

Abstract
Cryo-electron microscopy (cryo-EM), combined with image processing, is an increasingly powerful tool for structure determination of macromolecular protein complexes and assemblies. In fact, single particle electron microscopy1 and two-dimensional (2D) electron crystallography2 have become relatively routine methodologies and a large number of structures have been solved using these methods. At the same time, image processing and three-dimensional (3D) reconstruction of helical objects has rapidly developed, especially, the iterative helical real-space reconstruction (IHRSR) method3, which uses single particle analysis tools in conjunction with helical symmetry. Many biological entities function in filamentous or helical forms, including actin filaments4, microtubules5, amyloid fibers6, tobacco mosaic viruses7, and bacteria flagella8, and, because a 3D density map of a helical entity can be attained from a single projection image, compared to the many images required for 3D reconstruction of a non-helical object, with the IHRSR method, structural analysis of such flexible and disordered helical assemblies is now attainable.
In this video article, we provide detailed protocols for obtaining a 3D density map of a helical protein assembly (HIV-1 capsid9 is our example), including protocols for cryo-EM specimen preparation, low dose data collection by cryo-EM, indexing of helical diffraction patterns, and image processing and 3D reconstruction using IHRSR. Compared to other techniques, cryo-EM offers optimal specimen preservation under near native conditions. Samples are embedded in a thin layer of vitreous ice, by rapid freezing, and imaged in electron microscopes at liquid nitrogen temperature, under low dose conditions to minimize the radiation damage. Sample images are obtained under near native conditions at the expense of low signal and low contrast in the recorded micrographs. Fortunately, the process of helical reconstruction has largely been automated, with the exception of indexing the helical diffraction pattern. Here, we describe an approach to index helical structure and determine helical symmetries (helical parameters) from digitized micrographs, an essential step for 3D helical reconstruction. Briefly, we obtain an initial 3D density map by applying the IHRSR method. This initial map is then iteratively refined by introducing constraints for the alignment parameters of each segment, thus controlling their degrees of freedom. Further improvement is achieved by correcting for the contrast transfer function (CTF) of the electron microscope (amplitude and phase correction) and by optimizing the helical symmetry of the assembly.
Keywords: Immunology, Issue 54, cryo-electron microscopy, helical indexing, helical real-space reconstruction, tubular assemblies, HIV-1 capsid
Protocol
1. Frozen-hydrated EM specimen preparation
Because HIV-1 capsid protein (CA) assemblies9 are stable only in high salt (1M NaCl) buffer, which contributes strong background noise in cryo-EM images, we use a rapid dilution and back-side blotting method to transiently reduce the salt concentration when preparing the frozen-hydrated EM grid.
Glow discharge the carbon side of 200-mesh R2/1 Quantifoil copper grids under 25mA for 25 seconds.
Use a nebulizer to bring the humidity in the environmental chamber, a home-made manual gravity plunger, to 80%.
In the FEI Vitrobot Plunger dewar, cool liquid ethane using liquid nitrogen. Mount the plunge-freezing dewar onto the manual gravity plunger.
Apply 2.5 μl of preassembled CA solution onto the carbon side of the grid, which is mounted on forceps, and load the forceps onto the plunger with the carbon side of the grid facing away from you.
Add 3 μl of low salt dilution buffer (100 mM NaCl) to the back side of the grid and immediately blot the back side of the grid with a piece of filter paper. The whole back surface of the grid should be in close contact with the filter paper for approximately 6 seconds, before removing the filter paper. Immediately plunge the grid into liquid ethane after removing the filter paper.
Remove the forceps from the plunger and quickly transfer the grid into a grid storage box.
2. Cryo-electron microscopy of CA tubular assemblies
Load the frozen-hydrated grid into an FEI Polara G2 electron microscope operating at 200kv and equipped with a Gatan 4Kx4K CCD camera.
Under low-dose-search mode, at a magnification of ~200x and with a dose <0.001e-/Å2, screen the whole grid for areas with suitable ice and save the positions of these areas in a stage file.
Recall the saved positions and further screen these areas at a magnification of 3,900x in low-dose-search mode. Select the areas with a uniform, thin layer of ice containing well-separated, long tubes over holes, for data collection. Save the locations of these areas in a second stage file.
