

Abstract
The asymmetric unit of the title compound, C20H28N4 2+·2Br−, consists of half a 3,3′-di-n-propyl-1,1′-[p-phenylenenis(methylene)]diimidazolium cation and a bromide anion. The cation is located on an inversion center and adopts an ⋯AAA⋯ trans conformation. In the crystal, the cation is linked to the anions via weak C—H⋯Br hydrogen bonds.
Related literature
For details of N-heterocyclic carbenes, see: Herrmann et al. (1998 ▶); Zhang & Trudell (2000 ▶); Lee et al. (2004 ▶). For structures with similar ⋯AAA⋯ trans conformations, see: Chen et al. (2007 ▶); Cheng et al. (2009 ▶).
Experimental
Crystal data
C20H28N4 2+·2Br−
M r = 484.28
Monoclinic,
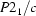
a = 8.9420 (2) Å
b = 11.2443 (2) Å
c = 11.3536 (2) Å
β = 109.716 (1)°
V = 1074.64 (4) Å3
Z = 2
Mo Kα radiation
μ = 3.78 mm−1
T = 296 K
0.37 × 0.35 × 0.30 mm
Data collection
Bruker SMART APEXII CCD area-detector diffractometer
Absorption correction: multi-scan (SADABS; Bruker, 2009 ▶) T min = 0.338, T max = 0.397
11799 measured reflections
3127 independent reflections
2409 reflections with I > 2σ(I)
R int = 0.030
Refinement
R[F 2 > 2σ(F 2)] = 0.030
wR(F 2) = 0.078
S = 1.04
3127 reflections
118 parameters
H-atom parameters constrained
Δρmax = 0.30 e Å−3
Δρmin = −0.59 e Å−3
Data collection: APEX2 (Bruker, 2009 ▶); cell refinement: SAINT (Bruker, 2009 ▶); data reduction: SAINT; program(s) used to solve structure: SHELXTL (Sheldrick, 2008 ▶); program(s) used to refine structure: SHELXTL; molecular graphics: SHELXTL; software used to prepare material for publication: SHELXTL and PLATON (Spek, 2009 ▶).
Supplementary Material
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536811026146/lh5273sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811026146/lh5273Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811026146/lh5273Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report
Table 1. Hydrogen-bond geometry (Å, °).
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
|---|---|---|---|---|
| C7—H7A⋯Br1 | 0.93 | 2.77 | 3.6222 (18) | 152 |
Acknowledgments
RAH and SFN thank Universiti Sains Malaysia (USM) for the FRGS fund (203/PKIMIA/671115), a short term grant (304/PKIMIA/639001) and an RU grant (1001/PKIMIA/813023). HKF and MH thank the Malaysian Government and USM for the Research University Grant No. 1001/PFIZIK/811160. MH also thanks Universiti Sains Malaysia for a post-doctoral research fellowship.
supplementary crystallographic information
Comment
N-Heterocyclic carbene (NHC) ligands have been shown to have wide applicability in coordination chemistry and catalysis. Current research efforts are devoted to the discovery of efficient metal NHC catalysts. For example, chelating palladium complexes of bis(NHC) carbenes have been found to be efficient catalysts in C–C coupling reactions (Herrmann et al., 1998; Zhang & Trudell, 2000). NHC ligands are generally accessible via the deprotonation of imidazolium salts. The preparation of chelating bis(NHC) ligands are also receiving much attention, since they can provide extra air and moisture stability for the metal centers. Several bis(imidazolium) halides, as bis(NHC) ligand precursors, have been synthesized and structurally characterized by us (Lee et al., 2004). We report here the structure of 3,3'-Di-n-propyl-1,1'-(p- phenylenedimethylene)diimidazolium dibromide, (I).
The asymmetric unit of the title compound, consists of a half of the 3,3'-Di-n-propyl-1,1'-(p-phenylenedimethylene) diimidazolium cation (located on a crystallographic inversion center) and a bromide anion (Fig. 1). The cation adopts the ···AAA··· trans conformation in the solid state. This conformation is the same as that found for the neutral N1,N2-di(2-pyridyl)adipoamide ligand which cocrystallizes with water and 2-{5-[N-(2-Pyridyl)carbamoyl]pentanamido} pyridinium hexafluorophosphate (Chen et al., 2007; Cheng et al., 2009).
