

Introduction of non-canonical amino acids into proteins is a powerful method for the creation of macromolecules with novel properties.[1–3] In particular, 5,5,5-trifluoroleucine (TFL, 2) has been utilized as a more hydrophobic surrogate of leucine in various contexts (Figure 1).[4, 5] When incorporated into the hydrophobic cores of certain coiled-coil proteins, TFL triggers an increase in stability, rendering proteins more resistant to thermal and chemical denaturation.[6, 7] Furthermore, despite the larger volume of CF3 compared to CH3, protein structure and activity can be retained upon replacement of leucine (Leu, 1) by TFL.[8, 9]
Figure 1.
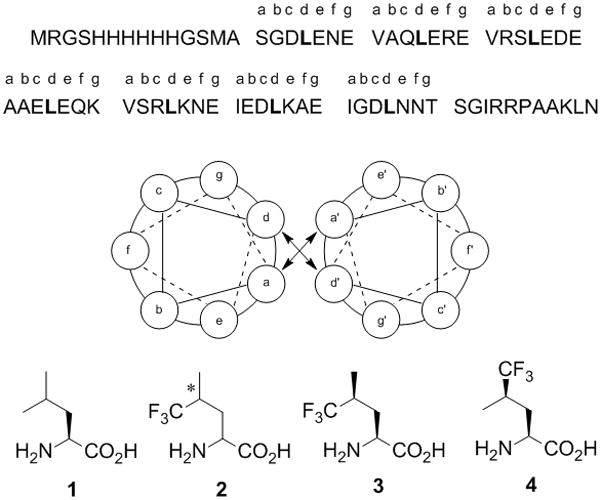
A1 peptide sequence and helical wheel diagram of the dimer in which leucines are highlighted in bold. Structures of leucine (1) and the trifluoromethyl analogs (2–4). The asterisk in structure 2 denotes unresolved stereochemistry at the 4-position.
Leu contains one stereocenter (Cα), which has the S-configuration. Replacement of a methyl group by a trifluoromethyl group at Cγ introduces an additional stereocenter and yields the two diastereoisomers (2S,4S)-5,5,5-trifluoroleucine (3) and (2S,4R)-5,5,5-trifluoroleucine (4) (Figure 1). Here we report the effects of TFL stereochemistry on coiled-coil peptide biosynthesis and stability. We demonstrate that both 3 and 4 are activated and incorporated into recombinant peptides prepared in Escherichia coli. Coiled-coil homodimers of peptides bearing 3 or 4 exhibit increased stability when compared to dimers of the Leu form of the peptide. An equimolar mixture of the two fluorinated peptides forms a heterodimer of modestly enhanced thermal stability relative to the homodimers.
The fidelity of translation is governed in large part by the activation of amino acids by their cognate aminoacyl-tRNA synthetases (AARS).[10] Although some AARSs are known to tolerate non-canonical substrates, amino acid activation can be acutely sensitive to side-chain stereochemistry. For example, isoleucine contains two stereocenters, both of S-configuration: one at Cα and another at Cβ. The (2S,3R)-isomer (allo-isoleucine) is not incorporated into proteins, although there is evidence that it is bound and activated by isoleucyl-tRNA synthetase (IleRS).[11–14] Stereochemistry can also determine the fate of non-canonical amino acids as possible substrates for protein synthesis in E. coli. For instance, the isoleucine analogue, 2-amino-3-methyl-4-pentenoic acid, is accepted only in its (2S,3S)-form, while the valine (or isoleucine) analogue, 4,4,4-trifluorovaline (TFV), is active only in its (2S,3R)-form.[15, 16] We sought to explore whether leucyl-tRNA synthetase (LeuRS) exhibits a stereochemical preference with respect to activation of TFL.
Coiled-coil peptides constitute simple model systems for use in investigations of protein biosynthesis and stability.[17–20] Stereoisomers 3 and 4 were prepared (Scheme 1S, supporting information) and evaluated for incorporation into coiled-coil peptide A1[18] (Figure 1) in an E. coli strain auxotrophic for leucine. A1 was also expressed in media supplemented with 1 or 2. Following purification on Ni–nitrilotriacetic acid affinity columns, protein yields were determined to be 18 ± 4 mg/L and 9 ± 3 mg/L upon incubation with 3 and 4, respectively, compared to 45 ± 6 mg/L for A1 prepared with 1. Peptides containing 3 and 4 were designated SS-A1 and SR-A1, respectively.