Switch to exposure mode at a magnification of 59,000x, inset a 100 μm objective aperture, and adjust the objective stigmatism and beam intensity for a dose of ˜15 e-/Å2 per exposure.
Return to the low-dose-search mode, move to a saved position and identify and center a good tube using the CCD camera. Switch to the focus mode, adjust the focus, and set a defocus value, normally between 0.5 to 2.5 μm. Switch to the exposure mode, set an exposure time of 0.3-0.5 seconds, for a dose of 15 e-/Å2, and collect an image. The images are collected on a plate camera, and the films should be allowed to settle for 10 seconds before an exposure is taken.
Move to the next saved position and repeat step 5 to collect more images.
The films are developed in full strength D19 for 12 minutes and digitized using a Nikon super coolscan 9000 ED scanner at a pixel size of 6.35 μm. The format of image files is TIFF.
3. Helical indexing
A helical object can be indexed by two parameters: the Bessel order, n, and layer line number, l. Each layer line in the Fourier transform, as characterized by (n, l), corresponds to a set of lines on the surface lattice of the helical object denoted by (h, k) indices, using the notation from a 2D lattice. For any (h, k), an (nhk, lhk) layer line is a linear combination of two basic vectors (n10, l10) and ( n01, l01), which are the n and l values of the two principal layer lines (1, 0) and (0, 1). l can be obtained from the layer line height measured along Z axis in the Fourier transform. The value of n can be estimated using the following equation10
2πRr ≈ Jn ≈ 1.1|n|+0.9 .....................(1)
where Jn is the Bessel function, which determines the intensity of the nth layer line, r is the radius of the helical object, and R is the radius of the maximum amplitude of the layer line. The layer line number, l, is related to n by the selection rule11
l = tn + um ....................(2)
where t and u are constants of the helix. For any given helix, there may be exactly u units in exactly (or very closely) t complete turns.
Box out a relatively straight and long tube with uniform diameter using the EMAN's program12 helixboxer and save the image in MRC format.
Determine the helical repeat distance, c, using a cross-correlation based program, such as 'imgccf', in the MRC package13.
Calculate the Fourier transform with a new box length that is an integral of the repeat distance, c.
Choose two principal layer lines that define two basic surface lattice vectors (1,0) and (0,1).
Measure the radius of the tube, r, and the height and radius of the two principal layer lines in Fourier transform, l10, R10, l01 R01, respectively (Fig. 1).
Calculate n10 and n01 according to equation (1).
Since n values are only estimation, a few combinations of n10 and n01 are tested in later steps (See step 4.2) to find the correct helical symmetry using the program package IHRSR.
Calculate the screw symmetry described by two real numbers: the rotation between subunits (Δφ) and the axial rise (Δz) of the one-star helix (n=1). Given l10, l01, n10, and n01 values, u and t can be obtained by testing a range of m values (for instance, -50<m<50) according to the selection rule. Finally, Δφ and Δz are calculated using Δφ =360t/u and Δz =c/u.
4. Three-dimensional reconstruction
- Particle segmentation
- Open a micrograph containing helical particles, using the graphical program Boxer, which is a program in the EMAN package.
- Cut the helical particle into overlapping segments. In the control panel of Boxer, choose Helix mode and set the parameters for boxing: the size of the box should be larger than the diameter of the particle and the value for 'Olap' should be ˜90% of the box size.
- After left-clicking on either end of the helical particle, Boxer will automatically generate a series of particle boxes along the helix length.
- Save boxed segments as well as their coordinates.
- Initial 3D reconstruction using IHRSR programs
- Invert the contrast of the cryo-EM images and apply low pass filtering14 (optional) prior to processing with the iterative helical real space reconstruction method (IHRSR).
- Open the graphical interface of IHRSR program by typing "Generator". Provide the graphical interface with all the information for the boxed particles stack, including the name and path of the stack, number of images in the stack, the values for symmetry parameters, etc. Click the button "Finish" to create the reconstruction script, b25.spi.
