

Crystals of FimP from A. oris were obtained. To facilitate selenomethionine labelling, three methionines were introduced by site-directed mutagenesis.
Keywords: fimbria, biofilms, mutagenesis
Abstract
Actinomyces oris is an oral bacterium important for the development of dental plaque. It expresses two forms of fimbriae: type 1 and type 2. FimP, which is the fimbrial protein that is polymerized into the stalk of the type 1 fimbriae, was cloned, overexpressed and crystallized. X-ray data were collected and processed to 2.2 Å resolution. The crystals belonged to space group P21212, with one molecule in the asymmetric unit. To facilitate structure determination using single anomalous dispersion, three methionines were introduced by site-directed mutagenesis. Crystals of selenomethionine-labelled protein were obtained by streak-seeding and diffracted to 2.0 Å resolution.
1. Introduction
Dental plaque is one of the most complex biofilms known and can consist of several hundred bacterial species (Jenkinson & Lamont, 2005 ▶). The development of dental plaque is initiated by the attachment of salivary proteins to the tooth surface, followed by adherence of early-colonizing bacteria, mostly oral streptococci and Actinomyces species. These Gram-positive bacteria form an initial biofilm that is used as an attachment surface for the late colonizers, many of which are Gram-negative. Surface adhesins and fimbriae are crucial factors for networking with other bacteria and cells. Actinomyces oris T14V (formerly A. naeslundii; Henssge et al., 2009 ▶) expresses two forms of fimbriae: type 1 (FimP) and type 2 (FimA). Using type 1 fimbriae the bacteria can bind to salivary proline-rich proteins attached to the tooth surface, while carbohydrate structures on the surface of bacteria and host cells are recognized using type 2 fimbriae. The two types of fimbria are related but are encoded by two separate operons (Yeung et al., 1998 ▶; Chen et al., 2007 ▶). Each operon consists of a putative adhesin followed by a shaft protein that is covalently polymerized into the fimbrial stalk and finally a sortase, a cysteine transpeptidase that is responsible for polymerization. In concurrence with other Gram-positive fimbria or pili, the sortase cleaves the shaft protein between a threonine and a glycine in an LPXTG motif near the C-terminus (Fig. 1 ▶). In the following step, a lysine side chain in the central part of the next shaft protein forms an amide linkage to the C-terminal threonine. Compared with the vast amount of structural information that is available for Gram-negative fimbriae/pili, understanding of the bioassembly of Gram-positive fimbriae is still in its infancy, with only a few structures being known (Izoré et al., 2010 ▶; Kang et al., 2009 ▶; Vengadesan et al., 2011 ▶). To gain further insight into oral biofilm formation and the structure of Gram-positive fimbriae, we aimed at solving the crystal structure of the FimP shaft protein. Native crystals were obtained after in situ proteolysis with α-chymotrypsin. The FimP protein contains only one methionine in 460 residues; therefore, selenomethionine (SeMet) labelling was unrealistic. When structure determination using molecular replacement was found to be unsuccessful, as was initial screening for heavy-atom derivatives, a triple isoleucine-to-methionine mutant (FimP-3M) was generated to facilitate structure determination using SeMet single anomalous dispersion.
Figure 1.

Schematic representation of FimP: signal peptide, structural subunit, LPXTG motif and TM helix. The positions of the Ile-to-Met mutations are shown as well as the naturally occurring Met. The central Lys predicted to be linked to the Thr of the LPXTG motif of another FimP subunit is also depicted. The construct used for crystallization (residues 31–491) is indicated by a bar.
2. Materials and methods
2.1. Cloning
The fimP gene encoding residues 31–491 of the protein was amplified from genomic DNA of A. oris strain T14V (GenBank accession code M32067). Preparation of the genomic DNA has been described previously (Hallberg et al., 1998 ▶). PCR primers were designed to exclude the signal peptide, the LPXTG motif and the C-terminal transmembrane helix. The forward primer was 5′-TTTTTCCATGGCACCCGCTGACCCGAACG-3′ and the reverse primer was 5′-AAAAAGGTACCTCAGTTCTTCGGGATGTCGGTGACG-3′ (restriction sites are shown in bold). The PCR product was digested with Acc65I and NcoI and ligated into the equivalent sites of the pET-M11 expression vector (kindly provided by G. Stier, EMBL, Germany). The final construct encodes MKHHHHHHPMSDYDIPTTENLYFQGAM-FimP31–491. The plasmids were transformed into Escherichia coli DH5α and subsequent colonies were selected for on kanamycin plates. Positive clones were verified by DNA sequencing.
