

Abstract
Engineering plasmonic nanostructures that simultaneously achieve high colloidal stability, high photothermal stability, low non-specific binding to biological specimens, and low toxicity is of significant interest to research in bionanotechnology. Using gold nanorods, we solved this problem by encapsulating them with a multilayer structure, silica, hydrophobic ligands, and amphiphilic-polymers. In comparison with nanorods covered with the conventional surface chemistries, such as surfactants, polyelectrolytes, thiolated polymers, and silica shells alone, the new nanorods remain single in various solutions and show remarkable stability against laser irradiation. We further demonstrated specific targeting and effective treatment of prostate tumor cells using nanorod–aptamer bioconjugates. This exquisitely formulated nanoencapsulation technology could potentially help stabilize other plasmonic nanostructures that are not in the most thermodynamically or chemically stable states, and should open exciting opportunities in nanotechnology-based imaging and therapeutics.
1. Introduction
Plasmonic nanostructures such as spherical nanoparticles, nanorods, nanoshells, and nanocages have captivated scientists, engineers, and lately physicians, owing to their fascinating size-dependent optical and electronic properties.1–17 These unique properties, unavailable in bulk materials and individual atoms, offer new opportunities in disease diagnosis, prognosis, and therapy. Among all, gold nanorods (GNRs) have become a model system for bionanotechnology and stimulated the development of nanoprobes for bioimaging, sensing, drug delivery, and photothermal therapy. Compared with spherical nanoparticles, GNRs are more sensitive to changes in local environments, manifested as a larger plasmon shift per refractive index unit (dλSPR/dnM), which, in addition to the narrow plasmon band width, is ideal for sensing applications.18,19 Furthermore, GNRs also offer stronger scattering and absorption efficiency per unit volume than their spherical counterparts and nanoshells,20 and thus provide the opportunity to integrate multiple imaging modes and therapeutic strategies, such as surface-enhanced Raman scattering,21 dark-field imaging,22 optical coherence tomography,23 multiphoton-induced luminescence,24,25 photoacoustic tomography,26 on-demand drug release,27–30 hyperthermia cancer therapy,31–33 and cellular photothermolysis.34
Despite these recent excitements, a number of outstanding issues must be addressed for widespread applications of GNRs in biology and medicine. First, high-quality GNRs are generally made in the presence of a cationic surfactant, cetyltrimethylammonium bromide (CTAB), which has been suggested to form bilayers on the surface of GNRs.35 CTAB stabilizes GNR colloids via strong electrostatic repulsion in water, due to its cationic quaternary-ammonium head group, which, on the other hand, also causes non-specific binding to biomolecules and cells for biological applications.32 The non-covalent nature of CTAB and GNR bonding results in a dynamic process between CTAB adsorption and desorption. Although gold is biologically inert and surface-bound CTAB is not highly cytotoxic either, detached CTAB is harmful to cells.36 A recent surge on studies of the GNR surface chemistry has largely solved the toxicity problems by replacing or covering the CTAB layer with thiolated polyethylene glycol (SH–PEG), phospholipids, silica shells, and polyelectrolytes such as poly(4-styrenesulfonic acid) (PSS).37–46 Among these new surface coating materials, GNRs coated with SH–PEG show the best colloidal stability. They remain single in a broad spectrum of buffers with various concentrations of salts and proteins, and pH values; whereas other surface coating materials often form aggregates.
The second problem is the stability of GNRs against laser illumination. This is particularly important for imaging and therapeutic modes that involve laser irradiation at high intensity or over extended periods, such as photoacoustic tomography, photothermal therapy, multiphoton microscopy, and optically modulated drug release. It has been shown that GNRs have large absorption cross-section but fairly low photoluminescence quantum efficiency (10−3–10−4).47 Thus, energy from absorbed photons is mainly converted to heat. When irradiated with high-intensity femtosecond or nanosecond lasers matching their plasma resonance bands, GNRs not only melt and change shape to spherical nanoparticles, but can also fragment over certain energy thresholds.48 This poor photothermal stability is highly undesirable for applications depending on photothermal conversion because the reshaped nanoparticles have significantly reduced absorbance compared with the original intact GNRs. Replacing the CTAB coating layer with silica shells allows faster heat dissipation, and thus renders GNR more photothermally stable.49,50 On the other hand, the silica shell-coated GNRs have poor colloidal stability in biological buffers and media. To solve all these problems, here, we report a new class of GNRs simultaneously achieving high colloidal stability, high photothermal stability, low non-specific binding to biological specimens, and low toxicity. We further demonstrate crosslinking of GNRs to aptamers, and probe the targeting and therapeutic capabilities of this new series of GNR bioconjugates on prostate tumor cells.
The logic behind the probe design is simple. Since the current surface coating technology is only capable of partially solving the aforementioned problems, intelligent selection and combination of surface coatings with complementary functionalities should address the problems in full. Therefore, we choose to encapsulate GNRs with a thin and uniform layer of silica shell for enhanced stability against laser irradiation and a layer of bifunctional PEG for biocompatibility, bioconjugation, and reduced non-specific binding. As shown in Fig. 1, water-soluble GNRs are first coated with a silica shell (GNR@SiO2). The resulting hydrophilic GNR@SiO2 are then converted to hydrophobic GNR@SiO2, thereby the solubilization approaches based on amphiphilic polymers initially developed for semiconductor nanoparticles51–54 can be utilized to functionalize GNRs. We demonstrate this concept using a representative amphiphilic polymer, 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy(polyethylene glycol)-2000] (PE-PEG).51 The terminal carboxylic acid groups allow crosslinking with aminated targeting molecules via the common carbodiimide reaction.
Fig. 1.
