

Abstract
In the title compound, C15H19ClN2OS, the dihedral angle between the amide and thiourea fragments is 58.07 (17)°. The cyclohexane group adopts a chair conformation and is twisted relative to the thiourea fragment, forming a dihedral angle of 87.32 (18)°. In the crystal, N—H⋯S hydrogen bond links the molecules into chains running parallel to the a-axis direction.
Related literature
For related structures and background references, see: Al-abbasi & Kassim (2011 ▶); Nasir et al. (2011 ▶). For further synthetic details, see: Hassan et al. (2008 ▶).
Experimental
Crystal data
C15H19ClN2OS
M r = 310.83
Triclinic,
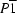
a = 5.042 (2) Å
b = 11.368 (4) Å
c = 15.139 (6) Å
α = 69.865 (7)°
β = 82.698 (8)°
γ = 80.702 (8)°
V = 801.7 (5) Å3
Z = 2
Mo Kα radiation
μ = 0.37 mm−1
T = 298 K
0.52 × 0.23 × 0.03 mm
Data collection
Bruker SMART APEX CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2000 ▶) T min = 0.906, T max = 0.989
9192 measured reflections
3149 independent reflections
1935 reflections with I > 2σ(I)
R int = 0.063
Refinement
R[F 2 > 2σ(F 2)] = 0.085
wR(F 2) = 0.192
S = 1.10
3149 reflections
182 parameters
H-atom parameters constrained
Δρmax = 0.37 e Å−3
Δρmin = −0.21 e Å−3
Data collection: SMART (Bruker, 2000 ▶); cell refinement: SAINT (Bruker, 2000 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: SHELXTL (Sheldrick, 2008 ▶); software used to prepare material for publication: SHELXTL, PARST (Nardelli, 1995 ▶) and PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811025013/hb5920sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811025013/hb5920Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811025013/hb5920Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H1⋯S1i | 0.86 | 2.73 | 3.411 (4) | 137 |
Symmetry code: (i) 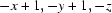 .
.
Acknowledgments
The authors thank Universiti Kebangsaan Malaysia for grants UKM-GUP-BTT-07–30–190 and UKM-OUP-TK-16–73/2010 and sabbatical leave for MBK, and the Kementerian Pengajian Tinggi, Malaysia, for the research fund No. UKM-ST-06-FRGS0111–2009. AAA thanks the Libyan Ministry of Higher Education and Sabha University for her PhD scholarship.
supplementary crystallographic information
Comment
The title compound, (I), is a thiourea derivative analogous to our previously reported compounds (Al-abbasi & Kassim, 2011; Nasir et al., 2011). The thiono S and the carbonyl O adopt a gauche conformation at a partially double N1—C8 bond with C7—N1—C8—S1 torsion angle of -124.4 (3)°. The dihedral angle between the mean planes of the thiourea (S1/N1/N2/C8) and the amide group (O1/N1/C1/C7/C8) is 58.07 (17)°. The cyclohexane has a chair corformation and the mean planes of (C9/C10/C11/C12/C13/C14) and the 4-chlorobenzoyl (Cl1/C1/C2/C3/C4/C5/C6/C7) fragments make an angle of 26.8 (2)°.
In the crystal, intermolecular N1—H···S1 hydrogen bond links the molecules into a one dimentional polymeric structure parallel to the a-axis.
Experimental
The title compound was prepared according to a previously reported compound (Hassan et al., 2008). Colourless plates of (I) were obtained by a slow evaporation of ethanolic solution at room temperature (yield 80%).
Refinement
All H atoms were postioned geometrically with C—H bond lengths in the range 0.93 - 0.97 Å and N—H bond of 0.86 Å,.and refined in the riding model approximation with Uiso(H)=1.2Ueq(C,N), except for methyl group where Uiso(H)= 1.5Ueq(C).
Figures
Fig. 1.

The molecular structure of the title compound with displacement ellipsoids drawn at the 50% probability level.
Fig. 2.

A packing diagram of the title compound down the c-axis showing the intermolecular hydrogen bonds N—H···S (-x+1, -y + 1, -z).