2.2. Generation of mutants
The triple mutant I121M, I204M, I347M was constructed following an existing protocol (Heckman & Pease, 2007 ▶). In brief, each mutant was produced by PCR-mediated overlap extension in two steps. Initial reactions generated two fragments with overlapping complementary 3′ ends where the mutations were introduced. In the second step the two PCR fragments were used to generate a full-length product. The triple mutant, FimP-3M, was generated by the stepwise introduction of mutations. The mutagenesis primers (only the forward primers are presented) were I121M, 5′-CCAGAAGATGACCACCGGT, I204M, 5′-GCGCGACATGACCTACACC, and I347M, 5′-CCCCGGCATGCCCACCACC. The methionine codons are shown in bold. The constructs were verified as above.
2.3. Overexpression and purification
The protein was overexpressed in E. coli BL21 (DE3) at 310 K in Luria broth supplemented with 50 µg ml−1 kanamycin. When the cultures reached an OD600 of 0.6, the temperature was lowered to 303 K and expression was induced with 0.4 mM IPTG, after which the cultures were grown for an additional 5 h. Cells were harvested by centrifugation at 5300g and the pellets were frozen at 193 K. Cell pellets were resuspended in 50 mM NaH2PO4 pH 7.5, 300 mM NaCl and 10 mM imidazole supplemented with EDTA-free protease-inhibitor cocktail (Roche). The suspension was lysed on ice by sonication and cellular debris was removed by centrifugation at 39 000g for 60 min. The supernatant was loaded onto a column packed with Ni–NTA agarose (Qiagen). The protein was washed in 50 mM NaH2PO4 pH 7.5, 300 mM NaCl and 20 mM imidazole and eluted with 50 mM NaH2PO4 pH 7.5, 300 mM NaCl and 300 mM imidazole. The buffer was exchanged to 20 mM Tris–HCl pH 8.0, 200 mM NaCl, 0.5 mM EDTA and 1 mM DTT. The protein was further purified by size-exclusion chromatography using a HiLoad 16/60 Superdex 200 prep-grade column (Amersham Biosciences). The protein purity was judged by SDS–PAGE and the protein was concentrated to 92 mg ml−1 in 20 mM Tris–HCl pH 8.5, 0.5 mM EDTA using an Amicon Ultra centrifugal filter device (Millipore).
To express SeMet-substituted FimP-3M, cells were grown in M9 medium supplemented with glucose at 310 K. At an optical density of ∼0.6 at 600 nm, lysine, threonine and phenylalanine at 100 mg l−1 and leucine, isoleucine, valine, proline and SeMet at 50 mg l−1 were added to downregulate the synthesis of methionine (Van Duyne et al., 1993 ▶). The SeMet-labelled protein protein was purified as described above with the exception that 0.5 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP) was present throughout in all steps. The SeMet protein was concentrated to 79 mg ml−1.
2.4. Crystallization and data collection
Initial crystallization trials were performed by the sitting-drop vapour-diffusion method in a 96-well MRC crystallization plate (Molecular Dimensions) using a Mosquito (TTP LabTech) pipetting robot. Both untreated protein and protein treated with 1%(w/w) α-chymotrypsin were used in the setups. The in situ proteolysis was performed as a means of trimming off flexible parts to facilitate crystal growth as described previously (Dong et al., 2007 ▶). In brief, α-chymotrypsin, stored at 2 mg ml−1 in 100 mM Tris pH 7.8, 2 mM CaCl2, was added to the FimP to a final ratio of 1%(w/w) immediately before crystallization. In both setups the FimP was diluted to a final concentration of 25 mg ml−1 in 20 mM Tris pH 8.0. Droplets of 0.1 µl protein solution were mixed with an equal volume of reservoir solution using screens from Hampton Research (Crystal Screen HT) and Molecular Dimensions (PACT) at 291 K. Crystal contents were analyzed on SDS–PAGE by comparing purified protein and dissolved crystals.
SeMet-labelled crystals grew in the same condition as the native crystals after streak-seeding. The seed stock was obtained by crushing 2–4 native crystals and resuspending them in 400 µl well solution. The mixture was vortexed for 20 s and spun in a microcentrifuge for 1 min. Streak-seeding was performed with a cat whisker on crystallization drops equilibrated for 1–2 d.
Crystals were soaked for 30 s in mother-liquor solution supplemented with 20% glycerol before being flash-cooled in liquid nitrogen and stored until data collection.
Diffraction data were collected from the native crystals to 2.2 Å resolution using a MAR 165 detector on beamline I911-5 at MAX-lab, Lund, Sweden. Data for the SeMet crystals were collected to 2.0 Å resolution using a MAR Mosaic 225 detector on beamline ID23-1 at the ESRF, Grenoble. Diffraction images were processed with XDS (Kabsch, 2010 ▶) and scaled with SCALA from the CCP4 program suite (Winn et al., 2011 ▶). Relevant processing statistics are summarized in Table 1 ▶.