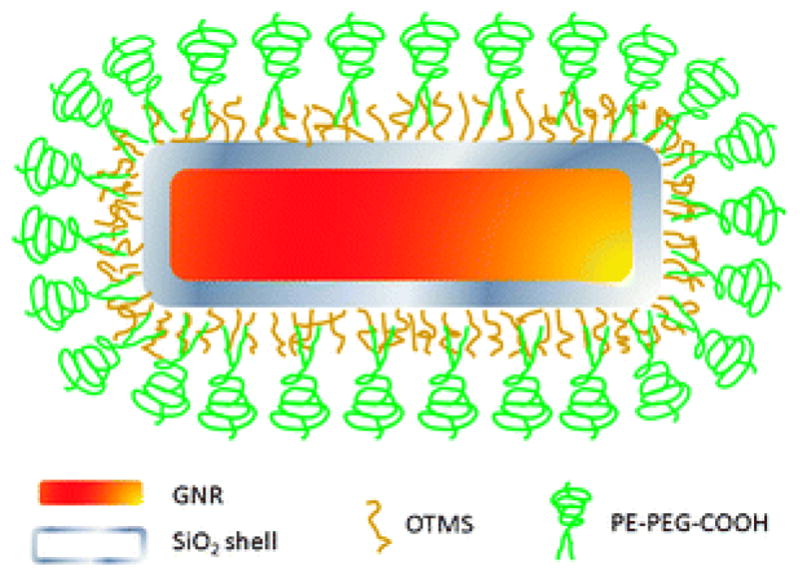
Schematic illustration of GNR@SiO2@PE-PEG. NRs are encapsulated within a thin layer of silica shell, followed by surface modification with a hydrophobic silane, OTMS. The hydrophobic GNR@SiO2–C18 is then solubilized with PE-PEG with terminal COOH groups for potential bioconjugation. Figure is not drawn to scale.
2. Experimental
Materials
Gold(III) chloride (HAuCl4·3H2O), AgNO3, cetyltrimethylammonium bromide, sodium borohydride (NaBH4), tetraethyl orthosilicate (TEOS), octadecyltrimethoxysilane (OTMS), 1-ethyl-3-[3-dimethylaminopropyl] carbodiimide hydrochloride (EDC), and ascorbic acid were purchased from Sigma-Aldrich (St. Louis, MO). Thiol-mPEG5000 (SH–PEG) was obtained from Laysan Bio (Arab, AL). 1,2-Distearoyl-sn-glycero-3-phosphoethanolamine-N-[carboxy (polyethylene glycol)-2000] was purchased from Avanti Polar Lipids (Alabaster, AL). The 41-base A10 RNA aptamer (sequence: 5′-NH2-GGG AGG AcG AuG cGG Auc AGc cAu Guu uAc Guc Acu ccu u (3′dT) 3′, lowercase letters indicate 2′fluoro RNA)55 was custom synthesized by IDT (San Diego, CA). All materials were used as received without further purification. Ultrapure water (18 MΩ cm−1) was obtained from a Milli-Q water purifier (Billerica, MA).
GNR preparation
GNRs were synthesized using the seed-mediated growth method invented by Murphy, El-Sayed, and coworkers.56,57 To prepare the nanoparticle seeds, ice cold NaBH4 (0.6 mL, 10 mM) was added to a mixture of CTAB (7.5 mL, 0.1 M) and HAuCl4 (0.1 mL, 25 mM) under vigorous stirring. The solution turned from yellow to brown indicating formation of gold nanoparticle seeds and was kept on stirring for a few minutes. The seed solution was then placed into a water bath kept at 27 °C. For GNR growth, CTAB (0.1 M, 50 mL) was mixed with HAuCl4 (1.0 mL, 25 mM) and AgNO3 (0.4 mL, 10 mM). A mild reducing agent, ascorbic acid (0.3 mL, 0.1 M), was then added, and the color of the solution rapidly changed from bright yellow to colorless. To initiate GNR growth, 60 μL of gold seed solution was added. The reaction mixture was swirled, and then allowed to sit overnight.
Silica coating of GNRs
Before silica coating, the as-prepared GNRs were purified by repeated centrifugation (10 000 g, 15 min, 3 times) to remove excess CTAB, and were redispersed in 50 mL water. SH–PEG (10 mg) was dissolved in water (1 mL), sonicated for 10 min, and treated with NaBH4 (50 μL, 0.1 M) under sonication for another 15 min to reduce dimerized SH–PEG (PEG–S–S–PEG), if any exists. GNR surface ligand exchange was conducted by incubating GNRs (10 mL, 0.7 nM) with the SH–PEG for 5 h. After purification with repeated centrifugation (9000g, 12 min, 2 times), GNRs were dispersed into 2.5 mL ethanol, mixed with H2O (0.4 mL), NH4OH (15 μL, 30%), and TEOS (4 μL), and stirred overnight. The resulting GNR@SiO2 was washed three times with ethanol.
Hydrophobic modification of the GNR@SiO2
To a 10 mL ethanol solution of GNR@SiO2, 100 μL of NH4OH (30%) was added to adjust the pH to 9, followed by drop-by-drop addition of an OTMS solution in CHCl3 (250 μL, 10%) under vigorous stirring. The reaction mixture was stirred for 18 h, and the modified GNR@SiO2–C18 nanoparticles were washed with ethanol for three times and dispersed in CHCl3 (3 mL).
Solubilization of hydrophobic GNR@SiO2–C18
To the chloroform solution of GNR@SiO2, PE-PEG (1 mg) was added and sonicated, followed by removal of chloroform by slow evaporation. A film of dried nanoparticles was hydrated with water. Excess PE-PEG were removed by repeated centrifugation (4500g × 15 min, 3 times), and the purified nanoparticles (GNR@SiO2@PE-PEG) were dispersed in 0.5 mL water.
Comparison of colloidal stability
Six aliquots of 50 μL of the GNR solution were diluted with 900 μL DI water, NaCl (100 mM), phosphate buffered saline (PBS, pH 7.4), OptiMEM, and RPMI 1640 cell medium, respectively. Following 1 h incubation, UV absorption, dynamic light scattering, and electron microscopy measurements were taken.
Comparison of photothermal stability
GNR@SiO2@PE-PEG, GNR@CTAB, GNR@SiO2, GNR@PSS, and GNR@SH–PEG nanoparticles solution were adjusted to have a longitudinal plasmon peak intensity of 1.5. Particle suspensions (1 mL) in a cuvette with a path length of 1 cm were illuminated with a doubled YAG pulsed laser with a pulse width of 7 ns and a repetition rate of 20 Hz at 812 nm (Surelite I-20, Continuum, Santa Clara, CA). Samples were subjected to 10 min irradiation at various laser fluencies followed by optical and TEM measurements.
Measurement of photothermal effect using continuous wave (CW) laser
Temperature elevation of GNR solutions (0.1 nM, 200 μL) induced by laser illumination (803 nm CW diode laser at 10 W cm−2) was measured for various illumination durations (0–60 s) using a homemade thermocouple.