In the crystal structure (Fig.2), the cations and anions are linked via C7—H7A···Br1 (Table 1) hydrogen bonds.
Experimental
To a solution of 1,4-bis((1H-imidazol-1-yl)methyl)benzene (1.0 g, 4.2 mmol) in 15 ml of acetonitrile, 1-bromopropane (1.0 g, 8.4 mmol) was added. The mixture was refluxed at 363 K for 24 h. The resulting white precipitate was filtered, washed with fresh acetonitrile (2 X 3 ml) and recrystallised from methanol to give colorless crystals. Yield :1.3 g, (93%); m.p: 521–523 K. Crystals suitable for X-ray diffraction studies were obtained by slow evaporation of the salt solution in methanol at ambient temperature.
Refinement
All hydrogen atoms were positioned geometrically [C–H = 0.93–0.97 Å] and were refined using a riding model, with Uiso(H) = 1.2 or 1.5 Ueq(C). A rotating group model was applied to the methyl groups.
Figures
Fig. 1.

The molecular structure of the title compound, showing 50% probability displacement ellipsoids. Only the unique anion is shown (symmetry code (A): -x, -y+1, -z+1).
Fig. 2.

The crystal packing of the title compound, showing weak hydrogen bonds as dashed lines.
Crystal data
| C20H28N42+·2Br− | F(000) = 492 |
| Mr = 484.28 | Dx = 1.497 Mg m−3 |
| Monoclinic, P21/c | Mo Kα radiation, λ = 0.71073 Å |
| Hall symbol: -P 2ybc | Cell parameters from 4287 reflections |
| a = 8.9420 (2) Å | θ = 2.4–29.8° |
| b = 11.2443 (2) Å | µ = 3.78 mm−1 |
| c = 11.3536 (2) Å | T = 296 K |
| β = 109.716 (1)° | Block, colourless |
| V = 1074.64 (4) Å3 | 0.37 × 0.35 × 0.30 mm |
| Z = 2 |
Data collection
| Bruker SMART APEXII CCD area-detector diffractometer | 3127 independent reflections |
| Radiation source: fine-focus sealed tube | 2409 reflections with I > 2σ(I) |
| graphite | Rint = 0.030 |
| φ and ω scans | θmax = 30.1°, θmin = 2.4° |
| Absorption correction: multi-scan (SADABS; Bruker, 2009) | h = −12→12 |
| Tmin = 0.338, Tmax = 0.397 | k = −15→15 |
| 11799 measured reflections | l = −11→16 |
Refinement
| Refinement on F2 | Primary atom site location: structure-invariant direct methods |
| Least-squares matrix: full | Secondary atom site location: difference Fourier map |
| R[F2 > 2σ(F2)] = 0.030 | Hydrogen site location: inferred from neighbouring sites |
| wR(F2) = 0.078 | H-atom parameters constrained |
| S = 1.04 | w = 1/[σ2(Fo2) + (0.0366P)2 + 0.234P] where P = (Fo2 + 2Fc2)/3 |
| 3127 reflections | (Δ/σ)max < 0.001 |
| 118 parameters | Δρmax = 0.30 e Å−3 |
| 0 restraints | Δρmin = −0.59 e Å−3 |
Special details