Crystal data
| C15H19ClN2OS | Z = 2 |
| Mr = 310.83 | F(000) = 328 |
| Triclinic, P1 | Dx = 1.288 Mg m−3 |
| Hall symbol: -P 1 | Melting point = 418–420 K |
| a = 5.042 (2) Å | Mo Kα radiation, λ = 0.71073 Å |
| b = 11.368 (4) Å | Cell parameters from 1114 reflections |
| c = 15.139 (6) Å | θ = 1.9–26.0° |
| α = 69.865 (7)° | µ = 0.37 mm−1 |
| β = 82.698 (8)° | T = 298 K |
| γ = 80.702 (8)° | Plate, colourless |
| V = 801.7 (5) Å3 | 0.52 × 0.23 × 0.03 mm |
Data collection
| Bruker SMART APEX CCD diffractometer | 3149 independent reflections |
| Radiation source: fine-focus sealed tube | 1935 reflections with I > 2σ(I) |
| graphite | Rint = 0.063 |
| ω scan | θmax = 26.0°, θmin = 1.9° |
| Absorption correction: multi-scan (SADABS; Bruker, 2000) | h = −6→6 |
| Tmin = 0.906, Tmax = 0.989 | k = −14→14 |
| 9192 measured reflections | l = −18→18 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.085 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.192 | H-atom parameters constrained |
| S = 1.10 | w = 1/[σ2(Fo2) + (0.0826P)2 + 0.0972P] where P = (Fo2 + 2Fc2)/3 |
| 3149 reflections | (Δ/σ)max < 0.001 |
| 182 parameters | Δρmax = 0.37 e Å−3 |
| 0 restraints | Δρmin = −0.21 e Å−3 |
Special details
| Geometry. All e.s.d.'s (except the e.s.d. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell e.s.d.'s are taken into account individually in the estimation of e.s.d.'s in distances, angles and torsion angles; correlations between e.s.d.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell e.s.d.'s is used for estimating e.s.d.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| S1 | 0.3915 (2) | 0.34433 (9) | 0.12184 (8) | 0.0545 (4) | |
| Cl1 | 1.2888 (4) | 0.63599 (15) | −0.45045 (10) | 0.1074 (6) | |
| N1 | 0.7565 (6) | 0.3373 (3) | −0.0182 (2) | 0.0470 (8) | |
| H1 | 0.7996 | 0.4111 | −0.0278 | 0.056* | |
| N2 | 0.7860 (6) | 0.1587 (3) | 0.1141 (2) | 0.0436 (8) | |
| O1 | 0.7203 (6) | 0.1941 (3) | −0.0894 (2) | 0.0647 (9) | |
| C1 | 0.9200 (8) | 0.3791 (3) | −0.1813 (3) | 0.0453 (10) | |
| C8 | 0.6580 (8) | 0.2720 (3) | 0.0727 (3) | 0.0434 (10) | |
| C7 | 0.7916 (8) | 0.2943 (4) | −0.0951 (3) | 0.0496 (10) | |
| C2 | 1.1049 (8) | 0.4556 (4) | −0.1799 (3) | 0.0511 (10) | |
| H2 | 1.1513 | 0.4549 | −0.1221 | 0.061* | |
| C9 | 0.6886 (8) | 0.0807 (3) | 0.2086 (3) | 0.0500 (10) | |
| H9 | 0.4977 | 0.1115 | 0.2179 | 0.060* | |
| C3 | 1.2217 (9) | 0.5326 (4) | −0.2615 (3) | 0.0593 (12) | |
| H3 | 1.3486 | 0.5823 | −0.2593 | 0.071* | |
| C10 | 0.8322 (9) | 0.0976 (4) | 0.2850 (3) | 0.0642 (12) | |
| H10A | 0.8101 | 0.1863 | 0.2789 | 0.077* | |
| H10B | 1.0233 | 0.0699 | 0.2772 | 0.077* | |
| C6 | 0.8558 (9) | 0.3803 (4) | −0.2693 (3) | 0.0614 (12) | |
| H6 | 0.7376 | 0.3271 | −0.2722 | 0.074* | |
| C15 | 1.0483 (8) | 0.1113 (4) | 0.0754 (3) | 0.0561 (11) | |
| H15A | 1.0200 | 0.0601 | 0.0393 | 0.084* | |
| H15B | 1.1572 | 0.0614 | 0.1262 | 0.084* | |
| H15C | 1.1382 | 0.1813 | 0.0354 | 0.084* | |
| C4 | 1.1485 (10) | 0.5351 (4) | −0.3466 (3) | 0.0668 (13) | |