Nanoparticle–aptamer bioconjugation
EDC (20 μL, 500 mM) was added to 0.5 mL GNR@SiO2@PE-PEG solution in PBS, followed by addition of the 5′-NH2-modified A10 aptamer targeting PSMA (6 μL, 100 μM) 10 min later. The reaction mixture was stirred for 3 h and another aliquot of fresh EDC solution was added. The reaction mixture was kept on stirring at 4 °C overnight. The aptamer-labeled GNRs were washed, and resuspended in RNase-free water.
Cell viability study based on the MTT assay
LNCaP cells were cultured in RPMI-1640 medium, supplemented with 10% fetal bovine serum (FBS) and antibiotics (100 μg mL−1 streptomycin and 100 U mL−1 penicillin) in a humidified atmosphere at 37 °C with 5% CO2. CellTiter-Blue Cell Viability Assay (Promega, San Luis Obispo, CA) was used to test the toxicity effects of the GNR samples in living cells. After seeding the LNCaP cells for 24 h, nanoparticles in water (20 μL) were added to the cell culture and incubated for another 24 h. Then, after adding 20 μL of the dye solution to each well and incubating for 4 h, fluorescence at 560 nm was recorded using a 96-well plate reader (Tecan Infinite M200).
Tumor cell photothermal therapy
Prostate tumor cells, LNCaP and PC3, were grown to 80% confluence and incubated with the GNR–aptamer bioconjugates (0.48 nM) for 3 h. After washing and changing with fresh culture media, the cells were exposed to NIR laser illumination (803 nm) for 7 min at a power density of 10 W cm−2. The treated cells were incubated for additional 2 h and then stained with a LIVE/DEAD® cell viability kit (Invitrogen, Carlsbad, CA) following the manufacturer’s manual. Live cells (green fluorescence) and dead cells (red fluorescence) were counted on a fluorescence microscope (Olympus, San Diego, CA) under 10× magnification.
3. Results and discussion
GNRs with an aspect ratio of 2.8 are synthesized via a seed-mediated growth method previously reported by Murphy and El-Sayed.56,57 The CTAB bilayer on the surface of GNRs renders GNRs highly positively charged with a zeta potential of +45 mV, which helps maintain the stability of the nanoparticle suspension, but, at the same time, introduces cytotoxicity due to detached CTAB molecules. Both non-porous and mesoporous silica have been previously reported to encapsulate GNRs to improve their biocompatibility.42,43,58 For example, Fernandez-Lopez and coworkers have developed a robust procedure for coating GNRs with silica shells of tunable thickness.43 The key is to first transfer GNRs in water to an ethanol solution by replacing CTAB with SH–PEG so that the classic Stöber chemistry can be applied.59 Indeed, using this procedure we were able to encapsulate GNRs with silica shells ranging from 7 to 30 nm thickness. Fig. 2a shows the transmission electron microscopy (TEM) image of GNR@SiO2 with a representative shell thickness of 7.5 nm. Although the GNRs, compared with other highly uniform spherical nanoparticles, have some size and shape variations, the coated silica shells appear uniform in thickness and smooth. For easy grafting of PEG molecules on the outer surface for optimal biocompatibility, the hydrophilic GNR@SiO2 was converted to hydrophobic first by reacting with a hydrophobic silane compound, OTMS. In spite of the three reactive sites (polymerizable), OTMS does not significantly increase the silica shell thickness, possibly due to steric hindrance caused by its long hydrocarbon chain. Nevertheless, the very thin layer of hydrocarbons efficiently converts the GNR@SiO2 hydrophobic, rendering them soluble in organic solvents such as chloroform. Previous research on semiconductor quantum dots has identified a series of simple and effective approaches based on amphiphilic polymers for solubilization of hydrophobic nanoparticles.51–54 These amphiphilic structures share a couple of common features. First, no ligand exchange is needed for nanoparticle solubilization (original hydrophobic ligands are kept and covered). Second, the amphiphilic polymers self-assemble onto inorganic nanoparticles via multivalent hydrophobic interactions whereas the hydrophilic segments in the polymers face outward. To demonstrate the concept, we used PE-PEG in the current work. The tail chains in PE and the hydrocarbons on GNR–SiO2 interdigitate, whereas the PEG block protrudes from the surface with terminal COOH groups as reactive sites for bioconjugation. The overall yield of this multistep procedure is typically above 70%.
Fig. 2.
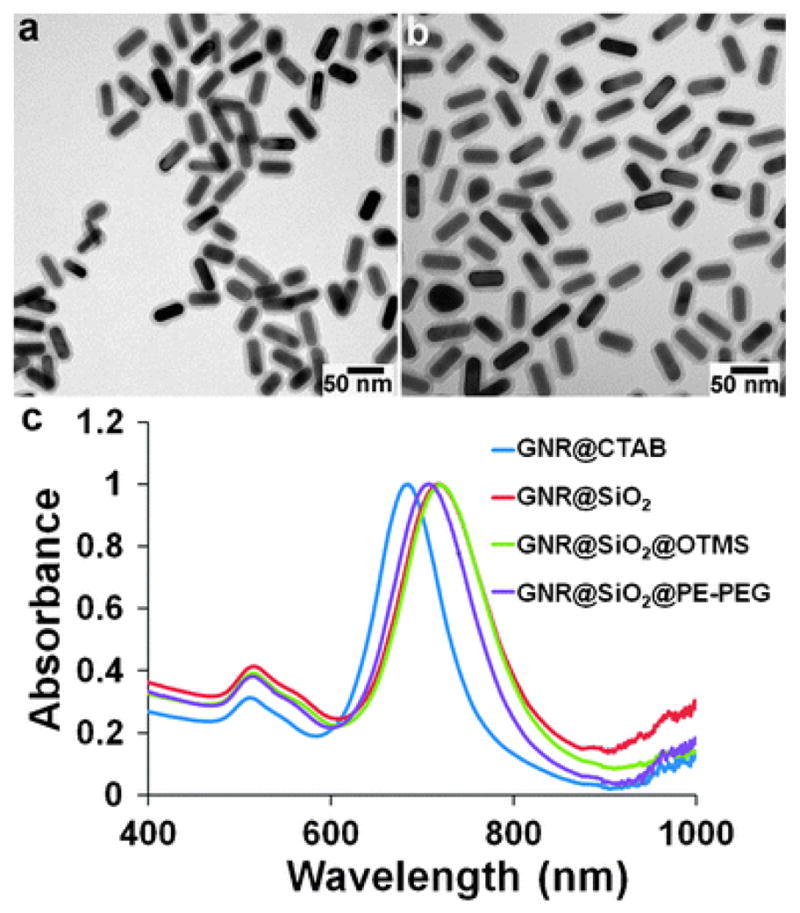
TEM and spectroscopic characterization of caged GNRs. (a and b) TEM images of GNR@SiO2 and GNR@SiO2@PE-PEG. The silica shell is 7.5 nm, and the organic molecules on the silica shell surface are not visible because they are not electron-dense materials. (c) Spectroscopic monitoring of GNR encapsulation. Initial CTAB capped GNRs dispersed in water show two extinction peaks at 515 (transverse) and 685 nm (longitudinal). Subsequent surface modifications with an SiO2 shell (dispersed in ethanol), C18 (dispersed in CHCl3) and PE-PEG (dispersed in water) shift the longitudinal peak to 719, 720, and 708 nm, respectively.
