

The crystallization of PBP4 from L. monocytogenes is reported.
Keywords: penicillin-binding proteins, Listeria monocytogenes
Abstract
Penicillin-binding proteins (PBPs), which catalyze peptidoglycan synthesis, have been extensively studied as a well established target of antimicrobial agents, including β-lactam derivatives. However, remarkable resistance to β-lactams has developed among pathogenic bacteria since the clinical use of penicillin began. Recently, the glycosyltransferase (GT) domain of class A PBPs has been proposed as an attractive target for antibiotic development as moenomycin-bound GT-domain structures have been determined. In this study, a class A PBP4 from Listeria monocytogenes was overexpressed, purified and crystallized using the hanging-drop vapour-diffusion method. Diffraction data were collected to 2.1 Å resolution using synchrotron radiation. The crystal belonged to the primitive orthorhombic space group P21212, with unit-cell parameters a = 84.6, b = 127.8, c = 54.9 Å. The structural information will contribute to the further development of moenomycin-derived antibiotics possessing broad-spectrum activity.
1. Introduction
The biosynthetic pathway of the bacterial cell wall has been the subject of intense research in the development of antibacterial agents since the discovery of β-lactams. The peptidoglycan, which is the major component of the bacterial cell wall, functions to preserve cell integrity by withstanding the internal osmotic pressure (Young, 2003 ▶; Popham & Young, 2003 ▶). The assembly of peptidoglycans is performed by multimodular penicillin-binding proteins (PBPs) that belong to either class A or class B depending on the structure and catalytic activity of their N-terminal module (Goffin & Ghuysen, 1998 ▶). The C-terminal penicillin-binding domain of both classes is endowed with transpeptidase (TP) activity that catalyzes peptide cross-linking between two adjacent glycan chains (van Heijenoort, 2001 ▶). In class A PBPs the N-terminal domain is responsible for a glycosyltransferase (GT) activity leading to glycan-chain elongation (Terrak et al., 1999 ▶). In class B PBPs the N-terminal domain is involved in interactions with other proteins involved in cell division (Höltje, 1998 ▶). Many current antibiotics are β-lactam derivatives that specifically inhibit the TP domain of PBPs. However, the remarkable ability of bacteria to develop resistance to β-lactams requires the identification of new antibiotic targets and the development of new antimicrobial agents (Macheboeuf et al., 2006 ▶). Recently, the GT domain of class A PBPs has been proposed as an attractive target for new antibacterial agents (Halliday et al., 2006 ▶).
Currently, structural information about class A PBPs, including both the GT and TP domains, is available for Staphylococcus aureus PBP2 (Wright, 2007 ▶; Lovering et al., 2008 ▶) and Escherichia coli PBP1b (Terrak et al., 2008 ▶), both in complex with moenomycin, the only known inhibitor of the GT domain. The structural and biochemical data shed light on the mechanism of glycan-chain elongation by the GT domain and provide a good starting point for structure-based drug design. The pentasaccharide of moenomycin forms extensive interactions, involving a number of conserved amino acids from the jaw and head subdomains, in the complex structures (Sung et al., 2009 ▶; Lovering et al., 2007 ▶). In addition, the orientation of the inhibitor in the enzyme active site also indicates that lipid II is the acceptor and the growing chain is the donor in glycosyl-bond formation (Lovering et al., 2007 ▶; Terrak et al., 2008 ▶). Specifically, the essential glutamate of motif I functions as the active-site general base and deprotonates the sugar head group of lipid II, acting as an acceptor for nucleophilic attack on the reducing end of the growing glycan chain, which acts as a donor. The reaction results in the formation of a β-1,4-glycosidic linkage with inversion of the configuration at the anomeric C1 C atom of MurNAc.
The multimodular class A PBP4 encoded by lmo2229 from Listeria monocytogenes is the major bifunctional peptidoglycan synthase, harbouring both GT and TP activities (Zawadzka-Skomial et al., 2006 ▶). The GT and TP activities of this enzyme are specifically inhibited by treatment with moenomycin A and penicillin, respectively. In addition, the overall fold of PBP4 may be most similar to that of PBP2 (PDB code 3dwk; Lovering et al., 2008) from S. aureus because PBP4 shows the highest sequence identity to PBP2 (33%) among solved PBP structures. However, PBP4 from L. monocytogenes belongs to a different subclass to PBP2 from S. aureus on the basis of amino-acid sequence alignment (Sauvage et al., 2008 ▶). Furthermore, the two proteins are different with respect to catalytic efficiency and substrate affinity, implying that their susceptibility to moenomycin might vary.