Table 1. Data-collection, refinement and model-quality statistics for FimP.
Values in parentheses are for the highest resolution shell.
| Native FimP | SeMet FimP-3M | |
|---|---|---|
| Space group | P21212 | P21212 |
| Unit-cell parameters (Å) | a = 77.2, b = 177.8, c = 40.4 | a = 76.3, b = 168.1, c = 39.8 |
| Wavelength (Å) | 1.500 | 0.9792 |
| Resolution (Å) | 29.9–2.2 (2.32–2.20) | 45.2–2.0 (2.11–2.00) |
| Total reflections | 196133 (26118) | 220706 (31846) |
| Unique reflections | 29152 (4029) | 35477 (5054) |
| 〈I/σ(I)〉 | 13.0 (8.1) | 29.9 (15.7) |
| Rmerge† (%) | 10.4 (18.8) | 4.0 (9.2) |
| Completeness (%) | 99.2 (96.0) | 99.8 (99.4) |
| Multiplicity | 6.7 (6.5) | 6.2 (6.3) |
R
merge = 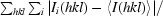
 , where Ii(hkl) is the intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the average over all observations of reflection hkl.
, where Ii(hkl) is the intensity of the ith observation of reflection hkl and 〈I(hkl)〉 is the average over all observations of reflection hkl.
3. Results and discussion
The FimP protein, comprising residues 31–491 (theoretical molecular weight 53.1 kDa, including the His-tag linker), was successfully cloned and overexpressed in E. coli BL21 (DE3) (Fig. 1 ▶). By the addition of 1% α-chymotrypsin, native crystals were obtained in solutions A11, C11 and D11 from the PACT screen. The crystallization conditions were further optimized to 20%(w/v) PEG 4000, 50 mM CaCl2 and 100 mM sodium acetate pH 5.5. Plate-shaped crystals grew to dimensions of 0.1 × 0.06 × 0.02 mm within 2 d (Fig. 2 ▶). Comparison of untreated protein and the crystal contents showed that the major part of the protein was intact after α-chymotrypsin treatment (Fig. 3 ▶). X-ray diffraction data from the native crystals were collected to a resolution of 2.2 Å with 99.2% completeness (96.0% completeness in the outer shell) and an R merge of 10.4%. Analysis of the symmetry and systematic absences in the recorded diffraction pattern indicated that the crystal belonged to the orthorhombic space group P21212, with unit-cell parameters a = 77.2, b = 177.8, c = 40.4 Å. A calculated Matthews coefficient of 2.8 Å3 Da−1 suggested 55.6% solvent content with one monomer in the asymmetric unit (Matthews, 1968 ▶). Crystals of SeMet-labelled FimP-3M were obtained after the addition of α-chymotrypsin following the same protocol. Crystal growth was initiated by streak-seeding with a suspension of crushed native crystals. Data were collected to 2.0 Å resolution with a completeness of 99.8% (99.4% in the outer shell) and an R merge of 4.0%. The SeMet crystals belonged to the same space group as the native crystals, with unit-cell parameters a = 76.3, b = 168.1, c = 39.8 Å.
Figure 2.

Crystal of FimP obtained in 0.2 M CaCl2, 0.1 M sodium acetate pH 5.5 and 20%(w/v) PEG 4000 with the addition of 1% α-chymotrypsin.
Figure 3.
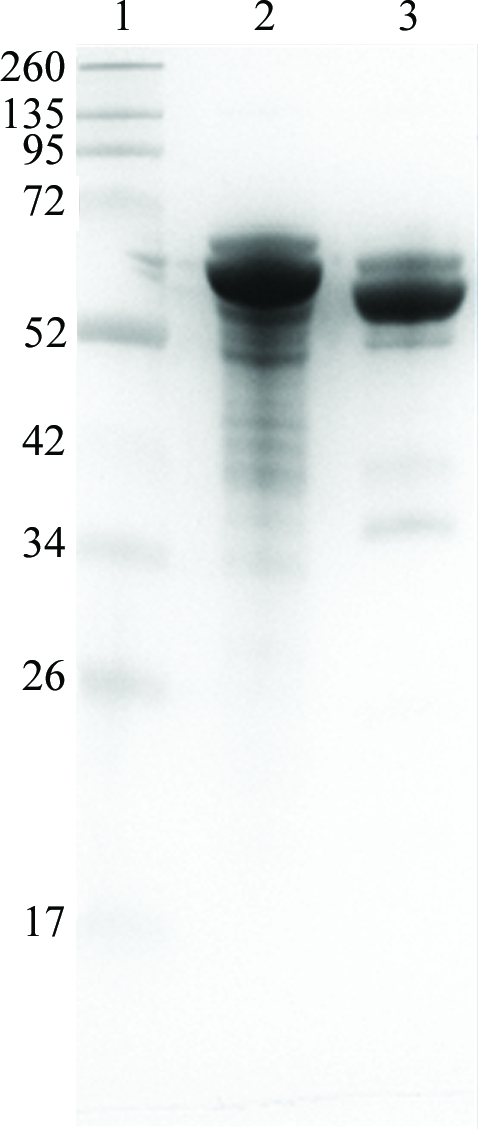
Analysis of FimP protein and crystals. Lane 1, marker (labelled in kDa); lane 2, untreated protein; lane 3, dissolved FimP crystals.