As shown in Fig. 2b, the resulting water-soluble GNR@SiO2@PE-PEG particles are well separated on the TEM grid, which also reflects their dispersity in solution, because if the particles were aggregated in solution, large clusters with particles piled on top of each other would be expected. To confirm the dispersity in solution, we also measured the hydrodynamic size of the particles using dynamic light scattering (DLS). In aqueous solution, the hydrodynamic diameters of GNR@SiO2 and GNR@SiO2@PE-PEG are 76 nm and 97 nm, respectively. The size discrepancy between these two samples in solution (‘dry’ sizes under TEM are similar) is likely resulted from the PEG surface coating and the negative charges on the surface of GNR@SiO2@PE-PEG (PEG terminated with COOH end group), which creates an electrical double layer surrounding the nanoparticles. It is also worth mentioning here that the DLS sizes of GNRs should be treated as an estimate because the numbers are actually expressed as GNRs’ spherical particle equivalents, spheres that have the same translational diffusion speeds as the non-spherical GNRs.
Besides the size measurements using TEM and DLS, the silica and PEG coating process can be monitored with spectroscopic measurements as well, because GNRs’ surface plasmon resonance (SPR) bands in particular the longitudinal band are sensitive to changes in local environments.18,19 As shown in Fig. 2c, the CTAB-coated GNRs in water initially have a transverse absorption band centered at 515 nm and a longitudinal band at 685 nm. Subsequent surface modifications with an SiO2 shell, C18 and PEG give rise to a significant red shift of the longitudinal band to 719, 720, and 708 nm, respectively. This spectral shift is commonly observed in plasmonic materials and has been previously attributed to changes in the refractive index of the surrounding environment.60 At the same time, the transverse band remains nearly unchanged in band location, but is slightly enhanced due to stronger Rayleigh scattering arising from the increased particle size after silica encapsulation.43
Next, we systematically compared the colloidal stability of our GNR@SiO2@PE-PEG with the conventional GNR@CTAB, GNR@SiO2, GNR@PSS, and GNR@SH–PEG dispersed in a variety of aqueous solutions: DI water, NaCl solution (100 mM), phosphate buffered saline (PBS, 1×), and cell culture media OptiMEM and RPMI. The DLS measurement allows a quick assessment of colloidal stability of the GNRs with different surface chemistry. As summarized in Table 1, the original GNR@CTAB and GNR@SiO2 form aggregates in NaCl and PBS solutions as well as OptiMEM, whereas GNR@PSS are not stable in NaCl and PBS solutions. In contrast, only GNR@SH–PEG and GNR@SiO2@PE-PEG remain single under all these conditions, manifested by minor size fluctuations possibly caused by the solvent change and adsorption of solutes (e.g., proteins in cell culture media). These results indicate the importance of the pegylation layer in nanoparticle stabilization. Note that the size fluctuation of GNR@SH–PEG is slightly less than that of GNR@SiO2@PE-PEG. This is because, in contrast to the PE-PEG terminated with COOH groups, the SH–PEG used in this study are terminated with neutral methoxy groups. When GNRs are coated with SH–PEG–COOH, they show a similar level of size fluctuation to that of GNR@SiO2@PE-PEG.
Table 1.
Hydrodynamic diameter of the encapsulated GNRs in different media
| Particle type/DLS diameter in buffers/nm | H2O | 100 mM NaCl | PBS | OptiMEM | RPMI |
|---|---|---|---|---|---|
| GNR@CTAB | 42.5 ± 0.6 | 497.9 ± 5.3 | 457.5 ± 22.5 | 619.9 ± 32.5 | 80.9 ± 2.0 |
| GNR@PSS | 51.5 ± 0.8 | 190.5 ± 23.9 | 348.3 ± 27.9 | 82.1 ± 1.9 | 92.2 ± 6.9 |
| GNR@SH–PEG | 77.0 ± 4.8 | 75.2 ± 0.2 | 78.5 ± 3.8 | 78.7 ± 1.1 | 75.7 ± 0.8 |
| GNR@SiO2 | 75.6 ± 1.3 | 1917.3 ± 123.7 | 1221 ± 110.6 | 1720.3 ± 183.3 | 112.7 ± 1.9 |
| GNR@SiO2@PE-PEG | 97.4 ± 2.2 | 103.6 ± 0.6 | 91.8 ± 3.1 | 104.3 ± 2.1 | 113.0 ± 1.0 |
The DLS data are also confirmed by measurements of GNRs’ SPR absorption, utilizing the sensitivity of the longitudinal bands to colloidal agglomeration (Fig. 3a–e). The spectral measurements are in nearly perfect agreement with the DLS results except for GNR@SiO2. The absorption peaks of GNR@SiO2 in various solutions show relatively small changes although DLS clearly suggests aggregation. This is due to the silica shells on their surfaces which effectively create spatial separations between adjacent GNRs in clusters. Representative TEM images were also taken for GNR@CTAB, GNR@SiO2, and GNR@SiO2@PE-PEG in 100 mM NaCl solution. It shows that GNR@SiO2@PE-PEG are well dispersed but not the other two samples (Fig. 3f–h). Taken together the DLS, TEM, and spectroscopic data, it is clear that direct ligand exchange with SH–PEG and encapsulation with SiO2 and PE-PEG offer the best colloidal stability for GNRs.
Fig. 3.