We have initiated determination of the three-dimensional structure of PBP4 from L. monocytogenes to provide structural information for the development of new antibiotic agents based on moenomycin. Here, we report the cloning, overexpression, purification, crystallization and preliminary X-ray crystallographic analysis of this enzyme.
2. Materials and methods
2.1. Cloning, expression and purification of PBP4
The gene encoding the putative penicillin-binding protein PBP4 (UniProt accession No. Q8Y547) was amplified by the polymerase chain reaction (PCR) with Pfu DNA polymerase from chromosomal DNA of L. monocytogenes EGD. The primers were 5′-GGAAG GATCCGGTTTAGAGTCAGCCACGATTATT-3′ for the forward primer and 5′-CCCGGCTCGAGTTAATTACCTATCGAATCGATTAAG-3′ for the reverse primer, containing BamHI and XhoI sites (bold). The PCR product was subcloned into the pProExHTb vector (Invitrogen, USA) to generate an expression plasmid encoding residues 73–714 of PBP4 with an N-terminal His6 tag. The lmo2229 gene insertion was confirmed by DNA sequencing. The resulting expression vector pProExHTb:lmo2229 was transformed into E. coli BL21(DE3)-RIL cells, which were then grown at 310 K in Luria–Bertani medium containing 100 µg ml−1 ampicillin and 34 µg ml−1 chloramphenicol until the OD600 reached ∼0.8. After induction with 0.5 mM isopropyl β-d-1-thiogalactopyranoside at 291 K for a further 12 h, the cells were harvested by centrifugation at 5000g at 277 K. The cell pellet was resuspended in ice-cold buffer A (30 mM Tris–HCl pH 8.0, 0.3 M NaCl) and lysed by sonication. The lysate was centrifuged at 15 000g for 30 min and the supernatant was loaded onto Ni–NTA resin (Qiagen, USA) equilibrated with buffer A. After washing with buffer A containing 20 mM imidazole, the bound protein was eluted using a solution consisting of 30 mM Tris–HCl pH 8.0, 50 mM NaCl and 200 mM imidazole. The eluted sample was digested overnight with TEV protease (Invitrogen, USA) at a ratio of 1:100(w:w) at 277 K to remove the hexahistidine tag. The target protein was bound to a 5 ml HiTrap Q column (GE Healthcare, Sweden) and eluted with a 20-column-volume linear gradient from 50 to 500 mM NaCl in a buffer consisting of 30 mM Tris–HCl pH 8.0. Finally, the protein was further purified using a Superdex 200 (GE Healthcare) column equilibrated in 10 mM Tris–HCl pH 8.0 and 100 mM NaCl. The fractions of PBP4 eluting as a monomer were pooled and concentrated to 20 mg ml−1. Aliquots were flash-frozen in liquid nitrogen and stored at 193 K for subsequent use in crystallization. The purity of the protein was greater than 95% based on SDS–PAGE analysis (data not shown).
2.2. Crystallization
Truncated PBP4 (residues 73–714) with an additional five amino acids (GHMGS) at the N-terminus was crystallized by the sitting-drop vapour-diffusion method in 96-well Intelli-Plates (Hampton Research, USA). Initial crystallization conditions were screened by the sparse-matrix method (Jancarik et al., 1991 ▶) using the commercial kits Index HT, PEG/Ion HT, Crystal Screen HT, SaltRX HT (Hampton Research, California, USA), Wizard I, II, Cryo I and II (Emerald BioSystems, Washington, USA). All crystallization trials were carried out at 295 K. For screening, 0.4 µl protein solution was mixed with 0.4 µl reservoir solution and equilibrated against 70 µl reservoir solution. To obtain crystals suitable for X-ray diffraction, initial crystals were further optimized by varying the concentration of PEG 3350, ammonium tartrate dibasic and protein.