| C5 | 0.9673 (10) | 0.4596 (5) | −0.3505 (3) | 0.0718 (14) | |
| H5 | 0.9201 | 0.4623 | −0.4086 | 0.086* | |
| C14 | 0.7051 (9) | −0.0591 (4) | 0.2195 (4) | 0.0708 (14) | |
| H14A | 0.6045 | −0.0679 | 0.1721 | 0.085* | |
| H14B | 0.8915 | −0.0930 | 0.2098 | 0.085* | |
| C12 | 0.7376 (12) | −0.1166 (6) | 0.3931 (4) | 0.108 (2) | |
| H12A | 0.6573 | −0.1624 | 0.4547 | 0.130* | |
| H12B | 0.9254 | −0.1518 | 0.3886 | 0.130* | |
| C13 | 0.5919 (11) | −0.1324 (5) | 0.3164 (5) | 0.100 (2) | |
| H13A | 0.6101 | −0.2212 | 0.3229 | 0.120* | |
| H13B | 0.4015 | −0.1031 | 0.3240 | 0.120* | |
| C11 | 0.7191 (12) | 0.0218 (6) | 0.3827 (4) | 0.0969 (18) | |
| H11A | 0.8191 | 0.0306 | 0.4303 | 0.116* | |
| H11B | 0.5322 | 0.0549 | 0.3927 | 0.116* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| S1 | 0.0603 (7) | 0.0371 (6) | 0.0621 (7) | 0.0054 (5) | −0.0001 (5) | −0.0182 (5) |
| Cl1 | 0.1343 (14) | 0.1065 (12) | 0.0655 (9) | −0.0256 (10) | 0.0032 (9) | −0.0077 (8) |
| N1 | 0.065 (2) | 0.0364 (17) | 0.045 (2) | −0.0057 (15) | −0.0051 (17) | −0.0215 (15) |
| N2 | 0.0444 (19) | 0.0371 (17) | 0.051 (2) | 0.0010 (14) | −0.0094 (16) | −0.0180 (15) |
| O1 | 0.090 (2) | 0.0534 (18) | 0.065 (2) | −0.0119 (16) | −0.0157 (17) | −0.0333 (16) |
| C1 | 0.049 (2) | 0.043 (2) | 0.046 (2) | 0.0083 (18) | −0.0065 (19) | −0.0231 (19) |
| C8 | 0.051 (2) | 0.038 (2) | 0.050 (2) | −0.0032 (18) | −0.010 (2) | −0.0238 (19) |
| C7 | 0.053 (3) | 0.043 (2) | 0.057 (3) | 0.0087 (19) | −0.016 (2) | −0.025 (2) |
| C2 | 0.051 (3) | 0.054 (2) | 0.055 (3) | 0.001 (2) | −0.006 (2) | −0.030 (2) |
| C9 | 0.042 (2) | 0.039 (2) | 0.067 (3) | −0.0003 (17) | −0.009 (2) | −0.016 (2) |
| C3 | 0.057 (3) | 0.056 (3) | 0.070 (3) | −0.003 (2) | −0.006 (2) | −0.029 (2) |
| C10 | 0.078 (3) | 0.057 (3) | 0.054 (3) | −0.015 (2) | −0.007 (2) | −0.010 (2) |
| C6 | 0.074 (3) | 0.066 (3) | 0.055 (3) | −0.004 (2) | −0.015 (2) | −0.033 (2) |
| C15 | 0.050 (3) | 0.056 (2) | 0.069 (3) | 0.011 (2) | −0.014 (2) | −0.033 (2) |
| C4 | 0.073 (3) | 0.064 (3) | 0.056 (3) | 0.002 (3) | −0.003 (3) | −0.017 (2) |
| C5 | 0.085 (4) | 0.087 (4) | 0.047 (3) | 0.002 (3) | −0.018 (3) | −0.028 (3) |
| C14 | 0.059 (3) | 0.037 (2) | 0.112 (4) | −0.002 (2) | −0.019 (3) | −0.016 (3) |
| C12 | 0.080 (4) | 0.093 (5) | 0.103 (5) | −0.011 (3) | 0.005 (4) | 0.024 (4) |
| C13 | 0.062 (3) | 0.045 (3) | 0.163 (6) | −0.013 (2) | −0.003 (4) | 0.003 (3) |
| C11 | 0.106 (5) | 0.104 (5) | 0.061 (3) | −0.018 (4) | −0.004 (3) | 0.000 (3) |
Geometric parameters (Å, °)
| S1—C8 | 1.687 (4) | C10—H10A | 0.9700 |
| Cl1—C4 | 1.739 (5) | C10—H10B | 0.9700 |
| N1—C8 | 1.391 (5) | C6—C5 | 1.365 (6) |
| N1—C7 | 1.391 (5) | C6—H6 | 0.9300 |
| N1—H1 | 0.8600 | C15—H15A | 0.9600 |
| N2—C8 | 1.321 (4) | C15—H15B | 0.9600 |
| N2—C9 | 1.470 (5) | C15—H15C | 0.9600 |
| N2—C15 | 1.474 (5) | C4—C5 | 1.370 (6) |
| O1—C7 | 1.221 (4) | C5—H5 | 0.9300 |
| C1—C2 | 1.381 (5) | C14—C13 | 1.507 (7) |
| C1—C6 | 1.406 (5) | C14—H14A | 0.9700 |
| C1—C7 | 1.474 (5) | C14—H14B | 0.9700 |