In situ proteolysis using trace amounts of protease is one of the methods that can be used to increase the chances of obtaining protein crystals. In general, flexible loops or termini are trimmed off, generating a protein that is more easily crystallized. Based on previous experiences α-chymotrypsin often cuts in the linker between the His tag and the protein, which has also been confirmed in silico by the ExPASy PeptideCutter tool (Gasteiger et al., 2003 ▶), indicating a cleavage site between positions 13 and 14 in the linker.
The three isoleucine-to-methionine mutants were designed to be evenly distributed along the protein and to not introduce unwanted NcoI restriction sites in the gene. SeMet-labelled FimP-3M purified at 295 K gave the same pattern during size-exclusion chomatography as the native protein, suggesting that the structure was not significantly destabilized by these mutations.
In summary, site-directed mutagenesis was used to introduce three methionines into FimP to facilitate SeMet labelling and structure determination using SAD/MAD methods. The choice of substituting isoleucines with methionines was based on the assumption that such conservative mutations would not affect the protein structure and that the methionines would be likely to be well ordered in hydrophobic environments. I am confident that the introduced methionines have not significantly affected the structure since the SeMet-labelled FimP-3M was as stable as the native protein during purification and could be crystallized under essentially the same conditions. Given the fast protocol for site-directed mutagenesis, the introduction of methionines and subsequent SeMet labelling can be a more time-effective way to solve a crystal structure than to screen for heavy-atom derivatives, especially if access to synchrotron radiation is limited.
Structure determination and refinement of FimP is under way and will be presented in a separate article.
Acknowledgments
I am grateful for access to beamline ID23-1 at the ESRF, Grenoble and beamline I911-5 at MAX-lab, Lund and for the support of the beamline scientists. I thank Gunter Stier, EMBL, Germany for cloning vectors. This work was supported by the Swedish Research Council and foundations from Umeå University.
References
- Chen, P., Cisar, J. O., Hess, S., Ho, J. T. & Leung, K. P. (2007). Infect. Immun. 75, 4181–4185. [DOI] [PMC free article] [PubMed]
- Dong, A. et al. (2007). Nature Methods, 4, 1019–1021. [DOI] [PMC free article] [PubMed]
- Gasteiger, E., Gattiker, A., Hoogland, C., Ivanyi, I., Appel, R. D. & Bairoch, A. (2003). Nucleic Acids Res. 31, 3784–3788. [DOI] [PMC free article] [PubMed]
- Hallberg, K., Holm, C., Ohman, U. & Strömberg, N. (1998). Infect. Immun. 66, 4403–4410. [DOI] [PMC free article] [PubMed]
- Heckman, K. L. & Pease, L. R. (2007). Nature Protoc. 2, 924–932. [DOI] [PubMed]
- Henssge, U., Do, T., Radford, D. R., Gilbert, S. C., Clark, D. & Beighton, D. (2009). Int. J. Syst. Evol. Microbiol. 59, 509–516. [DOI] [PMC free article] [PubMed]
- Izoré, T., Contreras-Martel, C., El Mortaji, L., Manzano, C., Terrasse, R., Vernet, T., Di Guilmi, A. M. & Dessen, A. (2010). Structure, 18, 106–115. [DOI] [PubMed]
- Jenkinson, H. F. & Lamont, R. J. (2005). Trends Microbiol. 13, 589–595. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kang, H. J., Paterson, N. G., Gaspar, A. H., Ton-That, H. & Baker, E. N. (2009). Proc. Natl Acad. Sci. USA, 106, 16967–16971. [DOI] [PMC free article] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- Van Duyne, G. D., Standaert, R. F., Karplus, P. A., Schreiber, S. L. & Clardy, J. (1993). J. Mol. Biol. 229, 105–124. [DOI] [PubMed]
- Vengadesan, K., Ma, X., Dwivedi, P., Ton-That, H. & Narayana, S. V. (2011). J. Mol. Biol. 407, 731–743. [DOI] [PMC free article] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Yeung, M. K., Donkersloot, J. A., Cisar, J. O. & Ragsdale, P. A. (1998). Infect. Immun. 66, 1482–1491. [DOI] [PMC free article] [PubMed]