- Use a solid or hollow cylinder as an initial reference and allow the procedure to cycle until there are no changes in the defined screw symmetry, which usually occurs after a few cycles. A right helical symmetry should give a stably converged reconstruction. The reconstruction generated in the last cycle will be used as an initial reference for further refinement. IHRSR performs 3D reconstruction using SPIDER programs.
- Reconstruction with iterative refinement15 The 3D reconstruction generated by IHRSR is now used as an initial reference for additional refinement. During the refinement, the helical symmetry is fixed at Δφ and Δz, which are determined from the IHRSR procedure.
- Determine the defocus and astigmatism present in the micrograph using programs CTFFIND3 and CTFTILT16.
- Multiply particle segments by the CTF using SPIDER programs17. The operation termed FT in the SPIDER suite is used to compute the Fourier transform (FFT) of the 2D image. Thereafter, the FFT is multiplied by CTF values, determined in the previous steps, using the operation termed MU within the SPIDER suite. The value obtained is then inverse transformed back to form a new image (CTF-corrected) in real space using the SPIDER operation, FT again.
- Perform projection matching by comparing projections of the reference volumes with the CTF-corrected images, using multi-reference alignment. The variation in the out-of-plane tilt angle is limited to +/-10° and sampled in 1° steps. Introduce constraints, such as high correlation coefficients, in-plane angles near 0° or 180°, and limited x-shifts, for the alignment parameters of each segment. Include in the reconstruction only those segments that satisfy the constraints.
- After each iterative refinement cycle, a 3D reconstruction is generated using back projection and divided by sum over the CTFˆ2.
- Impose the helical symmetry to generate a symmetrized volume. The iterative refinement is terminated when no further improvement in the resolution of the new 3D reconstruction occurs.
- The image processing package SPIDER is used for most of the refinement steps. A series of operations are controlled by SPIDER scripts, which are user-created batch control files containing sequences of operations and parameter values. The final reconstruction is calculated using programs in SPIDER suite.
5. Representative Results:
A single HIV-1 CA A92E tube (Fig. 1a) was boxed out and its Fourier transform (Fig. 1b) was calculated for helical indexing. For layer lines (1, 0) and (0, 1), l10=28, l01=37, R10=55, R01=44. Given a tube radius of 211.57Å, we approximated n10=-12, n01=11 (here, the handedness was predetermined). With a repeat distance of 5195.48Å, the screw symmetry of the tube was determined as Δz=6.8093Å, Δφ=328.88°. Δz and Δφ were refined to 7.1321Å and 328.86° using IHRSR (Fig. 2a) and the initial reconstruction is shown in Fig. 2b. The final reconstruction (Fig. 3), after iterative refinement, improved the density map significantly from the initial model calculated with IHRSR (Fig. 2b).
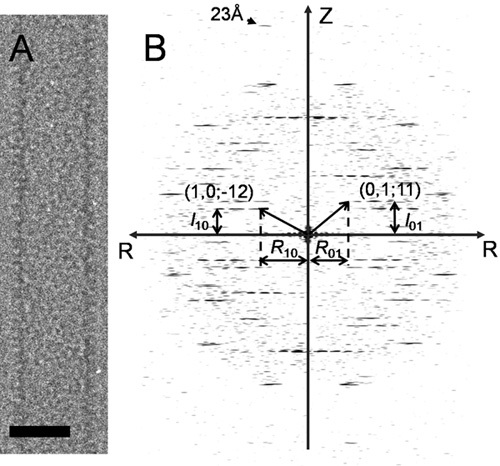 Figure 1. Indexing of HIV-1 CA helical tube. (A) A single HIV-1 CA A92E tube image. Scale bar, 30 nm. (B) The Fourier transform of the tube shown in (A). The helical indices (n10=-12, n01=11) are denoted. The arrowhead points to the layer line at 23Å resolution.
Figure 1. Indexing of HIV-1 CA helical tube. (A) A single HIV-1 CA A92E tube image. Scale bar, 30 nm. (B) The Fourier transform of the tube shown in (A). The helical indices (n10=-12, n01=11) are denoted. The arrowhead points to the layer line at 23Å resolution.