Spectral and TEM measurements of the colloidal stabilities of GNRs with various surface chemistries. (a–e) Vis-NIR spectra of GNR@CTAB, GNR@PSS, GNR@SH–PEG, GNR@SiO2, and GNR@SiO2@PE-PEG dispersed in water, PBS buffer, 100 mM NaCl solution, OptiMEM, and RPMI cell culture medium. Agglomeration can be detected based on the spectral shift of GNRs’ longitudinal bands. (f–h) Representative TEM images of GNR@CTAB, GNR@SiO2, and GNR@SiO2@PE-PEG dispersed in 100 mM NaCl. This comparison clearly shows that GNR@CTAB and GNR@SiO2 are clustered but GNR@SiO2@PE-PEG remain single.
As discussed above, GNRs’ photothermal stability is another issue that needs to be addressed for applications involving high-intensity laser illumination or extended photothermal conversion such as photoacoustic tomography, multiphoton microscopy, and optically modulated drug release. Systematic work by El-Sayed and coworkers shows that under pulsed laser irradiation, GNRs can melt into spherical particles of similar volumes or even fragment into smaller particles.7,48 High-resolution TEM also reveals that different from thermal melting, which starts from the nanomaterial surface, the photothermal melting process starts with the formation of defects inside nanorods.61 In our study, an 812 nm NIR laser was used to generate pulses of 7 ns with a repetition rate of 20 Hz, and its laser fluence was varied between 0–7.6 mJ cm−2. To match the photon energy with GNRs’ SPR absorption, another GNR sample with a longitudinal absorption band centered at 812 nm was used, thanks to the remarkable size tunability of GNRs. As demonstrated in Fig. 4, the silica-caged GNRs show significantly improved photothermal stability compared with CTAB, PSS, and SH–PEG coated GNRs, in agreement with prior reports.49,50 Based on the spectroscopic measurements, GNR@CTAB and GNR@PSS are already damaged at laser fluence as low as 1.3 mJ cm−2, indicated by a blue-shifted longitudinal peak (Fig. 4a–c). GNR@SH–PEG show slightly better stability, but still follow a similar trend as GNR@CTAB (Fig. 4a and d). For the silica-caged particles (GNR@SiO2 and GNR@SiO2@PE-PEG), their absorptions do not change at a laser fluence of 5.1 mJ cm −2, and drop less than 20% at a laser fluence of 7.6 mJ cm−2 after 10 min illumination (Fig. 4a, e and f). This improved stability likely comes from two effects. First, it is well known that GNR stability is directly linked with its surface ligands.62 It has been reported that the heat relaxation time for GNRs wrapped with CTAB bilayers (also applies for the PSS-coated GNRs, because PSS are adsorbed on top of CTAB via electrostatic interactions) is approximately 150 ps,63,64 significantly longer than the rod reshaping time (shape transformation from rods to spheres takes ca. 35 ps).65 In contrast, the heat relaxation time of GNRs in silica shells has been estimated to be 20 ps,63,64 which is competitive with the photothermal reshaping process. Second, the silica shell is also more rigid than most organic materials such as surfactants and polymers. The high sol–gel glass transition temperature of silica (1000–1500 K) helps keep GNRs in their original shape.
Fig. 4.

Comparison of photothermal stability of GNRs with various surface chemistries. (a) Peak intensity at 812 nm measured after exposure to nanosecond NIR laser at different laser fluencies. GNRs caged with silica are significantly more stable than coated with CTAB, PSS, and SH–PEG. (b–f) Spectra of GNR@CTAB, GNR@PSS, GNR@SH–PEG, GNR@SiO2, and GNR@SiO2@PE-PEG after laser treatments at different laser fluencies.
Before applying this new series of GNRs to tumor cell targeting and photothermal therapy, two additional properties need to be checked first: (i) whether GNR@SiO2@PE-PEG are toxic to cells; and (ii) whether the GNR@SiO2@PE-PEG maintain their capability of heating up the surrounding media when illuminated with a CW laser source. In contrast to pulsed laser that specifically heats GNRs and their very close proximity (10 nm), CW laser illumination warms the surrounding media and is thus frequently used in hyperthermia treatment of tumors. To probe the cytotoxicity and put it in context with other surface coating chemistry, we compared the cytotoxicity of GNR@SiO2@PE-PEG with GNR@CTAB, GNR@SiO2, GNR@PSS, and GNR@SH–PEG towards a prostate cancer cell line, LNCaP. We chose LNCaP because of its well characterized in vivo behavior and tumor biology such as profile of surface receptor expression, which can be used to develop specific targeting strategies.52 Plot of dose-dependent cytotoxicity (Fig. 5a) shows that after 24 hour exposure to cells GNR@SiO2@PE-PEG are essentially non-toxic in the concentration range of 0–0.6 nM, similar to GNR@SiO2, GNR@PSS, and GNR@SH–PEG. The only surface coating that is highly toxic to cells is CTAB, in agreement with the literatures.36–46 To investigate the photothermal conversion capability of the new GNR@SiO2@PE-PEG, we compare it with the original GNR@CTAB and control samples of water and a spherical gold nanoparticle solution. Under the same laser illumination condition, GNR@SiO2@PE-PEG and GNR@CTAB show virtually identical rate of temperature increase, which is approximately one order of magnitude higher than those of spherical particles and pure water (Fig. 5b).
Fig. 5.

Characterization of cytotoxicity and capability of photothermal conversion of GNR@SiO2@PE-PEG. (a) Dose-dependent cytotoxicity of GNR@SiO2@PE-PEG in comparison with GNR@CTAB, GNR@PSS, GNR@SH–PEG, GNR@SiO2 in LNCaP prostate tumor cells. In the concentrations probed (0–0.6 nM), GNR@CTAB show high toxicity but not the other samples (24 h incubation of cells with the various GNRs). (b) Rates of temperature increase (ΔT) for GNR@CTAB, GNR@SiO2@PE-PEG, spherical gold nanoparticles, and water during NIR-laser illumination. When fitted with linear trendlines, GNR@CTAB and GNR@SiO2@PE-PEG show nearly identical performance and their temperature increase rates are approximately 7 and 13 times faster than those of gold nanoparticles and water. (Inset) Vis-NIR extinction spectra of the GNR@SiO2@PE-PEG and spherical gold nanoparticles of 18 nm.