Matrix-assisted laser desorption/ionization (MALDI) mass spectrometry analysis of A1 fragments obtained by trypsin digestion was performed to assess the extent of substitution by 3 or 4. Fragment LKNEIEDLKAEIGDLNNTSGIR, corresponding to residues 46–67 in A1, contains three leucine residues (shown in bold type). Fragments that correspond to replacement at 0, 1, 2, and 3 sites by either 3 or 4 were observed (Figure 2). The expected mass increment of 54 Da was visible for each leucine residue replaced by TFL. Incomplete replacement of 1 most likely reflects a persistent pool of the natural amino acid replenished by cellular protein turnover.[19] The distribution of peak intensities (though not simply related to the relative abundances of fragments) is roughly consistent with unbiased substitution of 3 or 4, and suggests a slight preference for incorporation of 3 (90% replacement of Leu in SS-A1) versus 4 (82% in SR-A1). Quantitative amino acid analysis was consistent with the MALDI results, showing 91% replacement in SS-A1 and 80% in SR-A1.
Figure 2.

MALDI mass spectra of a tryptic fragment of A1 (residues 46–67) containing 3 leucine positions. A1 was expressed in media supplemented with either 1 (A), 3 (B), or 4 (C). Fragments corresponding to 0, 1, 2, and 3 sites of substitution are represented as ~2442, 2497, 2551 and 2605 Da, respectively.
The relative rates of activation of 1 and fluorinated analogues 2–4 by LeuRS were determined by an in vitro ATP-PPi exchange assay. The kinetic parameters are shown in Table 1. The relative kcat/Km values show that 3 is a slightly better substrate than 4, consistent with the modest differences in yield and incorporation levels described above. As expected, the apparent kcat/Km for 2 fell between the values for 3 and 4 (Table 1). The activation rates for both 3 and 4 are within the range of rates that have been shown to support protein synthesis in conventional hosts cultured with other non-canonical amino acids.[21] Although a slight stereochemical preference is observed with respect to activation of 3 vs. 4, this result stands in sharp contrast to the absolute selectivity imposed by IleRS and ValRS in the activation of TFV, in which only the 2S,3R isomer is tolerated.[15, 16] Furthermore, the fact that both 3 and 4 can be incorporated into proteins in E. coli is consistent with previous work showing that hexafluoroleucine is activated by LeuRS.[21] All of these results indicate that fluorination at either of the Cδ positions is tolerated by LeuRS.
Table 1.
Kinetic parameters for activation of 1 – 4 by E. coli LeuRS[a]
| Substrate | Km (μM) | kcat (s−1) | kcat/Km (rel) |
|---|---|---|---|
| 1 | 16.9 ± 4.5 | 4.22 ± 0.35 | 1 |
| 2 | 659 ± 103 | 0.40 ± 0.03 | 1/412 |
| 3 | 252 ± 92 | 0.59 ± 0.05 | 1/107 |
| 4 | 708 ± 280 | 0.19 ± 0.02 | 1/933 |
Substrate 1 was used as the L-isomer; 2 as a mixture of (2S,4S), (2S,4R), (2R,4S) and (2R,4R) forms; 3 as the (2S,4S) form and 4 as the (2S, 4R) form.
The secondary structures of all four A1 peptides were examined by circular dichroism (CD) spectroscopy. Because the peptides can form dimers, Leu-A1, SR-A1 and SS-A1 represent homodimers whereas the equimolar mixture of SR-A1 and SS-A1 can form either homo- or heterodimers. All four spectra were nearly identical at 1°C, and indicated ca. 90% helical content in each peptide as judged from the molar ellipticity at 222 nm (Figure 3A).[22, 23] CD provided no evidence that fluorination affects the secondary structure of A1.
Figure 3.

CD spectra of Leu-A1 (○), SR-A1 (△), SS-A1 (+), and an equimolar mixture of SR-A1 and SS-A1 (■). A) Wavelength scan at 1 °C, 10 μM protein concentration, PBS buffer, pH 7.4. B) Thermal denaturation (1.5 °C interval, 1 minute equilibration time, 10 second averaging time) at 10 μM protein concentration, PBS buffer, pH 7.4.
Previous studies have shown that incorporation of fluorinated amino acids into coiled-coil peptides and α-helical bundles results in enhanced stability;[7, 8, 19, 20, 24] the extent of stabilization varies depending on the identity of the fluorinated analogue.[19, 24] To determine whether the stereochemistry of TFL affects the extent of stabilization of A1, thermal denaturation was monitored by CD spectroscopy (Figure 3B). For both SR-A1 and SS-A1, the thermal melting temperature (Tm) was 65°C, 11°C higher than that of Leu-A1 (Tm = 55°C). The equimolar mixture of SR-A1 and SS-A1 exhibited a Tm of 68°C. This additional 3°C increase in Tm, suggested that SR-A1 and SS-A1 form heterodimers rather than a mixture of homodimers (Table 1S, supporting information). When a mixture of the (2S,4S)- and (2S,4R)-forms of TFL was used for expression of A1, ΔTm was 13 °C,[6] nearly identical to that observed for equimolar mixtures of SS-A1 and RR-A1.