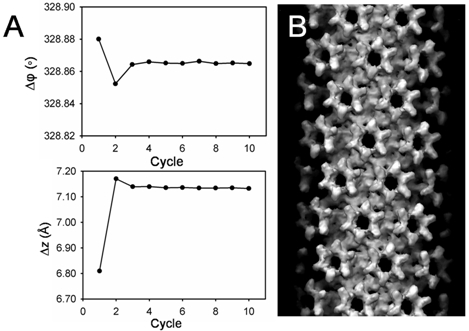 Figure 2. An initial reconstruction using IHRSR. (A) Screw symmetry determination for each iterative cycle. Δφ and Δz, starting from the initial values, converge to the stable values after 10 iterative refinement cycles. (B) The initial 3D density map after 10 iterative cycles.
Figure 2. An initial reconstruction using IHRSR. (A) Screw symmetry determination for each iterative cycle. Δφ and Δz, starting from the initial values, converge to the stable values after 10 iterative refinement cycles. (B) The initial 3D density map after 10 iterative cycles.
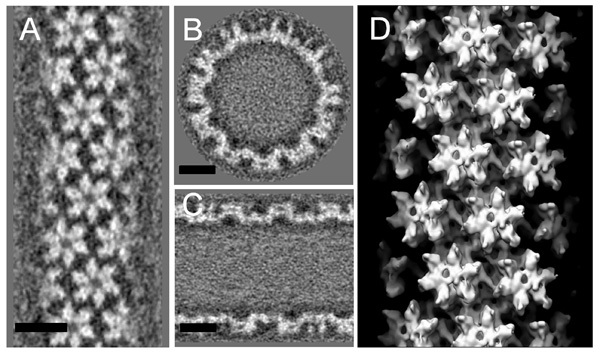 Figure 3. The 3D density map after the iterative refinement. (A-C) The density map of CA tubes is displayed as three orthogonal slices: parallel to the tube axis and close to the surface (A), perpendicular to the tube axis (B), and parallel to and through the tube axis (C). Scale bars, 10 nm. (D) Surface rendering of the 3D density map contoured at 1.8s enclosing 100% volume.
Figure 3. The 3D density map after the iterative refinement. (A-C) The density map of CA tubes is displayed as three orthogonal slices: parallel to the tube axis and close to the surface (A), perpendicular to the tube axis (B), and parallel to and through the tube axis (C). Scale bars, 10 nm. (D) Surface rendering of the 3D density map contoured at 1.8s enclosing 100% volume.
Discussion
We present a set of protocols to provide a straightforward approach to obtain 3D structures of helical objects. Using the described procedures, we acquired a 3D structure of HIV-1 capsid assembly from a single tube image (176 segments). Higher resolution structures can be achieved by including more image data.
There are several critical points for optimal data collection and analysis: First, during preparation of a cryo-EM specimen, the sample solution should be blotted away, leaving a uniform, thin layer of solution that is slightly thicker than the sample size. There are several different ways to blot the sample. For bacterial cells and tubular specimens, such as HIV-1 CA assembly, blotting from one-side, particularly from the back-side, is most appropriate.
Second, the handedness of the helix needs to be determined, as this cannot be done by helical indexing or reconstruction. A common practice is to use freeze-etching, followed by rotary shadowing18 to determine handedness. Handedness can also be determined post-reconstruction when the resolution of the density map is sufficiently high; the 3D atomic models of individual components should fit well into the density map when a correct handedness is assumed. Otherwise, the opposite handedness should be assumed.
Third, a Wiener filter should be used during image processing, for both phase and amplitude correction, to reduce noise amplification. Since the CTF from a single image always has zero crossings, part of the information in reciprocal space is lost. Therefore, it is necessary to have multiple projection data sets included for 3D reconstruction, each imaged at different defocus values.
Disclosures
No conflicts of interest declared.
Acknowledgments
The authors would like to thank Dr. Gongpu Zhao and Danxia Ke for technical support. We thank Drs. Edward Egelman and Niko Grigorieff for sharing their image processing software. We also acknowledge the staff who support the Structural Biology cryo-EM facility and Beowulf cluster and grid at the University of Pittsburgh School of Medicine. This work was supported by GM082251 and GM085043.