2.3. X-ray data collection
For X-ray data collection, a single crystal was immersed briefly into reservoir solution containing 15% glycerol as a cryoprotectant and immediately flash-cooled in a 100 K nitrogen stream. Native X-ray diffraction data were collected with an ADSC Q315r CCD detector on beamline 4A of Pohang Accelerator Laboratory (PAL; Republic of Korea) using 1° oscillations at a crystal-to-detector distance of 250 mm. The crystal was exposed for 3 s per image. A data set was collected to 2.1 Å resolution from a single crystal. The data were indexed and scaled with the HKL-2000 software package (Otwinowski & Minor, 1997 ▶). The data-collection statistics are summarized in Table 1 ▶.
Table 1. Diffraction statistics.
Values in parentheses are for the highest resolution shell. A 0σ cutoff filter was applied during the scaling process.
| X-ray source | Beamline 4A, PAL |
| Wavelength (Å) | 1.000 |
| Resolution (Å) | 30.0–2.1 (2.17–2.10) |
| Space group | P21212 |
| Unit-cell parameters (Å) | a = 84.6, b = 127.8, c = 54.9 |
| Completeness (%) | 96.7 (83.2) |
| Rmerge† (%) | 5.8 (32.1) |
| Multiplicity | 7.5 (3.1) |
| Average I/σ(I) | 21.3 (2.0) |
R
merge = 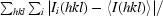
 , where Ii(hkl) and 〈I(hkl)〉 are the observed intensity and the mean intensity of related reflections, respectively.
, where Ii(hkl) and 〈I(hkl)〉 are the observed intensity and the mean intensity of related reflections, respectively.
3. Results and discussion
To facilitate purification and crystallization, the N-terminal 72 residues of PBP4, which contain the signal sequence and a putative transmembrane helix, were not included in the expression construct. The resulting construct is predicted to contain both the intact functional GT and TP domains as observed in the S. aureus PBP2 structures (Lovering et al., 2007 ▶). Recombinant PBP4 protein was successfully overexpressed and purified to homogeneity using sequential chromatographic steps. The yield of purified protein was ∼8 mg per litre of E. coli culture. Initial crystals were grown from PEG/Ion Screen condition No. 38 consisting of 0.2 M ammonium tartrate dibasic, 20%(w/v) PEG 3350 as clustered thin plates that were unsuitable for X-ray diffraction analysis. After optimization of the crystallization conditions, shiny single crystals were obtained by hanging-drop vapour diffusion by mixing equal volumes (2 µl) of protein solution (15 mg ml−1) and reservoir solution composed of 22%(w/v) PEG 3350 and 0.22 M ammonium tartrate dibasic pH 6.6. Thick plate-shaped crystals of PBP4 grew within 7 d to maximal dimensions of roughly 0.2 × 0.3 × 0.5 mm (Fig. 1 ▶). The crystals were transparent and colourless. A PBP4 crystal diffracted to a resolution of 2.1 Å (Fig. 2 ▶) and belonged to space group P21212, with unit-cell parameters a = 84.6, b = 127.8, c = 54.9 Å. The diffraction data set was 96.7% complete, with an R merge of 5.8% (Table 1 ▶). The asymmetric unit contained one PBP4 molecule of 69 kDa, resulting in a crystal volume per protein weight of 2.15 Å3 Da−1 and a solvent content of 43% (Matthews, 1968 ▶). The TP domains of PBP enzymes share a common fold, with root-mean-square deviations (r.m.s.d.s) ranging between 1.4 and 2.9 Å (Macheboeuf et al., 2006 ▶), indicating that molecular replacement would be the first choice for structure determination. Molecular replacement was attempted using the structure of PBP2 (PDB entry 3dwk; Lovering et al., 2008 ▶) as a search model in the programs MOLREP (Vagin & Teplyakov, 2010 ▶) and Phaser (McCoy, 2007 ▶), but these attempts were not successful. This result indicates that the relative orientation between the TP and GT domains might vary despite the two proteins sharing high sequence identity (33%). Sequence analysis revealed that PBP4 contains 18 nonterminal methionine residues. To solve the structure, selenomethionine-substituted PBP4 is being prepared for phasing by multiple anomalous dispersion (MAD).