With the systematic characterization studies on stability, biocompatibility and photothermal conversion discussed above, we proceeded to demonstrate specific targeting and thermal ablation of tumor cells using the new GNRs. Photothermal therapy of tumors using targeted plasmonic materials has the potential to improve current clinical tumor ablation approaches (e.g., laser, radio frequency, and high-intensity focused ultrasound or HIFU) that lack specificity, and to open new opportunities for treating small metastasis, and tumors with poorly defined boundaries or embedded in vital regions. Pioneer work by Halas et al. using gold nanoshells has produced encouraging results using cultured cells and lab animals, and the nanoshell agent is currently under clinical trials.66 GNRs have become an important alternative due to their higher absorption efficiency per unit volume. For specific targeting, we conjugated a RNA aptamer (A10) that recognizes a prostate specific membrane antigen (PSMA),67 one of the most specific biomarkers expressed in prostate tumor epithelial cells and attractive targets for imaging and therapeutic interventions (e.g., ProstaScint® scan).68 To evaluate specific targeting, PSMA-positive LNCaP cells and PSMA-negative PC3 cells were incubated with the GNR@SiO2@PE-PEG with or without the aptamer followed by exposure to a NIR laser (803 nm, 10 W cm−2). Cell viabilities were quantified using a dual-color labeling technique with calcein AM and ethidium homodimer 1 (EthD-1), where red fluorescence from EthD-1 indicates dead cells and green fluorescence from calcein AM indicates live cells. Control experiments (Fig. 6a–d), in which either NIR laser illumination or GNRs are absent, show that the cell viabilities are essentially unaffected (>95% live cells). When NIR illumination and GNR–aptamer bioconjugates are combined, majority of the LNCaP cells are killed with the cell viability dropping below 4% (Fig. 6e and f). In contrast, under the same experimental conditions, 99% of PSMA-negative PC3 cells survive the treatment (Fig. 6g and h), indicating remarkable targeting specificity.
Fig. 6.
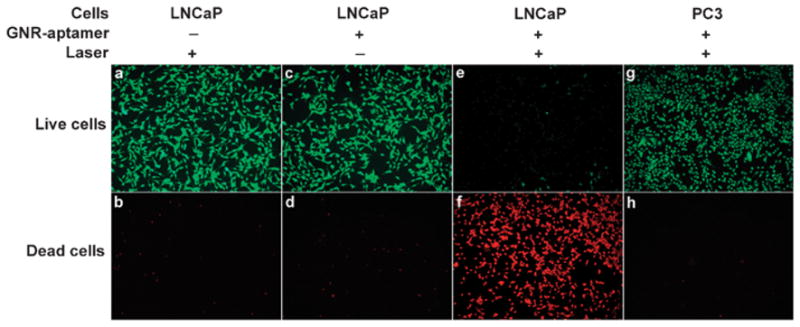
Photothermal therapy of tumor cells. (a–d) When either NIR laser illumination or GNRs targeted with A10 aptamers are missing, the viabilities of LNCaP cells remain high, 95.2% and 97.5% respectively. (e and f) Combined treatment of NIR laser illumination and GNR-A10 triggers a hyperthermia effect and efficiently kills tumor cells (viability 3.5%). (g and h) When PSMA-negative PC3 cells are used under the same condition as that in (e and f), a negligible therapeutic effect is observed (viability 99.6%), illustrating the specificity of the aptamer targeted GNRs.
Besides the targeting specificity and effective photothermal therapy, another important feature worth mentioning is the spectral insensitivity of GNR@SiO2@PE-PEG to aggregation. When plasmonic nanoparticles enter cells through endocytosis (one of the most common pathways for nanoparticle uptake), they often form clusters in endosome and lysosome, which leads to aggregation-induced spectral shifting. For example, when GNR@SH–PEG (targeted with the same PSMA aptamer) enter cells, the spectrum shifts by approximately 20 nm (Fig. S1, ESI†). The unpredictable nature of the aggregation-induced spectral shift renders photothermal therapy difficult because the nanoparticle absorption maximum no longer overlaps with the laser (thus new light source will have to be identified for effective treatment and minimized side effect). In contrast, because of the silica layer, even GNR@SiO2@PE-PEG particles form aggregates during the endocytosis process, the embedded GNRs are still spaced out by a distance at least twice of the silica shell thickness, which helps maintain the original spectrum.
4. Conclusion
In summary, we have developed a new class of GNRs by encapsulating them with a multi-layer coating composed of silica, hydrocarbons and amphiphilic polymer. In comparison with conventional GNRs capped with surfactant micelles, polyelectrolytes, thiol-PEG, and silica shell alone that solve either the toxicity, non-specific binding, colloidal instability, or photothermal instability problems, our nanoencapsulation technology based on intelligent selection of coating materials with complementary functionalities is capable of addressing all these problems simultaneously. As a result of these desirable properties, we were able to functionalize them with an aptamer and specifically target PSMA receptors on prostate tumor cells. In combination with NIR laser illumination, the cell viability of PSMA-positive LNCaP cells is reduced to 4% whereas that of PSMA-negative PC3 cells is virtually unaffected. Further development and optimization of this technology could help stabilize other plasmonic nanostructures that are not in the most thermodynamically or chemically stable states, and could enable sensitive detection and specific treatment of tumors in vivo.
Supplementary Material
Acknowledgments
This work was supported in part by NIH (R01CA131797, R01ES016189, U19ES019545), NSF (0645080), and the UW Department of Bioengineering. X.H.G. thanks the NSF for a Faculty Early Career Development award (CAREER). We are also grateful to Prof. Hong Shen and Hai Nguyen for their CW laser, Prof. Matt O’Donnell and Dr. Congxian Jia for their pulsed laser, and Prof. Terry Kavanagh for fruitful discussion on nanotoxicity.