The results reported here demonstrate that both the SR- and SS-isomers of TFL are incorporated into proteins expressed in E. coli; the S- isomer is activated at a slightly higher rate by LeuRS. The higher activation rate leads to higher protein yields for SS-A1 relative to SR-A1 and to increased levels of incorporation of the fluorinated analogue. Neither stereoisomer appears to alter the coiled-coil structure of A1. Replacement of Leu by either isomer enhances the thermostability of A1; the heterodimer of SS- and SR-A1 shows an additional modest increase in stability. Experiments are underway to extend these findings and explore more fully the influence of side-chain fluorination on protein stability.
Experimental Section
Synthesis of 3 and 4
Amino acids 3 and 4 were prepared as described previously (see supporting information).[25–27]
Protein biosynthesis and purification
Leucine auxotrophic strain LAM1000 transformed with pREP4 (Qiagen) was used as the E. coli host to express A1, which was encoded within pQEA1 under the control of a lac promoter. Protein expression and purification were performed as described previously.[7] Protein concentrations were determined via UV spectroscopy.
Protein characterization
CD data were collected on an Aviv 62DS spectropolarimeter (Lakewood, NJ) using a 1 mm pathlength cell. Wavelength scans were taken from 195 to 250 nm, with points taken every 1 nm. Temperature scans were performed from 0–95°C in 1.5°C steps. Each plot represents an average of 3 scans.
Activation kinetics
An N-terminal His6-LeuRS fusion was expressed and purified as previously reported.[21] Measurement of the rates of activation of leucine and analogues was performed by an ATP-PPi exchange assay. The assay buffer conditions were 30 mM HEPES, pH 7.4, 10 mM MgCl2, 1 mM DTT, 2 mM ATP and 2 mM [32P]-PPi (0.5 TBq/mol). A fixed concentration of 75 nM of His6-LeuRS was used. Depending upon the activity of the enzyme toward the substrate, the following substrate concentrations ranges were used (1: 0.6–312.5 μM; 2, 3, 4: 6.1–6250 μM). Once the reaction was completed, the reaction mixture was quenched by addition of 200 mM PPi, 7% w/v HClO4 and 3% activated charcoal. The charcoal was washed twice and measured on a scintillation counter. Kinetic data were fit using non-linear regression analysis.
Supplementary Material
Acknowledgments
This work was supported by the NIH GM 62523 and 5FM GM67375-2 (DAT and JKM), GM65500 (KK), NSF graduate fellowship (SS), GAANN fellowship (GAC) and a NSF CAREER award (KK).
Footnotes
Supporting information for this article is available
References
- 1.a) Link AJ, Mock ML, Tirrell DA. Curr Op Biotech. 2003;14:603–609. doi: 10.1016/j.copbio.2003.10.011. [DOI] [PubMed] [Google Scholar]; b) Connor RE, Tirrrell DA. Polymer Rev. 2007;47:9–28. [Google Scholar]; Budisa N. Angew Chem Int Ed. 2004;43:6426–6463. doi: 10.1002/anie.200300646. [DOI] [PubMed] [Google Scholar]
- 2.a) Hendrickson TL, de Crecy-Lagard V, Schimmel P. Ann Rev Biochem. 2004;73:147–176. doi: 10.1146/annurev.biochem.73.012803.092429. [DOI] [PubMed] [Google Scholar]; b) Wang L, Schultz PG. Angew Chem Int Ed. 2005;44:34–66. doi: 10.1002/anie.200460627. [DOI] [PubMed] [Google Scholar]
- 3.a) Bilgicer B, Kumar K. J Chem Ed. 2003;80:1275–1281. [Google Scholar]; b) Jäckel C, Koksch B. Eur J Org Chem. 2005;21:4483–4503. [Google Scholar]
- 4.Rennert OM, Anker HS. Biochemistry. 1963;2:471–476. doi: 10.1021/bi00903a013. [DOI] [PubMed] [Google Scholar]
- 5.Marsh ENG. Chem Biol. 2000;7:R153–R157. doi: 10.1016/s1074-5521(00)00139-3. [DOI] [PubMed] [Google Scholar]
- 6.Tang Y, Ghirlanda G, Petka WA, Nakajima T, DeGrado WF, Tirrell DA. Angew Chem Int Ed. 2001;40:1494–1496. [PubMed] [Google Scholar]
