

Abstract
The molecule of the title compound, C8H8INO2, amide-typical resonance shortens the nominal C—N single bond to 1.322 (7) Å. In the crystal, hydrogen bonds involving both nitrogen-bound H atoms as well as C—H⋯O contacts connect the molecules into double layers approximately perpendicular to the crystallographic b axis. No π-stacking is apparent in the crystal structure.
Related literature
For the crystal structure of 2-(4-nitrophenoxy)acetamide, see: Lakshmi Rao et al. (1987 ▶) and of 2-(4-chloro-2-methylphenoxy)acetamide, see: Rao et al. (1987 ▶). For graph-set analysis of hydrogen bonds, see: Etter et al. (1990 ▶); Bernstein et al. (1995 ▶). For a description of the Cambridge Structural Database, see: Allen (2002 ▶). For the preparation, see: Glover et al. (1973 ▶).
Experimental
Crystal data
C8H8INO2
M r = 277.05
Monoclinic,

a = 5.1411 (4) Å
b = 26.473 (2) Å
c = 7.2960 (7) Å
β = 109.564 (3)°
V = 935.66 (14) Å3
Z = 4
Mo Kα radiation
μ = 3.38 mm−1
T = 200 K
0.55 × 0.18 × 0.10 mm
Data collection
Bruker APEXII CCD diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2008 ▶) T min = 0.824, T max = 1.000
8332 measured reflections
2282 independent reflections
2089 reflections with I > 2σ(I)
R int = 0.020
Refinement
R[F 2 > 2σ(F 2)] = 0.064
wR(F 2) = 0.120
S = 1.29
2282 reflections
109 parameters
H-atom parameters constrained
Δρmax = 1.50 e Å−3
Δρmin = −1.58 e Å−3
Data collection: APEX2 (Bruker, 2010 ▶); cell refinement: SAINT (Bruker, 2010 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXS97 (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXL97 (Sheldrick, 2008 ▶); molecular graphics: ORTEP-3 (Farrugia, 1997 ▶) and Mercury (Macrae et al., 2006 ▶); software used to prepare material for publication: SHELXL97 and PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811025840/aa2016sup1.cif
Supplementary material file. DOI: 10.1107/S1600536811025840/aa2016Isup2.cdx
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811025840/aa2016Isup3.hkl
Supplementary material file. DOI: 10.1107/S1600536811025840/aa2016Isup4.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| N1—H71⋯O1i | 0.88 | 2.00 | 2.881 (6) | 178 |
| N1—H72⋯O1ii | 0.88 | 2.28 | 2.954 (6) | 133 |
| C2—H21⋯O1iii | 0.99 | 2.48 | 3.422 (8) | 158 |
Symmetry codes: (i)  ; (ii)
; (ii) 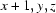 ; (iii)
; (iii)  .
.
Acknowledgments
The authors thank Mrs Vida Maqoko for helpful discussions.
supplementary crystallographic information
Comment
Unlike carboxylic acids, their pertaining amides have not been studied extensively as ligands in coordination chemistry. Owing to their versatility in terms of denticity, keto-enol tautomerism as well as their possible use as neutral – or upon deprotonation – anionic ligands we set out to investigate the coordination behaviour of substituted acetamide derivatives to elucidate the rules guiding the formation of complex compounds. To enable comparative studies with envisioned reaction products, we determined the molecular and crystal structure of the title compound. So far, only the structures of 2-(4-nitrophenoxy)acetamide (Lakshmi Rao et al., 1987) and of 2-(4-chloro-2-methylphenoxy)acetamide (Rao et al., 1987) have been discussed as examples of phenoxy-substituted derivatives of acetamide.
The C–N single bond is shortened to 1.322 (7) Å due to the amide-typical resonance. This value is in good agreement with other derivatives of acetamide whose crystallographic data has been deposited with the Cambridge Structural Database (Allen, 2002) and whose ketonic oxygen atom is not involved in donor action towards transition metals. Intracyclic C–C–C angles hardly deviate from the ideal value of 120 °. The least-squares planes defined by the acetamide moiety and the oxygen atom of the phenoxy-derivative substituent on the one hand and the carbon atoms of the carbocycle on the other hand intersect at an angle of 24.91 (29) ° (Fig. 1 and Fig. 2).