References
- 1.Chen JY, Wiley B, Li ZY, Campbell D, Saeki F, Cang H, Au L, Lee J, Li XD, Xia YN. Adv Mater. 2005;17:2255–2261. [Google Scholar]
- 2.Daniel MC, Astruc D. Chem Rev (Washington, DC, U S) 2004;104:293–346. doi: 10.1021/cr030698+. [DOI] [PubMed] [Google Scholar]
- 3.Huang XH, Jain PK, El-Sayed IH, El-Sayed MA. Nanomedicine. 2007;2:681–693. doi: 10.2217/17435889.2.5.681. [DOI] [PubMed] [Google Scholar]
- 4.Jain PK, El-Sayed IH, El-Sayed MA. Nano Today. 2007;2:18–29. [Google Scholar]
- 5.Jain PK, Huang XH, El-Sayed IH, El-Sayed MA. Acc Chem Res. 2008;41:1578–1586. doi: 10.1021/ar7002804. [DOI] [PubMed] [Google Scholar]
- 6.Lal S, Clare SE, Halas NJ. Acc Chem Res. 2008;41:1842–1851. doi: 10.1021/ar800150g. [DOI] [PubMed] [Google Scholar]
- 7.Link S, El-Sayed MA. Int Rev Phys Chem. 2000;19:409–453. [Google Scholar]
- 8.Murphy CJ, Gole AM, Hunyadi SE, Stone JW, Sisco PN, Alkilany A, Kinard BE, Hankins P. Chem Commun. 2008:544–557. doi: 10.1039/b711069c. [DOI] [PubMed] [Google Scholar]
- 9.Murphy CJ, Gole AM, Stone JW, Sisco PN, Alkilany AM, Goldsmith EC, Baxter SC. Acc Chem Res. 2008;41:1721–1730. doi: 10.1021/ar800035u. [DOI] [PubMed] [Google Scholar]
- 10.Murphy CJ, San TK, Gole AM, Orendorff CJ, Gao JX, Gou L, Hunyadi SE, Li T. J Phys Chem B. 2005;109:13857–13870. doi: 10.1021/jp0516846. [DOI] [PubMed] [Google Scholar]
- 11.Niemeyer CM. Angew Chem, Int Ed. 2001;40:4128–4158. doi: 10.1002/1521-3773(20011119)40:22<4128::AID-ANIE4128>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 12.Qian XM, Nie SM. Chem Soc Rev. 2008;37:912–920. doi: 10.1039/b708839f. [DOI] [PubMed] [Google Scholar]
- 13.Rosi NL, Mirkin CA. Chem Rev. 2005;105:1547–1562. doi: 10.1021/cr030067f. [DOI] [PubMed] [Google Scholar]
- 14.Sperling RA, Rivera gil P, Zhang F, Zanella M, Parak WJ. Chem Soc Rev. 2008;37:1896–1908. doi: 10.1039/b712170a. [DOI] [PubMed] [Google Scholar]
- 15.West JL, Halas NJ. Annu Rev Biomed Eng. 2003;5:285–292. doi: 10.1146/annurev.bioeng.5.011303.120723. [DOI] [PubMed] [Google Scholar]
- 16.Jin Y, Jia C, Huang S-W, O’Donnell M, Gao X. Nat Commun. 2010;1:41. doi: 10.1038/ncomms1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin YD, Gao XH. Nat Nanotechnol. 2009;4:571–576. doi: 10.1038/nnano.2009.193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee KS, El-Sayed MA. J Phys Chem B. 2006;110:19220–19225. doi: 10.1021/jp062536y. [DOI] [PubMed] [Google Scholar]
- 19.Yu CX, Irudayaraj J. Anal Chem. 2007;79:572–579. doi: 10.1021/ac061730d. [DOI] [PubMed] [Google Scholar]
- 20.Jain PK, Lee KS, El-Sayed IH, El-Sayed MA. J Phys Chem B. 2006;110:7238–7248. doi: 10.1021/jp057170o. [DOI] [PubMed] [Google Scholar]
- 21.Oyelere AK, Chen PC, Huang X, El-Sayed IH, El-Sayed MA. Bioconjugate Chem. 2007;18:1490–1497. doi: 10.1021/bc070132i. [DOI] [PubMed] [Google Scholar]
- 22.Ding H, Yong KT, Roy I, Pudavar HE, Law WC, Bergey EJ, Prasad PN. J Phys Chem C. 2007;111:12552–12557. [Google Scholar]
- 23.Oldenburg AL, Hansen MN, Zweifel DA, Wei A, Boppart SA. Opt Express. 2006;14:6724–6738. doi: 10.1364/oe.14.006724. [DOI] [PubMed] [Google Scholar]
- 24.Imura K, Nagahara T, Okamoto H. J Am Chem Soc. 2004;126:12730–12731. doi: 10.1021/ja047836c. [DOI] [PubMed] [Google Scholar]
- 25.Wang H, Huff TB, Zweifel DA, He W, Low PS, Wei A, Cheng JX. Proc Natl Acad Sci U S A. 2005;102:15752–15756. doi: 10.1073/pnas.0504892102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim K, Huang SW, Ashkenazi S, O’Donnell M, Agarwal A, Kotov NA, Denny MF, Kaplan MJ. Appl Phys Lett. 2007;90:223901–223903. [Google Scholar]
- 27.Takahashi H, Niidome Y, Yamada S. Chem Commun. 2005:2247–2249. doi: 10.1039/b500337g. [DOI] [PubMed] [Google Scholar]
- 28.Chanda N, Shukla R, Katti KV, Kannan R. Nano Lett. 2009;9:1798–1805. doi: 10.1021/nl8037147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee SE, Liu GL, Kim F, Lee LP. Nano Lett. 2009;9:562–570. doi: 10.1021/nl802689k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wijaya A, Schaffer SB, Pallares IG, Hamad-Schifferli K. ACS Nano. 2008;3:80–86. doi: 10.1021/nn800702n. [DOI] [PubMed] [Google Scholar]
- 31.Huang YF, Sefah K, Bamrungsap S, Chang HT, Tan W. Langmuir. 2008;24:11860–11865. doi: 10.1021/la801969c. [DOI] [PubMed] [Google Scholar]
- 32.Huang X, El-Sayed IH, Qian W, El-Sayed MA. J Am Chem Soc. 2006;128:2115–2120. doi: 10.1021/ja057254a. [DOI] [PubMed] [Google Scholar]
- 33.von Maltzahn G, Park JH, Agrawal A, Bandaru NK, Das SK, Sailor MJ, Bhatia SN. Cancer Res. 2009;69:3892–3900. doi: 10.1158/0008-5472.CAN-08-4242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tong L, Zhao Y, Huff T, Hansen M, Wei A, Cheng JX. Adv Mater. 2007;19:3136–3141. doi: 10.1002/adma.200701974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Nikoobakht B, El-Sayed MA. Langmuir. 2001;17:6368–6374. [Google Scholar]