Figure 1.

A crystal of L. monocytogenes PBP4. The crystal grew within one week at 295 K to maximum dimensions of approximately 0.2 × 0.3 × 0.5 mm.
Figure 2.

Representative X-ray diffraction image for L. monocytogenes PBP4. The crystal was exposed for 3 s over a 1° oscillation range. The circles superimposed onto the diffraction pattern correspond to resolutions of 6.0, 3.5 and 2.0 Å. In the magnified inset, diffraction spots extending to 2.1 Å resolution can be observed.
Acknowledgments
This work was supported by the Marine Extreme Genome Research Center Program of the Ministry of Maritime of Land, Transportation and Maritime Affairs, Republic of Korea.
References
- Goffin, C. & Ghuysen, J. M. (1998). Microbiol. Mol. Biol. Rev. 62, 1079–1093. [DOI] [PMC free article] [PubMed]
- Halliday, J., McKeveney, D., Muldoon, C., Rajaratnam, P. & Meutermans, W. (2006). Biochem. Pharmacol. 71, 957–967. [DOI] [PubMed]
- Heijenoort, J. van (2001). Glycobiology, 11, 25R–36R. [DOI] [PubMed]
- Höltje, J. V. (1998). Microbiol. Mol. Biol. Rev. 62, 181–203. [DOI] [PMC free article] [PubMed]
- Jancarik, J., Scott, W. G., Milligan, D. L., Koshland, D. E. & Kim, S.-H. (1991). J. Mol. Biol. 221, 31–34. [DOI] [PubMed]
- Lovering, A. L., de Castro, L. H., Lim, D. & Strynadka, N. C. (2007). Science, 315, 1402–1405. [DOI] [PubMed]
- Lovering, A. L., De Castro, L. & Strynadka, N. C. (2008). J. Mol. Biol. 383, 167–177. [DOI] [PubMed]
- Macheboeuf, P., Contreras-Martel, C., Job, V., Dideberg, O. & Dessen, A. (2006). FEMS Microbiol. Rev. 30, 673–691. [DOI] [PubMed]
- Matthews, B. W. (1968). J. Mol. Biol. 33, 491–497. [DOI] [PubMed]
- McCoy, A. J. (2007). Acta Cryst. D63, 32–41. [DOI] [PMC free article] [PubMed]
- Otwinowski, Z. & Minor, W. (1997). Methods Enzymol. 276, 307–326. [DOI] [PubMed]
- Popham, D. L. & Young, K. D. (2003). Curr. Opin. Microbiol. 6, 594–599. [DOI] [PubMed]
- Sauvage, E., Kerff, F., Terrak, M., Ayala, J. A. & Charlier, P. (2008). FEMS Microbiol. Rev. 32, 234–258. [DOI] [PubMed]
- Sung, M.-T., Lai, Y.-T., Huang, C.-Y., Chou, L.-Y., Shih, H.-W., Cheng, W.-C., Wong, C.-H. & Ma, C. (2009). Proc. Natl Acad. Sci. USA, 106, 8824–8829. [DOI] [PMC free article] [PubMed]
- Terrak, M., Ghosh, T. K., van Heijenoort, J., Van Beeumen, J., Lampilas, M., Aszodi, J., Ayala, J. A., Ghuysen, J. M. & Nguyen-Distèche, M. (1999). Mol. Microbiol. 34, 350–364. [DOI] [PubMed]
- Terrak, M., Sauvage, E., Derouaux, A., Dehareng, D., Bouhss, A., Breukink, E., Jeanjean, S. & Nguyen-Distèche, M. (2008). J. Biol. Chem. 283, 28464–28470. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Wright, G. D. (2007). Science, 315, 1373–1374. [DOI] [PubMed]
- Young, K. D. (2003). Mol. Microbiol. 49, 571–580. [DOI] [PubMed]
- Zawadzka-Skomial, J., Markiewicz, Z., Nguyen-Distèche, M., Devreese, B., Frère, J.-M. & Terrak, M. (2006). J. Bacteriol. 188, 1875–1881. [DOI] [PMC free article] [PubMed]