In the crystal structure, both nitrogen-bonded H atoms participate in hydrogen bonds which invariably have the carbonyl oxygen atom as acceptor. While one of the H atoms of the amino group connects the molecules to centrosymmetric dimeric subunits, the other H atom of the NH2 group connects these dimers to chains along [1 0 0]. Additionally, one of the hydrogen atoms of the methylene group forms a C–H···O contact whose range falls by more than 0.2 Å below the sum of van-der-Waals radii of the respective atoms. Again, the double-bonded O atom acts as acceptor. In terms of graph-set analysis (Etter et al., 1990; Bernstein et al., 1995), the hydrogen bonds stemming from the amino group can be described by a C11(4)R22(8) descriptor on the unitary level while the C–H···O contacts necessitate a R22(4) descriptor on the same level. In total, the molecules are connected to double layers approximately perpendicular to the crystallographic b-axis (Fig. 3). No π-stacking is apparent in the crystal structure.
The packing of the compound is shown in Figure 4.
Experimental
The compound was prepared upon reacting 2-phenoxyacetamide with tert-butyl hypochlorite and iodine according to a published procedure (Glover et al., 1973).
Refinement
Carbon-bound H-atoms were placed in calculated positions (C—H 0.99 Å for the methylene group and C—H 0.95 Å for aromatic carbon atoms) and were included in the refinement in the riding model approximation, with U(H) set to 1.2Ueq(C). Nitrogen-bound H-atoms were placed in calculated positions (N—H 0.88 Å) and were included in the refinement in the riding model approximation, with U(H) set to 1.2Ueq(N).
Figures
Fig. 1.

The molecular structure of the title compound, with atom labels and anisotropic displacement ellipsoids (drawn at 50% probability level).
Fig. 2.

Observed distribution of C–N bond lengths in the O=C—NH2 moiety of acetamide derivatives in which the O atom is not acting as a donor atom in coordination compounds (data based on CSD search including all deposited crystal structures up to November 2010).
Fig. 3.

Intermolecular contacts, viewed along [0 0 - 1]. Symmetry operators: ix - 1, y, z; ii -x + 1, -y, -z + 1; iii -x + 2, -y, -z + 2; ivx + 1, y, z.
Fig. 4.

Molecular packing of the title compound, viewed along [-1 0 0] (anisotropic displacement ellipsoids drawn at 50% probability level).
Crystal data
| C8H8INO2 | F(000) = 528 |
| Mr = 277.05 | Dx = 1.967 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 5293 reflections |
| a = 5.1411 (4) Å | θ = 3.1–28.2° |
| b = 26.473 (2) Å | µ = 3.38 mm−1 |
| c = 7.2960 (7) Å | T = 200 K |
| β = 109.564 (3)° | Rod, colourless |
| V = 935.66 (14) Å3 | 0.55 × 0.18 × 0.10 mm |
| Z = 4 |
Data collection
| Bruker APEXII CCD diffractometer | 2282 independent reflections |
| Radiation source: fine-focus sealed tube | 2089 reflections with I > 2σ(I) |
| graphite | Rint = 0.020 |
| φ and ω scans | θmax = 28.2°, θmin = 3.1° |