- 36.Connor E, Mwamuka J, Gole A, Murphy C, Wyatt M. Small. 2005;1:325–327. doi: 10.1002/smll.200400093. [DOI] [PubMed] [Google Scholar]
- 37.Takahashi H, Niidome Y, Niidome T, Kaneko K, Kawasaki H, Yamada S. Langmuir. 2005;22:2–5. doi: 10.1021/la0520029. [DOI] [PubMed] [Google Scholar]
- 38.Hauck T, Ghazani A, Chan W. Small. 2008;4:153–159. doi: 10.1002/smll.200700217. [DOI] [PubMed] [Google Scholar]
- 39.Niidome T, Yamagata M, Okamoto Y, Akiyama Y, Takahashi H, Kawano T, Katayama Y, Niidome Y. J Controlled Release. 2006;114:343–347. doi: 10.1016/j.jconrel.2006.06.017. [DOI] [PubMed] [Google Scholar]
- 40.Wang C, Ma Z, Wang T, Su Z. Adv Funct Mater. 2006;16:1673–1678. [Google Scholar]
- 41.Niidome Y, Honda K, Higashimoto K, Kawazumi H, Yamada S, Nakashima N, Sasaki Y, Ishida Y, Kikuchi J-I. Chem Commun. 2007:3777–3779. doi: 10.1039/b706671f. [DOI] [PubMed] [Google Scholar]
- 42.Pastoriza-Santos I, Pérez-Juste J, Liz-Marzán LM. Chem Mater. 2006;18:2465–2467. [Google Scholar]
- 43.Fernández-López C, Mateo-Mateo C, Álvarez-Puebla RnA, Pérez-Juste J, Pastoriza-Santos I, Liz-Marzán LM. Langmuir. 2009;25:13894–13899. doi: 10.1021/la9016454. [DOI] [PubMed] [Google Scholar]
- 44.Gole A, Murphy CJ. Chem Mater. 2005;17:1325–1330. [Google Scholar]
- 45.Leonov AP, Zheng J, Clogston JD, Stern ST, Patri AK, Wei A. ACS Nano. 2008;2:2481–2488. doi: 10.1021/nn800466c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang S, Lu W, Tovmachenko O, Rai US, Yu H, Ray PC. Chem Phys Lett. 2008;463:145–149. doi: 10.1016/j.cplett.2008.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mohamed MB, Volkov V, Link S, El-Sayed MA. Chem Phys Lett. 2000;317:517–523. [Google Scholar]
- 48.Link S, Burda C, Mohamed MB, Nikoobakht B, El-Sayed MA. J Phys Chem A. 1999;103:1165–1170. [Google Scholar]
- 49.Chen LC, Wei CW, Souris JS, Cheng SH, Chen CT, Yang CS, Li PC, Lo LW. J Biomed Opt. 2010;15:016010. doi: 10.1117/1.3292574. [DOI] [PubMed] [Google Scholar]
- 50.Chen YS, Frey W, Kim S, Homan K, Kruizinga P, Sokolov K, Emelianov S. Opt Express. 2010;18:8867–8877. doi: 10.1364/OE.18.008867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Dubertret B, Skourides P, Norris DJ, Noireaux V, Brivanlou AH, Libchaber A. Science. 2002;298:1759–1762. doi: 10.1126/science.1077194. [DOI] [PubMed] [Google Scholar]
- 52.Gao XH, Cui YY, Levenson RM, Chung LWK, Nie SM. Nat Biotechnol. 2004;22:969–976. doi: 10.1038/nbt994. [DOI] [PubMed] [Google Scholar]
- 53.Pellegrino T, Manna L, Kudera S, Liedl T, Koktysh D, Rogach AL, Keller S, Radler J, Natile G, Parak WJ. Nano Lett. 2004;4:703–707. [Google Scholar]
- 54.Wu X, Liu H, Liu J, Haley KN, Treadway JA, Larson JP, Ge N, Peale F, Bruchez MP. Nat Biotechnol. 2003;21:41–46. doi: 10.1038/nbt764. [DOI] [PubMed] [Google Scholar]
- 55.Dassie JP, Liu XY, Thomas GS, Whitaker RM, Thiel KW, Stockdale KR, Meyerholz DK, McCaffrey AP, McNamara JO, Giangrande PH. Nat Biotechnol. 2009;27:839–846. doi: 10.1038/nbt.1560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jana NR, Gearheart L, Murphy CJ. Adv Mater. 2001;13:1389–1393. [Google Scholar]
- 57.Nikoobakht B, El-Sayed MA. Chem Mater. 2003;15:1957–1962. [Google Scholar]
- 58.Gorelikov I, Matsuura N. Nano Lett. 2008;8:369–373. doi: 10.1021/nl0727415. [DOI] [PubMed] [Google Scholar]
- 59.Stöber W, Fink A, Bohn E. J Colloid Interface Sci. 1968;26:62–69. [Google Scholar]
- 60.Huang X, Neretina S, El-Sayed MA. Adv Mater. 2009;21:4880–4910. doi: 10.1002/adma.200802789. [DOI] [PubMed] [Google Scholar]
- 61.Link S, Wang ZL, El-Sayed MA. J Phys Chem B. 2000;104:7867–7870. [Google Scholar]
- 62.Alpert J, Hamad-Schifferli K. Langmuir. 2010;26:3786–3789. doi: 10.1021/la904855s. [DOI] [PubMed] [Google Scholar]
- 63.Chon JWM, Bullen C, Zijlstra P, Gu M. Adv Funct Mater. 2007;17:875–880. [Google Scholar]
- 64.Horiguchi Y, Honda K, Kato Y, Nakashima N, Niidome Y. Langmuir. 2008;24:12026–12031. doi: 10.1021/la800811j. [DOI] [PubMed] [Google Scholar]
- 65.Link S, Burda C, Nikoobakht B, El-Sayed MA. Chem Phys Lett. 1999;315:12–18. [Google Scholar]
- 66.Hirsch LR, Stafford RJ, Bankson JA, Sershen SR, Rivera B, Price RE, Hazle JD, Halas NJ, West JL. Proc Natl Acad Sci U S A. 2003;100:13549–13554. doi: 10.1073/pnas.2232479100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lupold SE, Hicke BJ, Lin Y, Coffey DS. Cancer Res. 2002;62:4029–4033. [PubMed] [Google Scholar]
- 68.Feneley MR, Jan H, Granowska M, Mather SJ, Ellison D, Glass J, Coptcoat M, Kirby RS, Ogden C, Oliver RTD, Badenoch DF, Chinegwundoh FI, Nargund VH, Paris AMI, Britton KE. Prostate Cancer Prostatic Dis. 2000;3:47–52. doi: 10.1038/sj.pcan.4500390. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.