| Geometry. All s.u.'s (except the s.u. in the dihedral angle between two l.s. planes) are estimated using the full covariance matrix. The cell s.u.'s are taken into account individually in the estimation of s.u.'s in distances, angles and torsion angles; correlations between s.u.'s in cell parameters are only used when they are defined by crystal symmetry. An approximate (isotropic) treatment of cell s.u.'s is used for estimating s.u.'s involving l.s. planes. |
| Refinement. Refinement of F2 against ALL reflections. The weighted R-factor wR and goodness of fit S are based on F2, conventional R-factors R are based on F, with F set to zero for negative F2. The threshold expression of F2 > 2σ(F2) is used only for calculating R-factors(gt) etc. and is not relevant to the choice of reflections for refinement. R-factors based on F2 are statistically about twice as large as those based on F, and R- factors based on ALL data will be even larger. |
Fractional atomic coordinates and isotropic or equivalent isotropic displacement parameters (Å2)
| x | y | z | Uiso*/Ueq | ||
| Br1 | 0.24338 (3) | 0.428391 (17) | 0.069769 (19) | 0.04870 (9) | |
| N1 | 0.06730 (17) | 0.72483 (12) | 0.28704 (13) | 0.0340 (3) | |
| N2 | 0.28760 (18) | 0.76133 (14) | 0.25525 (15) | 0.0389 (3) | |
| C1 | −0.0905 (2) | 0.60075 (16) | 0.49714 (17) | 0.0358 (4) | |
| H1A | −0.1508 | 0.6684 | 0.4960 | 0.043* | |
| C2 | 0.0537 (2) | 0.47667 (16) | 0.40181 (16) | 0.0350 (4) | |
| H2A | 0.0907 | 0.4608 | 0.3360 | 0.042* | |
| C3 | −0.03759 (19) | 0.57747 (14) | 0.39781 (16) | 0.0316 (3) | |
| C4 | −0.0764 (2) | 0.66197 (17) | 0.28836 (18) | 0.0383 (4) | |
| H4A | −0.1216 | 0.6179 | 0.2110 | 0.046* | |
| H4B | −0.1547 | 0.7193 | 0.2941 | 0.046* | |
| C5 | 0.1346 (2) | 0.82204 (16) | 0.35896 (17) | 0.0401 (4) | |
| H5A | 0.0933 | 0.8639 | 0.4116 | 0.048* | |
| C6 | 0.2712 (2) | 0.84497 (16) | 0.33885 (18) | 0.0427 (4) | |
| H6A | 0.3420 | 0.9061 | 0.3747 | 0.051* | |
| C7 | 0.1620 (2) | 0.68965 (16) | 0.22476 (17) | 0.0370 (4) | |
| H7A | 0.1434 | 0.6259 | 0.1695 | 0.044* | |
| C8 | 0.4185 (2) | 0.75295 (18) | 0.2048 (2) | 0.0456 (4) | |
| H8A | 0.4123 | 0.6772 | 0.1625 | 0.055* | |
| H8B | 0.5189 | 0.7557 | 0.2732 | 0.055* | |
| C9 | 0.4132 (2) | 0.85232 (19) | 0.1143 (2) | 0.0469 (5) | |
| H9A | 0.4181 | 0.9283 | 0.1558 | 0.056* | |
| H9B | 0.3139 | 0.8488 | 0.0446 | 0.056* | |
| C10 | 0.5517 (3) | 0.8419 (2) | 0.0657 (2) | 0.0586 (6) | |
| H10A | 0.5471 | 0.9058 | 0.0085 | 0.088* | |
| H10B | 0.5457 | 0.7672 | 0.0234 | 0.088* | |