| Absorption correction: multi-scan (SADABS; Bruker, 2008) | h = −6→6 |
| Tmin = 0.824, Tmax = 1.000 | k = −34→35 |
| 8332 measured reflections | l = −8→9 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.064 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.120 | H-atom parameters constrained |
| S = 1.29 | w = 1/[σ2(Fo2) + (0.0042P)2 + 6.0795P] where P = (Fo2 + 2Fc2)/3 |
| 2282 reflections | (Δ/σ)max < 0.001 |
| 109 parameters | Δρmax = 1.50 e Å−3 |
| 0 restraints | Δρmin = −1.58 e Å−3 |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| I1 | 0.81735 (14) | 0.20849 (2) | −0.25506 (9) | 0.0685 (2) | |
| O1 | 0.6872 (8) | 0.01765 (17) | 0.7947 (6) | 0.0390 (10) | |
| O2 | 1.0024 (8) | 0.08086 (17) | 0.4969 (6) | 0.0416 (10) | |
| N1 | 1.1340 (9) | 0.0300 (2) | 0.8286 (7) | 0.0367 (11) | |
| H71 | 1.1917 | 0.0152 | 0.9432 | 0.044* | |
| H72 | 1.2543 | 0.0422 | 0.7781 | 0.044* | |
| C1 | 0.8667 (10) | 0.0339 (2) | 0.7322 (8) | 0.0278 (11) | |
| C2 | 0.7756 (11) | 0.0587 (2) | 0.5354 (9) | 0.0359 (13) | |
| H21 | 0.6883 | 0.0332 | 0.4343 | 0.043* | |
| H22 | 0.6365 | 0.0850 | 0.5302 | 0.043* | |
| C11 | 0.9411 (12) | 0.1079 (2) | 0.3245 (9) | 0.0345 (12) | |
| C12 | 1.1409 (13) | 0.1428 (2) | 0.3187 (10) | 0.0399 (14) | |
| H12 | 1.3013 | 0.1474 | 0.4296 | 0.048* | |
| C13 | 1.1073 (13) | 0.1706 (2) | 0.1538 (11) | 0.0425 (15) | |
| H13 | 1.2466 | 0.1936 | 0.1489 | 0.051* | |
| C14 | 0.8721 (14) | 0.1650 (2) | −0.0038 (10) | 0.0408 (14) | |
| C15 | 0.6678 (15) | 0.1314 (3) | 0.0021 (10) | 0.0440 (15) | |
| H15 | 0.5036 | 0.1281 | −0.1069 | 0.053* | |
| C16 | 0.7036 (13) | 0.1029 (3) | 0.1658 (10) | 0.0435 (15) | |
| H16 | 0.5647 | 0.0797 | 0.1699 | 0.052* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| I1 | 0.0910 (5) | 0.0588 (3) | 0.0581 (3) | −0.0160 (3) | 0.0282 (3) | 0.0173 (3) |
| O1 | 0.0201 (19) | 0.062 (3) | 0.038 (2) | −0.0006 (18) | 0.0138 (17) | 0.007 (2) |
| O2 | 0.027 (2) | 0.056 (3) | 0.042 (2) | −0.0059 (19) | 0.0109 (18) | 0.014 (2) |
| N1 | 0.022 (2) | 0.055 (3) | 0.035 (3) | 0.001 (2) | 0.012 (2) | 0.011 (2) |
| C1 | 0.022 (3) | 0.035 (3) | 0.029 (3) | 0.001 (2) | 0.013 (2) | −0.001 (2) |
| C2 | 0.022 (3) | 0.049 (3) | 0.038 (3) | −0.002 (2) | 0.012 (2) | 0.003 (3) |
| C11 | 0.030 (3) | 0.037 (3) | 0.041 (3) | 0.007 (2) | 0.018 (3) | 0.010 (2) |
| C12 | 0.031 (3) | 0.035 (3) | 0.051 (4) | −0.002 (2) | 0.012 (3) | 0.005 (3) |
| C13 | 0.039 (3) | 0.031 (3) | 0.063 (4) | −0.004 (3) | 0.024 (3) | 0.008 (3) |
| C14 | 0.052 (4) | 0.032 (3) | 0.046 (4) | 0.001 (3) | 0.026 (3) | 0.000 (3) |
| C15 | 0.050 (4) | 0.047 (4) | 0.035 (3) | −0.007 (3) | 0.014 (3) | −0.001 (3) |
| C16 | 0.038 (3) | 0.054 (4) | 0.044 (4) | −0.010 (3) | 0.020 (3) | 0.000 (3) |
Geometric parameters (Å, °)
| I1—C14 | 2.102 (6) | C11—C16 | 1.380 (9) |
| O1—C1 | 1.236 (6) | C11—C12 | 1.392 (8) |
| O2—C11 | 1.389 (7) | C12—C13 | 1.371 (9) |
| O2—C2 | 1.415 (6) | C12—H12 | 0.9500 |
| N1—C1 | 1.322 (7) | C13—C14 | 1.370 (10) |
| N1—H71 | 0.8800 | C13—H13 | 0.9500 |