- 7.Bilgicer B, Fichera A, Kumar K. J Am Chem Soc. 2001;123:4393–4399. doi: 10.1021/ja002961j. [DOI] [PubMed] [Google Scholar]
- 8.Tang Y, Ghirlanda G, Vaidehi N, Kua J, Mainz DT, Goddard WA, DeGrado WF, Tirrell DA. Biochemistry. 2001;40:2790–2796. doi: 10.1021/bi0022588. [DOI] [PubMed] [Google Scholar]
- 9.a) Panchenko T, Zhu WW, Montclare JK. Biotech Bioeng. 2006;94:921–930. doi: 10.1002/bit.20910. [DOI] [PubMed] [Google Scholar]; b) Montclare JK, Tirrell DA. Angew Chem Int Ed. 2006;45:4518–4521. doi: 10.1002/anie.200600088. [DOI] [PubMed] [Google Scholar]; c) Voloshchuk N, Lee MX, Tanrikulu IC, Montclare JK. Bioorg Med Chem Lett. 2007;17:5907–5911. doi: 10.1016/j.bmcl.2007.07.107. [DOI] [PubMed] [Google Scholar]
- 10.Cusack S, Yaremchuk A, Tukalo M. EMBO J. 2000;19:2351–2361. doi: 10.1093/emboj/19.10.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Loftfield RB. Biochem J. 1963;89:82–92. doi: 10.1042/bj0890082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flossdorf J, Kula MR. Eur J Biochem. 1973;36:534–540. doi: 10.1111/j.1432-1033.1973.tb02940.x. [DOI] [PubMed] [Google Scholar]
- 13.Loftfield RB, Eigner EA. Biochim Biophys Acta. 1966;130:426–448. doi: 10.1016/0304-4165(66)90239-x. [DOI] [PubMed] [Google Scholar]
- 14.Polet H, Conrad ME. Proc Soc Exp Biol Med. 1969;130:581–586. doi: 10.3181/00379727-130-33612. [DOI] [PubMed] [Google Scholar]
- 15.Mock ML, Michon T, van Hest JCM, Tirrell DA. ChemBioChem. 2006;7:83–87. doi: 10.1002/cbic.200500201. [DOI] [PubMed] [Google Scholar]
- 16.Wang P, Fichera A, Kumar K, Tirrell DA. Angew Chem Int Ed. 2004;43:3664–3666. doi: 10.1002/anie.200454036. [DOI] [PubMed] [Google Scholar]
- 17.O’Shea EK, Klemm JD, Kim PS, Alber T. Science. 1991;254:539–344. doi: 10.1126/science.1948029. [DOI] [PubMed] [Google Scholar]
- 18.Petka WA, Harden JL, McGrath KP, Wirtz D, Tirrell DA. Science. 1998;281:389–392. doi: 10.1126/science.281.5375.389. [DOI] [PubMed] [Google Scholar]
- 19.a) Tang Y, Tirrell DA. J Am Chem Soc. 2001;123:11089–11090. doi: 10.1021/ja016652k. [DOI] [PubMed] [Google Scholar]; b) Son S, Tanrikulu IC, Tirrell DA. ChemBioChem. 2006;7:1251–1257. doi: 10.1002/cbic.200500420. [DOI] [PubMed] [Google Scholar]
- 20.a) Jäckel C, Seufert W, Thust S, Koksch B. ChemBioChem. 2004;5:717–720. doi: 10.1002/cbic.200300840. [DOI] [PubMed] [Google Scholar]; b) Jäckel C, Salwiczek M, Koksch B. Angew Chem Int Ed. 2006;45:4198–4203. doi: 10.1002/anie.200504387. [DOI] [PubMed] [Google Scholar]
- 21.Kiick KL, Weberskirch R, Tirrell DA. FEBS Lett. 2001;505:25–30. doi: 10.1016/s0014-5793(01)02657-6. [DOI] [PubMed] [Google Scholar]
- 22.The software CDNN was employed to calculate the % helical content from the CD wavelength data. This program is available at http://bioinformatik.biochemtech.uni-halle.de/.
- 23.Protein concentrations were determined by Bradford Assay (BioRad, Hercules, CA).
- 24.a) Lee KH, Lee HY, Slutsky MM, Anderson JT, Marsh ENG. Biochemistry. 2004;43:16277–16284. doi: 10.1021/bi049086p. [DOI] [PubMed] [Google Scholar]; b) Lee HY, Lee KH, Al-Hashimi HM, Marsh ENG. J Am Chem Soc. 2006;128:337–343. doi: 10.1021/ja0563410. [DOI] [PubMed] [Google Scholar]; c) Gottler LM, de la Salud-Bea R, Marsh ENG. Biochemistry. 2008;47:4484–4490. doi: 10.1021/bi702476f. [DOI] [PubMed] [Google Scholar]
- 25.Xing X, Fichera A, Kumar K. J Org Chem. 2002;67:1722–1725. doi: 10.1021/jo011097q. [DOI] [PubMed] [Google Scholar]
- 26.Weinges K, Kromm E. Liebigs Ann Chem. 1985:90–102. [Google Scholar]
- 27.Yamada S, Hongo C, Yoshioka R, Chibata I. J Org Chem. 1983;48:843–846. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.