References
- Frank J, Radermacher M. Three-dimensional reconstruction of single particles negatively stained or in vitreous ice. Ultramicroscopy. 1992;46:241–262. doi: 10.1016/0304-3991(92)90018-f. [DOI] [PubMed] [Google Scholar]
- Henderson R. Structure of Purple Membrane from Halobacterium-Halobium - Analysis of X-Ray-Diffraction Pattern. J Mol Biol. 1975;93:123–128. doi: 10.1016/0022-2836(75)90123-0. [DOI] [PubMed] [Google Scholar]
- Egelman EH. The iterative helical real space reconstruction method: surmounting the problems posed by real polymers. J Struct Biol. 2007;157:83–94. doi: 10.1016/j.jsb.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Egelman EH, Francis N, Derosier DJ. F-Actin Is a Helix with a Random Variable Twist. Nature. 1982;298:131–135. doi: 10.1038/298131a0. [DOI] [PubMed] [Google Scholar]
- Nogales E, Whittaker M, Milligan RA, Downing KH. High-resolution model of the microtubule. Cell. 1999;96:79–88. doi: 10.1016/s0092-8674(00)80961-7. [DOI] [PubMed] [Google Scholar]
- Jimenez JL. Cryo-electron microscopy structure of an SH3 amyloid fibril and model of the molecular packing. Embo J. 1999;18:815–821. doi: 10.1093/emboj/18.4.815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler PJ, Klug A. Assembly of the particle of tobacco mosaic virus from RNA and disks of protein. Nat New Biol. 1971;229:47–50. doi: 10.1038/newbio229047a0. [DOI] [PubMed] [Google Scholar]
- Namba K, Yamashita I, Vonderviszt F. Structure of the core and central channel of bacterial flagella. Nature. 1989;342:648–654. doi: 10.1038/342648a0. [DOI] [PubMed] [Google Scholar]
- Byeon IJL. Structural Convergence between Cryo-EM and NMR Reveals Intersubunit Interactions Critical for HIV-1 Capsid Function. Cell. 2009;139:780–790. doi: 10.1016/j.cell.2009.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima C, Unwin N. Three-dimensional structure of the acetylcholine receptor by cryoelectron microscopy and helical image reconstruction. J Cell Biol. 1990;111:2623–2635. doi: 10.1083/jcb.111.6.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyoshima C. Structure determination of tubular crystals of membrane proteins. I. Indexing of diffraction patterns. Ultramicroscopy. 2000;84:1–14. doi: 10.1016/s0304-3991(00)00010-3. [DOI] [PubMed] [Google Scholar]
- Ludtke SJ, Baldwin PR, Chiu W. EMAN: semiautomated software for high-resolution single-particle reconstructions. J Struct Biol. 1999;128:82–97. doi: 10.1006/jsbi.1999.4174. [DOI] [PubMed] [Google Scholar]
- Yonekura K, Toyoshima C. Structure determination of tubular crystals of membrane proteins. IV. Distortion correction and its combined application with real-space averaging and solvent flattening. Ultramicroscopy. 2007;107:1141–1158. doi: 10.1016/j.ultramic.2007.01.013. [DOI] [PubMed] [Google Scholar]
- van Heel M. Single-particle electron cryo-microscopy: towards atomic resolution. Q Rev Biophys. 2000;33:307–369. doi: 10.1017/s0033583500003644. [DOI] [PubMed] [Google Scholar]
- Sachse C. High-resolution electron microscopy of helical specimens: A fresh look at Tobacco Mosaic Virus. J Mol Biol. 2007;371:812–835. doi: 10.1016/j.jmb.2007.05.088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mindell JA, Grigorieff N. Accurate determination of local defocus and specimen tilt in electron microscopy. J Struct Biol. 2003;142:334–347. doi: 10.1016/s1047-8477(03)00069-8. [DOI] [PubMed] [Google Scholar]
- Frank J. SPIDER and WEB: Processing and visualization of images in 3D electron microscopy and related fields. J Struct Biol. 1996;116:190–199. doi: 10.1006/jsbi.1996.0030. [DOI] [PubMed] [Google Scholar]
- Zhang PJ, Hinshaw JE. Three-dimensional reconstruction of dynamin in the constricted state. Nat Cell Biol. 2001;3:922–926. doi: 10.1038/ncb1001-922. [DOI] [PubMed] [Google Scholar]