| N1—H72 | 0.8800 | C14—C15 | 1.388 (9) |
| C1—C2 | 1.503 (8) | C15—C16 | 1.372 (9) |
| C2—H21 | 0.9900 | C15—H15 | 0.9500 |
| C2—H22 | 0.9900 | C16—H16 | 0.9500 |
| C11—O2—C2 | 116.2 (4) | C13—C12—C11 | 120.3 (6) |
| C1—N1—H71 | 120.0 | C13—C12—H12 | 119.8 |
| C1—N1—H72 | 120.0 | C11—C12—H12 | 119.8 |
| H71—N1—H72 | 120.0 | C14—C13—C12 | 119.7 (6) |
| O1—C1—N1 | 123.2 (5) | C14—C13—H13 | 120.1 |
| O1—C1—C2 | 118.2 (5) | C12—C13—H13 | 120.1 |
| N1—C1—C2 | 118.6 (5) | C13—C14—C15 | 120.5 (6) |
| O2—C2—C1 | 110.9 (4) | C13—C14—I1 | 119.7 (5) |
| O2—C2—H21 | 109.5 | C15—C14—I1 | 119.8 (5) |
| C1—C2—H21 | 109.5 | C16—C15—C14 | 119.8 (6) |
| O2—C2—H22 | 109.5 | C16—C15—H15 | 120.1 |
| C1—C2—H22 | 109.5 | C14—C15—H15 | 120.1 |
| H21—C2—H22 | 108.0 | C15—C16—C11 | 120.0 (6) |
| C16—C11—O2 | 125.4 (5) | C15—C16—H16 | 120.0 |
| C16—C11—C12 | 119.6 (6) | C11—C16—H16 | 120.0 |
| O2—C11—C12 | 115.1 (5) | ||
| C11—O2—C2—C1 | −175.7 (5) | C12—C13—C14—C15 | −0.1 (10) |
| O1—C1—C2—O2 | 172.4 (5) | C12—C13—C14—I1 | 179.0 (5) |
| N1—C1—C2—O2 | −8.5 (8) | C13—C14—C15—C16 | −1.1 (10) |
| C2—O2—C11—C16 | −20.4 (9) | I1—C14—C15—C16 | 179.8 (5) |
| C2—O2—C11—C12 | 159.1 (5) | C14—C15—C16—C11 | 0.5 (10) |
| C16—C11—C12—C13 | −2.6 (9) | O2—C11—C16—C15 | −179.2 (6) |
| O2—C11—C12—C13 | 177.9 (6) | C12—C11—C16—C15 | 1.3 (10) |
| C11—C12—C13—C14 | 2.0 (10) |
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| N1—H71···O1i | 0.88 | 2.00 | 2.881 (6) | 178 |
| N1—H72···O1ii | 0.88 | 2.28 | 2.954 (6) | 133 |
| C2—H21···O1iii | 0.99 | 2.48 | 3.422 (8) | 158 |
Symmetry codes: (i) −x+2, −y, −z+2; (ii) x+1, y, z; (iii) −x+1, −y, −z+1.
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: AA2016).
References
- Allen, F. H. (2002). Acta Cryst. B58, 380–388. [DOI] [PubMed]
- Bernstein, J., Davis, R. E., Shimoni, L. & Chang, N.-L. (1995). Angew. Chem. Int. Ed. Engl. 34, 1555–1573.
- Bruker (2008). SADABS Bruker Inc., Madison, Wisconsin, USA.
- Bruker (2010). APEX2 and SAINT Bruker AXS Inc., Madison, Wisconsin, USA.
- Etter, M. C., MacDonald, J. C. & Bernstein, J. (1990). Acta Cryst. B46, 256–262. [DOI] [PubMed]
- Farrugia, L. J. (1997). J. Appl. Cryst. 30, 565.
- Glover, S. A., Goosen, A. & Laue, H. A. H. (1973). J. S. Afr. Chem. Inst. 26, 77–81.
- Lakshmi Rao, Bh., Seshadri, T. P. & Rao, L. M. (1987). Acta Cryst. C43, 1924–1927.
- Macrae, C. F., Edgington, P. R., McCabe, P., Pidcock, E., Shields, G. P., Taylor, R., Towler, M. & van de Streek, J. (2006). J. Appl. Cryst. 39, 453–457.
- Rao, B. L., Seshadri, T. P. & Rao, L. M. (1987). Z. Kristallogr. C180, 37–42.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) I, global. DOI: 10.1107/S1600536811025840/aa2016sup1.cif
Supplementary material file. DOI: 10.1107/S1600536811025840/aa2016Isup2.cdx
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811025840/aa2016Isup3.hkl
Supplementary material file. DOI: 10.1107/S1600536811025840/aa2016Isup4.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report