profiling of the yeast genome. Molecular cell. 2004;16:487–496. doi: 10.1016/j.molcel.2004.09.035. [DOI] [PubMed] [Google Scholar]
- Schuldiner M. Exploration of the Function and Organization of the Yeast Early Secretory Pathway through an Epistatic Miniarray Profile. Cell. 2005;123:507–519. doi: 10.1016/j.cell.2005.08.031. [DOI] [PubMed] [Google Scholar]
- Deutschbauer AM. Mechanisms of haploinsufficiency revealed by genome-wide profiling in yeast. Genetics. 2005;169:1915–1925. doi: 10.1534/genetics.104.036871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giaever G. Chemogenomic profiling: identifying the functional interactions of small molecules in yeast. Proceedings of the National Academy of Sciences of the United States of America. 2004;101:793–798. doi: 10.1073/pnas.0307490100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum PY. Discovering modes of action for therapeutic compounds using a genome-wide screen of yeast heterozygotes. Cell. 2004;116:121–137. doi: 10.1016/s0092-8674(03)01035-3. [DOI] [PubMed] [Google Scholar]
- Hillenmeyer ME. The chemical genomic portrait of yeast: uncovering a phenotype for all genes. Science. 2008;320:362–365. doi: 10.1126/science.1150021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J. A universal TagModule collection for parallel genetic analysis of microorganisms. Nucleic acids research. 2010;38:e146–e146. doi: 10.1093/nar/gkq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh J. Gene annotation and drug target discovery in Candida albicans with a tagged transposon mutant collection. PLoS pathogens. 2010;6 doi: 10.1371/journal.ppat.1001140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nislow C, Giaever G. Chapter 387. In: Stark I, Stansfields MJR, editors. Yeast Gene Analysis. 2nd Edition. Elsevier Ltd; 2007. pp. 387–414. [Google Scholar]
- Pierce SE, Davis RW, Nislow C, Giaever G. Genome-wide analysis of barcoded Saccharomyces cerevisiae gene-deletion mutants in pooled cultures. Nature protocols. 2007;2:2958–2974. doi: 10.1038/nprot.2007.427. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Russell DW Cold Spring Harbor Laboratory. Molecular cloning : a laboratory manual. 3rd. edn. Cold Spring Harbor Laboratory; 2001. [Google Scholar]
- Smith AM. Quantitative phenotyping via deep barcode sequencing. Genome Res. 2009. [DOI] [PMC free article] [PubMed]
- Hamady M, Walker JJ, Harris JK, Gold NJ, Knight R. Error-correcting barcoded primers for pyrosequencing hundreds of samples in multiplex. Nature. 2008;5:235–237. doi: 10.1038/nmeth.1184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gresham D. System-Level Analysis of Genes and Functions Affecting Survival During Nutrient Starvation in Saccharomyces cerevisiae. Genetics. 2011;187:299–317. doi: 10.1534/genetics.110.120766. [DOI] [PMC free article] [PubMed] [Google Scholar]