| H10C | 0.6499 | 0.8462 | 0.1347 | 0.088* |
Atomic displacement parameters (Å2)
| U11 | U22 | U33 | U12 | U13 | U23 | |
| Br1 | 0.06110 (15) | 0.03539 (11) | 0.05110 (14) | 0.00544 (9) | 0.02086 (10) | 0.00409 (8) |
| N1 | 0.0411 (8) | 0.0308 (7) | 0.0319 (7) | −0.0010 (6) | 0.0149 (6) | 0.0013 (6) |
| N2 | 0.0424 (8) | 0.0354 (7) | 0.0417 (9) | −0.0021 (6) | 0.0176 (7) | −0.0004 (6) |
| C1 | 0.0370 (9) | 0.0318 (8) | 0.0416 (10) | 0.0037 (7) | 0.0173 (8) | 0.0001 (7) |
| C2 | 0.0386 (9) | 0.0355 (8) | 0.0352 (9) | 0.0020 (7) | 0.0181 (8) | −0.0010 (7) |
| C3 | 0.0301 (8) | 0.0311 (8) | 0.0337 (8) | −0.0039 (6) | 0.0109 (7) | 0.0001 (7) |
| C4 | 0.0357 (9) | 0.0406 (9) | 0.0382 (10) | −0.0003 (7) | 0.0119 (7) | 0.0036 (8) |
| C5 | 0.0552 (11) | 0.0329 (8) | 0.0333 (9) | 0.0016 (8) | 0.0165 (8) | −0.0023 (7) |
| C6 | 0.0522 (11) | 0.0349 (9) | 0.0397 (10) | −0.0072 (8) | 0.0138 (9) | −0.0035 (8) |
| C7 | 0.0456 (10) | 0.0319 (8) | 0.0361 (9) | −0.0015 (7) | 0.0171 (8) | −0.0009 (7) |
| C8 | 0.0416 (10) | 0.0456 (10) | 0.0545 (12) | 0.0018 (8) | 0.0224 (9) | 0.0055 (9) |
| C9 | 0.0471 (11) | 0.0496 (11) | 0.0484 (11) | 0.0004 (9) | 0.0219 (9) | 0.0047 (9) |
| C10 | 0.0566 (13) | 0.0693 (15) | 0.0598 (14) | −0.0037 (11) | 0.0326 (11) | 0.0048 (12) |
Geometric parameters (Å, °)
| N1—C7 | 1.333 (2) | C4—H4B | 0.9700 |
| N1—C5 | 1.376 (2) | C5—C6 | 1.341 (3) |
| N1—C4 | 1.471 (2) | C5—H5A | 0.9300 |
| N2—C7 | 1.330 (2) | C6—H6A | 0.9300 |
| N2—C6 | 1.379 (2) | C7—H7A | 0.9300 |
| N2—C8 | 1.470 (2) | C8—C9 | 1.508 (3) |
| C1—C3 | 1.387 (2) | C8—H8A | 0.9700 |
| C1—C2i | 1.388 (3) | C8—H8B | 0.9700 |
| C1—H1A | 0.9300 | C9—C10 | 1.521 (3) |
| C2—C1i | 1.388 (3) | C9—H9A | 0.9700 |
| C2—C3 | 1.389 (2) | C9—H9B | 0.9700 |
| C2—H2A | 0.9300 | C10—H10A | 0.9600 |
| C3—C4 | 1.509 (2) | C10—H10B | 0.9600 |
| C4—H4A | 0.9700 | C10—H10C | 0.9600 |
| C7—N1—C5 | 108.76 (15) | C5—C6—N2 | 107.47 (16) |
| C7—N1—C4 | 125.17 (15) | C5—C6—H6A | 126.3 |
| C5—N1—C4 | 125.78 (14) | N2—C6—H6A | 126.3 |
| C7—N2—C6 | 108.44 (15) | N2—C7—N1 | 108.29 (15) |
| C7—N2—C8 | 124.99 (16) | N2—C7—H7A | 125.9 |
| C6—N2—C8 | 126.56 (16) | N1—C7—H7A | 125.9 |
| C3—C1—C2i | 120.18 (16) | N2—C8—C9 | 111.85 (16) |
| C3—C1—H1A | 119.9 | N2—C8—H8A | 109.2 |
| C2i—C1—H1A | 119.9 | C9—C8—H8A | 109.2 |
| C1i—C2—C3 | 120.77 (16) | N2—C8—H8B | 109.2 |
| C1i—C2—H2A | 119.6 | C9—C8—H8B | 109.2 |
| C3—C2—H2A | 119.6 | H8A—C8—H8B | 107.9 |
| C1—C3—C2 | 119.05 (16) | C8—C9—C10 | 110.34 (17) |
| C1—C3—C4 | 120.26 (15) | C8—C9—H9A | 109.6 |