| C2—C3 | 1.370 (6) | C12—C11 | 1.515 (8) |
| C2—H2 | 0.9300 | C12—C13 | 1.524 (8) |
| C9—C10 | 1.520 (6) | C12—H12A | 0.9700 |
| C9—C14 | 1.530 (5) | C12—H12B | 0.9700 |
| C9—H9 | 0.9800 | C13—H13A | 0.9700 |
| C3—C4 | 1.375 (6) | C13—H13B | 0.9700 |
| C3—H3 | 0.9300 | C11—H11A | 0.9700 |
| C10—C11 | 1.524 (6) | C11—H11B | 0.9700 |
| C8—N1—C7 | 126.1 (3) | N2—C15—H15A | 109.5 |
| C8—N1—H1 | 117.0 | N2—C15—H15B | 109.5 |
| C7—N1—H1 | 117.0 | H15A—C15—H15B | 109.5 |
| C8—N2—C9 | 120.4 (3) | N2—C15—H15C | 109.5 |
| C8—N2—C15 | 122.6 (3) | H15A—C15—H15C | 109.5 |
| C9—N2—C15 | 116.5 (3) | H15B—C15—H15C | 109.5 |
| C2—C1—C6 | 118.3 (4) | C5—C4—C3 | 120.9 (4) |
| C2—C1—C7 | 123.2 (4) | C5—C4—Cl1 | 119.9 (4) |
| C6—C1—C7 | 118.5 (4) | C3—C4—Cl1 | 119.2 (4) |
| N2—C8—N1 | 116.8 (3) | C6—C5—C4 | 120.3 (4) |
| N2—C8—S1 | 125.5 (3) | C6—C5—H5 | 119.9 |
| N1—C8—S1 | 117.8 (3) | C4—C5—H5 | 119.9 |
| O1—C7—N1 | 121.5 (4) | C13—C14—C9 | 110.6 (4) |
| O1—C7—C1 | 124.2 (4) | C13—C14—H14A | 109.5 |
| N1—C7—C1 | 114.4 (3) | C9—C14—H14A | 109.5 |
| C3—C2—C1 | 121.6 (4) | C13—C14—H14B | 109.5 |
| C3—C2—H2 | 119.2 | C9—C14—H14B | 109.5 |
| C1—C2—H2 | 119.2 | H14A—C14—H14B | 108.1 |
| N2—C9—C10 | 111.3 (3) | C11—C12—C13 | 110.4 (5) |
| N2—C9—C14 | 113.4 (4) | C11—C12—H12A | 109.6 |
| C10—C9—C14 | 110.9 (4) | C13—C12—H12A | 109.6 |
| N2—C9—H9 | 107.0 | C11—C12—H12B | 109.6 |
| C10—C9—H9 | 107.0 | C13—C12—H12B | 109.6 |
| C14—C9—H9 | 107.0 | H12A—C12—H12B | 108.1 |
| C2—C3—C4 | 119.0 (4) | C14—C13—C12 | 111.1 (4) |
| C2—C3—H3 | 120.5 | C14—C13—H13A | 109.4 |
| C4—C3—H3 | 120.5 | C12—C13—H13A | 109.4 |
| C9—C10—C11 | 110.8 (4) | C14—C13—H13B | 109.4 |
| C9—C10—H10A | 109.5 | C12—C13—H13B | 109.4 |
| C11—C10—H10A | 109.5 | H13A—C13—H13B | 108.0 |
| C9—C10—H10B | 109.5 | C12—C11—C10 | 111.0 (5) |
| C11—C10—H10B | 109.5 | C12—C11—H11A | 109.4 |
| H10A—C10—H10B | 108.1 | C10—C11—H11A | 109.4 |
| C5—C6—C1 | 120.0 (4) | C12—C11—H11B | 109.4 |
| C5—C6—H6 | 120.0 | C10—C11—H11B | 109.4 |
| C1—C6—H6 | 120.0 | H11A—C11—H11B | 108.0 |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H1···S1i | 0.86 | 2.73 | 3.411 (4) | 137 |
Symmetry codes: (i) −x+1, −y+1, −z.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: HB5920).
References
- Al-abbasi, A. A. & Kassim, M. B. (2011). Acta Cryst. E67, o611. [DOI] [PMC free article] [PubMed]
- Bruker (2000). SADABS, SMART and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Hassan, I. N., Yamin, B. M. & Kassim, M. B. (2008). Acta Cryst. E64, o1727. [DOI] [PMC free article] [PubMed]
- Nardelli, M. (1995). J. Appl. Cryst. 28, 659.
- Nasir, M. F. M., Hassan, I. N., Wan Daud, W. R., Yamin, B. M. & Kassim, M. B. (2011). Acta Cryst. E67, o1218. [DOI] [PMC free article] [PubMed]
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811025013/hb5920sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811025013/hb5920Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811025013/hb5920Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report