| C2—C3—C4 | 120.68 (15) | C10—C9—H9A | 109.6 |
| N1—C4—C3 | 110.61 (14) | C8—C9—H9B | 109.6 |
| N1—C4—H4A | 109.5 | C10—C9—H9B | 109.6 |
| C3—C4—H4A | 109.5 | H9A—C9—H9B | 108.1 |
| N1—C4—H4B | 109.5 | C9—C10—H10A | 109.5 |
| C3—C4—H4B | 109.5 | C9—C10—H10B | 109.5 |
| H4A—C4—H4B | 108.1 | H10A—C10—H10B | 109.5 |
| C6—C5—N1 | 107.04 (16) | C9—C10—H10C | 109.5 |
| C6—C5—H5A | 126.5 | H10A—C10—H10C | 109.5 |
| N1—C5—H5A | 126.5 | H10B—C10—H10C | 109.5 |
| C2i—C1—C3—C2 | −0.7 (3) | N1—C5—C6—N2 | −0.3 (2) |
| C2i—C1—C3—C4 | −179.54 (16) | C7—N2—C6—C5 | 0.5 (2) |
| C1i—C2—C3—C1 | 0.7 (3) | C8—N2—C6—C5 | 179.06 (17) |
| C1i—C2—C3—C4 | 179.54 (16) | C6—N2—C7—N1 | −0.4 (2) |
| C7—N1—C4—C3 | 92.7 (2) | C8—N2—C7—N1 | −179.02 (16) |
| C5—N1—C4—C3 | −80.3 (2) | C5—N1—C7—N2 | 0.2 (2) |
| C1—C3—C4—N1 | 110.19 (18) | C4—N1—C7—N2 | −173.88 (15) |
| C2—C3—C4—N1 | −68.6 (2) | C7—N2—C8—C9 | 107.5 (2) |
| C7—N1—C5—C6 | 0.1 (2) | C6—N2—C8—C9 | −70.9 (2) |
| C4—N1—C5—C6 | 174.13 (16) | N2—C8—C9—C10 | 179.05 (18) |
Symmetry codes: (i) −x, −y+1, −z+1.
Hydrogen-bond geometry (Å, °)
| D—H···A | D—H | H···A | D···A | D—H···A |
| C7—H7A···Br1 | 0.93 | 2.77 | 3.6222 (18) | 152 |
Footnotes
Supplementary data and figures for this paper are available from the IUCr electronic archives (Reference: LH5273).
References
- Bruker (2009). APEX2, SAINT and SADABS Bruker AXS Inc., Madison, Wisconsin, USA.
- Chen, H.-C., Hu, H.-L., Chan, Z.-K., Yeh, C.-W., Jia, H.-W., Wu, C.-P., Chen, J.-D. & Wang, J.-C. (2007). Cryst. Growth Des. 7, 698–704.
- Cheng, P.-C., Wu, C.-J., Chen, H.-C., Chen, J.-D. & Wang, J.-C. (2009). Acta Cryst. E65, o1825. [DOI] [PMC free article] [PubMed]
- Herrmann, W. A., Reisinger, C.-P. & Spiegler, M. (1998). J. Organomet. Chem. 557, 93–96.
- Lee, H. M., Lu, C. Y., Chen, C. Y., Chen, W. L., Lin, H. C., Chiu, P. L. & Cheng, P. Y. (2004). Tetrahedron, 60, 5807–5825.
- Sheldrick, G. M. (2008). Acta Cryst. A64, 112–122. [DOI] [PubMed]
- Spek, A. L. (2009). Acta Cryst. D65, 148–155. [DOI] [PMC free article] [PubMed]
- Zhang, C. & Trudell, M. L. (2000). Tetrahedron Lett. 41, 595–598.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Crystal structure: contains datablock(s) global, I. DOI: 10.1107/S1600536811026146/lh5273sup1.cif
Structure factors: contains datablock(s) I. DOI: 10.1107/S1600536811026146/lh5273Isup2.hkl
Supplementary material file. DOI: 10.1107/S1600536811026146/lh5273Isup3.cml
Additional supplementary materials: crystallographic information; 3D view; checkCIF report